

Abstract
Neuropeptide Y (NPY) is one of the most abundant neuropeptides in the mammalian brain and exerts a variety of physiological processes in humans via four different receptor subtypes Y1, Y2, Y4 and Y5. Y2 receptor is the most abundant Y subtype receptor in the central nervous system and implicated with food intake, bone formation, affective disorders, alcohol and drugs of abuse, epilepsy, pain, and cancer. The lack of small molecule non-peptidic Y2 receptor modulators suitable as in vivo pharmacological tools hampered the progress to uncover the precise pharmacological role of Y2. Only in recent years, several potent, selective and non-peptidic Y2 antagonists have been discovered providing the tools to validate Y2 receptor as a therapeutic target. This article reviews Y2 receptor modulators mainly non-peptidic antagonists and their structure-activity relationships.
Keywords: Neuropeptide Y, Y2 receptor, Antagonists, Agonists, Structure activity relationships, Brain-penetration, Non-peptidic
Introduction
Neuropeptide Y (NPY) is a highly conserved 36-amino acid peptide neurotransmitter, structurally and functionally related to the 36-amino acid pancreatic peptide (PYY) and pancreatic polypeptide (PP) and one of the most abundant neuropeptides in the mammalian brain. NPY is involved in a variety of physiological processes and exerts its actions in humans via four different receptor subtypes (Y1, Y2, Y4, Y5) that are expressed in both the central and peripheral nervous systems.1, 2 All NPY receptor subtypes belong to the family of G protein coupled receptors (GPCR) and mediate their biological responses via Gαi signaling pathways. Y2 receptors are the most abundant Y subtype receptor in the central nervous system (CNS) and widely expressed in the brain including hypothalamus, hippocampus, brain stem, amygdala and lateral septum.3-5 Y2 receptors are peripherally found in the liver, intestine, spleen, muscle and adipose tissue.6 Y2 receptors primarily act as presynaptic autoreceptors modulating endogenous NPY release and as heteroreceptors regulating the release of other neurotransmitters such as γ–amino butyric acid (GABA) and glutamate.7, 8 Several articles reviewed the role of Y2 receptors in various physiological and pathological processes and the potential therapeutic use of Y2 modulators.9-16 Y2 receptors are implicated with food intake, bone formation, affective disorders, alcohol and drugs of abuse, epilepsy, pain, and cancer.10 The pharmacological role of Y2 receptors are mostly investigated by knockout experiments, using peptidic Y2 agonists such as NPY, PYY(3-36) and Y2 selective antagonist BIIE0246. Several studies demonstrated the gut peptide PYY(3-36), a preferring Y2 agonist, reduced food intake and body-weight in animal models of rodents and primates.17, 18 The anorexigenic actions of PYY(3-36) were abolished in Y2 knockout mice and blocked by Y2 antagonist BIIE0246 in rats.18, 19 However, the anorexigenic actions of PYY(3-36) could not be repeated by some research groups and the potential use of PYY(3-36) as anti-obesity agent is a debate.17 Compelling evidence suggests NPY acts as an anti-convulsant and its actions are mediated via presynaptic Y2 receptors in the hippocampus by suppressing the glutamatergic synaptic transmission.14, 20, 21 The deletion of Y2 receptors or the blockade of Y2 receptors, by Y2 antagonist BIIE0246, completely abolished the anti-convulsant actions of NPY.22 Intracerebroventricular (i.c.v) injection of Y2 selective agonist AcPYY(24-36)-L31 suppressed seizures similarly to NPY in animal models of absence seizures.21 Mice lacking Y2 receptors displayed reduced anxiety and antidepressant-like behavior compared to wild type.23 The site-specific deletion of Y2 gene in the basolateral and central amygdala resulted in anxiolytic profile.24 BIIE0246 decreased anxiety in rats and induced antidepressant-like effects in mice following i.c.v administration.25, 26 NPY is anxiolytic and exerts potent actions via Y1 receptor. Antagonists of Y2 receptors are envisaged to increase endogenous NPY release, by blocking the Y2 receptor negative-feedback mechanism, which augments the activation of Y1 receptors and are thus believed to be anxiolytics.27 Studies also indicated the possible role of Y2 in alcohol intake.28 BIIE0246 administered i.c.v. suppressed operant self-administration of ethanol intake by rats with a history of alcohol dependence.29, 30 The studies demonstrated the hypothalamic Y2 receptors play a key role in the central regulation of bone formation.31 The specific deletion of Y2 receptors in the hypothalamus increases the rate of bone mineralization and formation by stimulating the osteoblast activity. Neuroblastomas release high levels of NPY that induces neuroblastoma cell proliferation and angiogenesis via Y2 receptors.32 BIIE0246 reduced tumor growth and decreased tumor vascularization.33 Despite Y2 role, the potential therapeutic use of NPY Y2 modulators, antagonists and agonists, are not yet clinically validated. The lack of small molecule non-peptidic Y2 receptor modulators suitable as in vivo pharmacological tools hampered the progress to uncover the precise pharmacological role of Y2 and to validate Y2 receptor as a potential therapeutic target. Only in recent years, several selective, non-peptidic and systemically active Y2 receptor antagonists were discovered, providing tools to elucidate the pharmacological role of Y2. This article reviews currently known Y2 modulators, mainly non-peptidic antagonists, and their structure-activity relationships (SAR).
NPY Y2 receptor antagonists
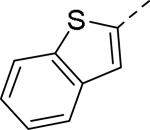
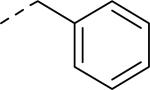
Doods et al. reported the first highly potent and selective peptide-like Y2 antagonist BIIE0246 (Fig. 1) that exhibited high affinity for Y2 receptor with an IC50 of 3.3 nM in a radio-ligand binding assay using [125I]NPY.34 In functional Ca+2 mobilization assay, when pre-incubated for 30 min, BIIE0246 behaved as an insurmountable antagonist.35 Recently, a selective radioligand [3H]UR-PLN196 (Fig. 1) for Y2 receptor was identified through modification of the guanidine moeity of BIIE0246.36, 37 The cold UR-PLN196 (Ki=9.9 nM) also inhibited agonist responses in an insurmountable fashion when pre-incubated for 20 min. BIIE0246 did not show binding for Y1, Y4, Y5 and a variety of receptors or enzymes up to 1 μM.38 However, Brothers et al. recently reported BIIE0246 had significant binding at α1a- adrenergic, μ- and κ-opioid receptors with Ki values of 360, 323 and 948 nM, respectively.39 Despite highly potent and selective (>100-fold), the use of BIIE0246 as a potential therapeutic agent and as in vivo pharmacological tool is limited due to its pseudo-peptidic nature, high molecular weight (896 Da), poor brain-penetration and off-target activity.39 Many efforts have been consequently focused on discovery of highly potent, selective and brain-penetrant non-peptidic Y2 receptor antagonists (Fig. 1). Bristol-Myