

Abstract
Novel α,β-CH2 and β,γ-NH (1a) or α,β-NH and β,γ-CH2 (1b) “Met-Im” dTTPs were synthesized via monodemethylation of triethyl-dimethyl phosphorimido-bisphosphonate synthons (4a, 4b), formed via a base-induced [1,3]-rearrangement of precursors (3a, 3b) in a reaction with dimethyl or diethyl phosphochloridate. Anomerization during final bromotrimethylsilane (BTMS) deprotection after Mitsunobu conjugation with dT was avoided by microwave conditions. 1a was 9-fold more potent in inhibiting DNA polymerase β, attributed to an NH-group interaction with R183 in the active site.
DNA polymerase (pol) β fills the short gaps in DNA during base excision repair (BER) of damaged DNA.1 Pol β is tightly regulated in normal cells, but is often mutated and/or overexpressed in cancer cells.2 Selective inhibition of pol β to prevent repair of drug-damaged DNA is therefore a promising approach for cancer therapy.3
dNTP analogues are critically important tools to study the mechanism and fidelity of DNA processing enzymes such as DNA polymerases and also are a basis for drug design targeting these enzymes in viral or cancer cells. Modifications of natural dNTP substrates in their triphosphate moiety have been recently utilized to probe the fidelity and catalytic mechanism of pol β.4 Typically, when modification is carried out at the β,γ-bridge position, the resulting analogue is a substrate of pol β with altered enzyme affinity and leaving group properties. By varying the stereoelectronic properties of a substituted β,γ-methylene bridge (Figure 1A), correlations of log kpol with leaving group pKa4 were used to identify the rate-determining step (RDS) in the catalysis of nucleotide insertion by the enzyme.5 Alternatively, if CH2, CXY, or NH replaces the Pα–O–Pβ bridging oxygen, insertion catalyzed by pol β is prevented, and the analogue is an inhibitor, but not a substrate (Figure 1B).6
Figure 1.
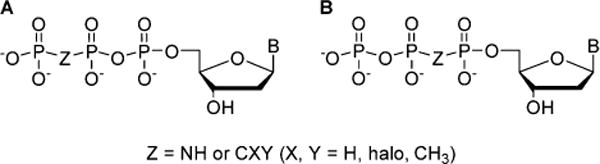
Singly bridge-modified dNTP analogues as (A) substrates and (B) inhibitors of pol β.5,6
The bond lengths and angles of the P–N–P moiety more closely resemble those of the natural P–O–P link in comparison with the P–C–P parameters,7 which are significantly different. Thus, using CH2 as both the α,β and β,γ linkage may perturb the geometry of the ternary complex with the enzyme.8 A further concern with a doubled CH2 linkage is the divergence in polarity of methylene from oxygen.9 One might consider a double CF2 linkage to address this problem; however, in an analogue wherein a CF2 group replaced both oxygen bridges in dTTP, the pol β affinity was drastically decreased.10 Analysis of crystal structures of the ternary complexes of (α,β)- or (β,γ)-CH2-dNTP with pol β and DNA11 suggests that the structural perturbations introduced by a single CH2 linkage can be accommodated.6a,b Substituting both the (α,β)- and (β,γ)-linking oxygens with imido linkages limits modification relative to the more synthetically versatile methylene group. We therefore considered two new scaffolds to explore selective inhibition of pol β, in which the Pα–O–Pβ and Pβ–O–Pγ oxygens are replaced by either a methylene (CH2) or an imido (NH) group in alternation: “Met-Im” nucleotides (Figure 2).
Figure 2.
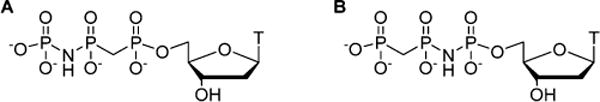
(A) (α,β)-CH2-(β,γ)-NH-dTTP (1a) and (B) (α,β)-NH-(β,γ)-CH2-dTTP (1b) “Met-Im” nucleotides.
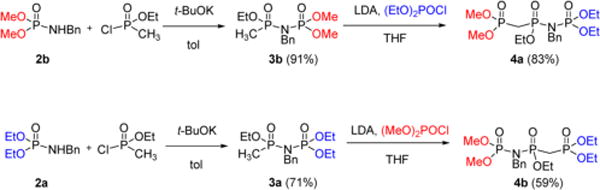
Our approach to a general pairwise synthesis of “Met-Im” nucleotides centered on construction of the isomeric pentaalkyl phosphorimido-bisphosphonate synthons 4a and 4b (Scheme 1) which have terminal (EtO)2P(O) or (MeO)2P(O) groups joined via a NBn or CH2 bridge to a central EtOP(O). Regioselective removal of one terminal methyl would expose a free P(O)OH, which would become the Pα group in the final nucleotide analogue product (1a or 1b) after coupling to dT and deprotection.
Scheme 1.
Flip Synthesis of 4a/b
To synthesize 4b, we initially prepared precursor 3b from dimethyl benzylphosphoramidate 2b12 and ethyl methylphosphonochloridate13 in the presence of t-BuOK (Scheme 1). We envisioned next converting the lithium salt of 3b into 4b by reaction with diethyl chlorophosphate. In the event, the LDA-promoted reaction of 3b proceeded with the formation of the isomeric precursor 4a (Scheme 1), apparently via an anionic 1,3-rearrangement14 of the initially formed carbanion. The driving force of this N → C rearrangement14a–e is likely formation of the 3b carbanion, followed by the rapid migration of the dimethoxyphosphoryl group to the anionic carbon and then reaction of the resulting amido anion with diethyl chlorophosphate, yielding the “flipped” product 4a (Scheme 2).
Scheme 2.
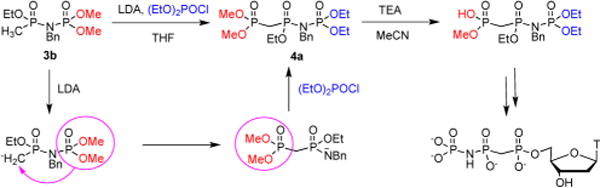
[1,3] N → C Migration Mechanism
This result suggested that the original intermediate target, 4b, should be accessible via the same “flip” route simply by swapping the ethyl groups in (EtO)2P(O)Cl with the methyl groups in 3b, resulting in 3a which indeed reacted with (MeO)2P(O)Cl under similar conditions forming 4b (Scheme 1).
The isomers 4a and 4b were characterized by their 1H, 31P, and 13C NMR spectra and by MS. The 31P resonance at δ 22.7 ppm is assigned to a (MeO)2P(O)CH2 in 4a (Δδ +3 ppm relative to (EtO)2P(O)CH2 in 4b), both coupled (JPP = 4 Hz) to the same phosphorus at δ ~23 ppm, assigned to the central P(O)OEt. The most upfield resonance (δ 3 ppm) is then (EtO)2P(O)NBn in 4a, with the (MeO)2P(O)NBn resonance in 4b again observed 3 ppm downfield (both with JPP = 20 Hz to the central phosphorus nucleus).
The structures of 4a and 4b were further established by 1H–31P gHMBC NMR. For 4a (Figure S14) a strong cross-peak is observed for the protons of two OCH3 groups (3.84 and 3.79 ppm) and the phosphonate 31P resonance (22.7 ppm), while no cross-peak is observed for the terminal phosphorimide 31P resonance (2.79 ppm). In contrast, the 1H–31P gHMBC NMR of 4b (Figure S18) reveals a strong cross-peak for the OCH3 protons (3.70 and 3.54 ppm) and phosphorimide 31P resonance (6 ppm), while no cross-peak is observed for these protons and the terminal phosphonate 31P resonance (19.65 ppm).
The same procedure was utilized for the synthesis of 1a and 1b (Scheme 3). After selective removal of a single methyl group from 4a or 4b with TEA,15 the resulting salt was converted by DOWEX H+ to the corresponding acid (5a or 5b) which was coupled to thymidine under Mitsunobu conditions16 to form 6a or 6b. Debenzylation by catalytic hydrogenolysis then gave 7a or 7b (Scheme 3).5e
Scheme 3.
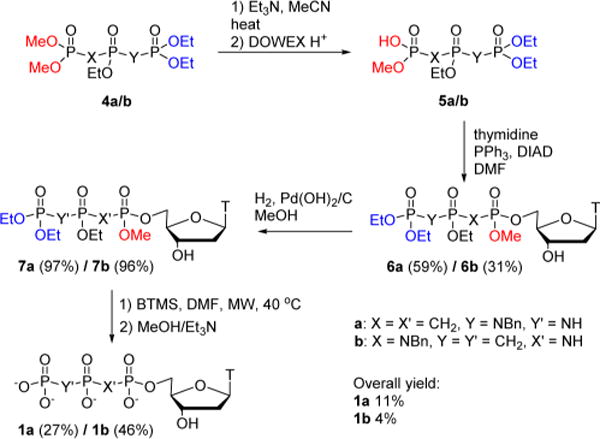
Synthesis of “Met-Im” Nucleotides 1a/b
The final deprotection step, simultaneous removal of the remaining methyl and three ethyl groups, was effected by silyldealkylation with BTMS followed by neutral hydrolysis.17 The major product had the anticipated MS ([M – H]− = 478), but in a preliminary pol β dTTP incorporation assay exhibited very low (Ki > 1 mM) inhibitory potency. Careful analysis of the NMR (Figure S28) suggested that the major product had undergone extensive anomerization (the undesired α-anomer is usually not the main product when β-ribonucleotides are treated with BTMS;18 however, anomerization has been observed for some β-deoxyribonucleotides).19
Fortunately, we discovered that microwave (MW) acceleration20 significantly reduced anomerization: treatment of 7a with microwave irradiation for 30 min at 40 °C followed by hydrolysis gave 80% of the desired β-anomer 1a (Scheme 4), which was purified using two-stage preparative (strong anion exchange (SAX) followed by C18) HPLC.5b
Scheme 4.
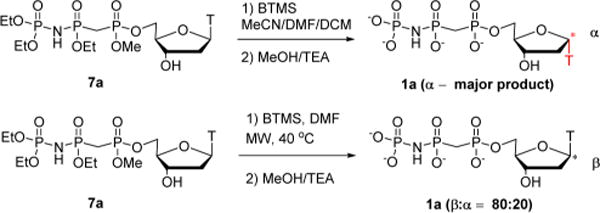
Microwave Reaction Abated Anomerization of Nucleotides 1 during BTMS Reaction
Similarly, deprotection of 7b was achieved in 7 min at 40 °C, with minor α-anomer formation, and the pure β-product 1b could again be isolated by preparative HPLC.
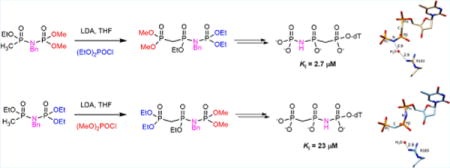
Inhibition of pol β by 1a or 1b at variable concentrations for a constant concentration of dTTP was determined using a standard gel assay (Figure 3A). For each inhibitor concentration, an observed rate was determined and then plotted, and the data were fit to a hyperbolic decay curve from which the Ki was calculated (Figure 3; see Supporting Information for details).
Figure 3.
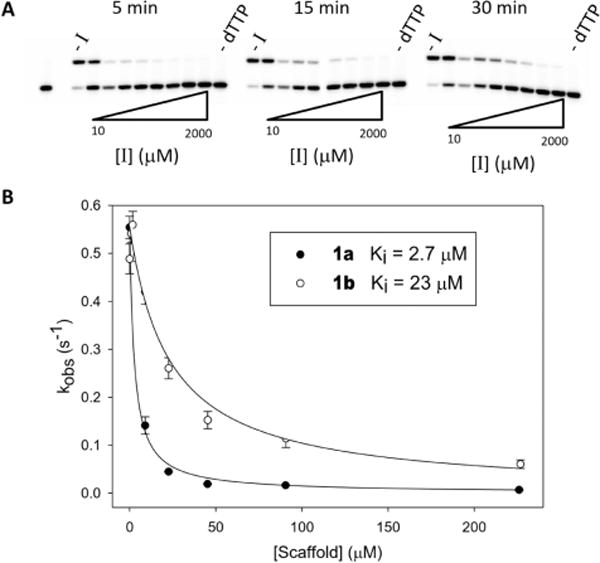
Inhibition of pol β by “Met-Im” dTTP analogues. (A) Representative gels for inhibition by 1a. Aliquots were quenched at 5, 15, and 30 min, and the DNA was run on denaturing polyacrylamide gels to separate unextended from extended primer. The first lane corresponds to reaction without inhibitor. The final lane in each set is the reaction without dTTP, showing that the inhibitor is not incorporated into DNA by pol β. (B) Hyperbolic decay fits of data from inhibition reactions. Filled circles represent 1a, and open circles, 1b. Error bars represent σ for three independent experiments.
The Ki values for the two inhibitors differed by 9-fold (1a, 2.7 μM; 1b, 23 μM).
The X-ray crystal structures of 1a and 1b in ternary complexes with pol β and DNA (Table S1) reveal that both bound inhibitors assume conformations similar to those of the natural nucleotide (Figure 4). However, the inhibitor having its NH group at the β,γ-bridging position (1a) has a water-mediated H-bonding interaction between its NH and R183 in the active site. This interaction is unavailable for 1b which has a β,γ-CH2 at the β,γ-bridge, suggesting that some or all of the observed Ki difference could be attributed to it (the α,β-NH in 1b has no apparent interactions with proximal active site residues).
Figure 4.
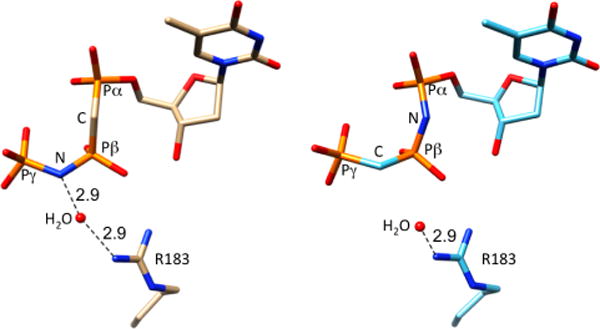
1a (left) and 1b (right) in ternary complex with pol β and DNA. R183 makes a water-mediated H-bond with the β,γ-NH in 1a. A corresponding interaction between R183 and the α,β-NH of 1b is not possible.
In summary, we have described the synthesis of two novel (α,β):(β,γ)-CH2/NH-dTTP isomers that will permit exploration of active site interactions of DNA polymerases both near the β,γ-bridge atom of the enzyme bound nucleotide and in a more interior region of the active site adjacent to the locus of catalysis. Furthermore, they constitute isomeric scaffolds for the future introduction of stereodefined substituents5e at either CH2 position to modulate inhibitor activity.
X-ray crystallographic studies of the inhibitor-enzyme ternary complex (with bound DNA primer and template) reveal that (β,γ)-NH is uniquely capable of a structural water-mediated H-bond interaction with Arg183, which is a promising “anchor” to bind selective inhibitors of DNA polymerase β.5,6
The synthetic strategy devised to prepare 1a and 1b should be broadly applicable to the synthesis of other “Met-Im” nucleotides by simply coupling the appropriate nucleoside with synthons 4a and 4b. Their synthesis takes advantage of a [1,3] N → C P(O)(OR2) migration, which neatly provides access to both compounds by a simple swap of terminal alkyl ester groups. The observed suppression of unwanted anomerization by microwave acceleration in the BTMS-mediated deprotection of 7a and 7b may also be of general utility and further extends the scope of BTMS for preparation of phosphonic acids incorporating sensitive functional groups.
Supplementary Material
Acknowledgments
This work was supported by NIH Grant U19CA177547, and, in part, by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (project numbers Z01-ES050158 and Z01-ES050159). The authors would like to thank Ms. Inah Kang for assistance in preparing the manuscript.
Footnotes
Experimental details and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.(a) Beard WA, Wilson SH. Chem Rev. 2006;106:361–382. doi: 10.1021/cr0404904. [DOI] [PubMed] [Google Scholar]; (b) Wilson SH. Mutat Res. 1998;407:203–215. doi: 10.1016/s0921-8777(98)00002-0. [DOI] [PubMed] [Google Scholar]
- 2.(a) Albertella MR, Lau A, O’Connor MJ. DNA Repair. 2005;4:583–593. doi: 10.1016/j.dnarep.2005.01.005. [DOI] [PubMed] [Google Scholar]; (b) Bergoglio V, Canitrot Y, Hogarth L, Minto L, Howell SB, Cazaux C, Hoffmann J-S. Oncogene. 2001;20:6181–6187. doi: 10.1038/sj.onc.1204743. [DOI] [PubMed] [Google Scholar]; (c) Chan K, Houlbrook S, Zhang Q-M, Harrison M, Hickson ID, Dianov GL. Mutagenesis. 2007;22:183–188. doi: 10.1093/mutage/gel070. [DOI] [PubMed] [Google Scholar]; (d) Dalal S, Hile S, Eckert KA, Sun K-w, Starcevic D, Sweasy JB. Biochemistry. 2005;44:15664–15673. doi: 10.1021/bi051179z. [DOI] [PubMed] [Google Scholar]; (e) Louat T, Servant L, Rols M-P, Bieth A, Teissie J, Hoffmann J-S, Cazaux C. Mol Pharmacol. 2001;60:553–558. [PubMed] [Google Scholar]; (f) Servant L, Bieth A, Hayakawa H, Cazaux C, Hoffmann J-S. J Mol Biol. 2002;315:1039–1047. doi: 10.1006/jmbi.2001.5307. [DOI] [PubMed] [Google Scholar]; (g) Starcevic D, Dalal S, Sweasy JB. Cell Cycle. 2004;3:998–1001. [PubMed] [Google Scholar]; (h) Sweasy JB, Lauper JM, Eckert KA. Radiat Res. 2006;166:693–714. doi: 10.1667/RR0706.1. [DOI] [PubMed] [Google Scholar]
- 3.(a) Lange SS, Takata K-i, Wood RD. Nat Rev Cancer. 2011;11:96–110. doi: 10.1038/nrc2998. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sharma RA, Dianov GL. Mol Aspects Med. 2007;28:345–374. doi: 10.1016/j.mam.2007.06.002. [DOI] [PubMed] [Google Scholar]; (c) Barakat KH, Gajewski MM, Tuszynski JA. Drug Discovery Today. 2012;17:913–920. doi: 10.1016/j.drudis.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 4.McKenna CE, Kashemirov BA, Peterson LW, Goodman MF. Biochim Biophys Acta, Proteins Proteomics. 2010;1804:1223–1230. doi: 10.1016/j.bbapap.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 5.(a) Batra VK, Pedersen LC, Beard WA, Wilson SH, Kashemirov BA, Upton TG, Goodman MF, McKenna CE. J Am Chem Soc. 2010;132:7617–7625. doi: 10.1021/ja909370k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McKenna CE, Kashemirov BA, Upton TG, Batra VK, Goodman MF, Pedersen LC, Beard WA, Wilson SH. J Am Chem Soc. 2007;129:15412–15413. doi: 10.1021/ja072127v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Oertell K, Chamberlain BT, Wu Y, Ferri E, Kashemirov BA, Beard WA, Wilson SH, McKenna CE, Goodman MF. Biochemistry. 2014;53:1842–1848. doi: 10.1021/bi500101z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Sucato CA, Upton TG, Kashemirov BA, Osuna J, Oertell K, Beard WA, Wilson SH, Florian J, Warshel A, McKenna CE, Goodman MF. Biochemistry. 2008;47:870–879. doi: 10.1021/bi7014162. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wu Y, Zakharova VM, Kashemirov BA, Goodman MF, Batra VK, Wilson SH, McKenna CE. J Am Chem Soc. 2012;134:8734–8737. doi: 10.1021/ja300218x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Upton TG, Kashemirov BA, McKenna CE, Goodman MF, Prakash GKS, Kultyshev R, Batra VK, Shock DD, Pedersen LC, Beard WA, Wilson SH. Org Lett. 2009;11:1883–1886. doi: 10.1021/ol701755k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chamberlain BT, Batra VK, Beard WA, Kadina AP, Shock DD, Kashemirov BA, McKenna CE, Goodman MF, Wilson SH. ChemBioChem. 2012;13:528–530. doi: 10.1002/cbic.201100738. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Batra VK, Beard WA, Shock DD, Krahn JM, Pedersen LC, Wilson SH. Structure. 2006;14:757–766. doi: 10.1016/j.str.2006.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yount RG, Babcock D, Ballantyne W, Ojala D. Biochemistry. 1971;10:2484–2489. doi: 10.1021/bi00789a009. [DOI] [PubMed] [Google Scholar]
- 8.Labataille P, Pelicano H, Maury G, Imbach J-L, Gosselin G. Bioorg Med Chem Lett. 1995;5:2315–2320. [Google Scholar]
- 9.(a) McKenna CE, Shen PD. J Org Chem. 1981;46:4573–4576. [Google Scholar]; (b) Blackburn GM, Kent DE, Kolkmann F. J Chem Soc, Chem Commun. 1981:1188–1190. [Google Scholar]; (c) Leswara ND, Shen PD, McKenna CE. Fed Proc. 1982;41:860–860. [Google Scholar]; (d) Berkowitz DB, Bose M. J Fluorine Chem. 2001;112:13–33. [Google Scholar]
- 10.Surya Prakash GK, Zibinsky M, Upton TG, Kashemirov BA, McKenna CE, Oertell K, Goodman MF, Batra VK, Pedersen LC, Beard WA, Shock DD, Wilson SH, Olah GA. Proc Natl Acad Sci USA. 2010;107:15693–15698. doi: 10.1073/pnas.1007430107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Sucato CA, Upton TG, Kashemirov BA, Batra VK, Martinek V, Xiang Y, Beard WA, Pedersen LC, Wilson SH, McKenna CE, Florian J, Warshel A, Goodman MF. Biochemistry. 2007;46:461–471. doi: 10.1021/bi061517b. [DOI] [PubMed] [Google Scholar]; (b) Batra VK, Beard WA, Shock DD, Pedersen LC, Wilson SH. Mol Cell. 2008;30:315–324. doi: 10.1016/j.molcel.2008.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glidewell C, Pogorzelec PJ. J Chem Educ. 1980;57:740–741. [Google Scholar]
- 13.Briseno-Roa L, Hill J, Notman S, Sellers D, Smith AP, Timperley CM, Wetherell J, Williams NH, Williams GR, Fersht AR, Griffiths AD. J Med Chem. 2006;49:246–255. doi: 10.1021/jm050518j. [DOI] [PubMed] [Google Scholar]
- 14.(a) Hammerschmidt F, Hanbauer M. J Org Chem. 2000;65:6121–6131. doi: 10.1021/jo000585f. [DOI] [PubMed] [Google Scholar]; (b) He Z, Modro TA. Synthesis. 2000:565–570. [Google Scholar]; (c) Hodgson DM, Humphreys PG, Xu Z, Ward JG. Angew Chem, Int Ed. 2007;46:2245–2248. doi: 10.1002/anie.200604920. [DOI] [PubMed] [Google Scholar]; (d) Hodgson DM, Xu Z. Beilstein J Org Chem. 2010;6:978–983. doi: 10.3762/bjoc.6.110. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Jardine AM, Vather SM, Modro TA. J Org Chem. 1988;53:3983–3985. [Google Scholar]; (f) Calogeropoulou T, Hammond GB, Wiemer DF. J Org Chem. 1987;52:4185–4190. [Google Scholar]
- 15.Ahlmark MJ, Vepsalainen JJ. Tetrahedron. 1997;53:16153–16160. [Google Scholar]
- 16.Mitsunobu O, Yamada M. Bull Chem Soc Jpn. 1967;40:2380–2382. [Google Scholar]
- 17.McKenna CE, Higa MT, Cheung NH, McKenna MC. Tetrahedron Lett. 1977:155–158. [Google Scholar]
- 18.(a) Taylor SD, Mirzaei F, Bearne SL. Org Lett. 2006;8:4243–4246. doi: 10.1021/ol0615432. [DOI] [PubMed] [Google Scholar]; (b) Taylor SD, Mirzaei F, Bearne SL. J Org Chem. 2008;73:1403–1412. doi: 10.1021/jo702249j. [DOI] [PubMed] [Google Scholar]
- 19.(a) Brossette T, Le Faou A, Goujon L, Valleix A, Creminon C, Grassi J, Mioskowski C, Lebeau L. J Org Chem. 1999;64:5083–5090. doi: 10.1021/jo982502p. [DOI] [PubMed] [Google Scholar]; (b) Van Poecke S, Sinnaeve D, Martins JC, Balzarini J, Van Calenbergh S. Nucleosides Nucleotides Nucleic Acids. 2012;31:256–272. doi: 10.1080/15257770.2012.654876. [DOI] [PubMed] [Google Scholar]
- 20.Seamon KJ, Hansen EC, Kadina AP, Kashemirov BA, McKenna CE, Bumpus NN, Stivers JT. J Am Chem Soc. 2014;136:9822–9825. doi: 10.1021/ja5035717. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.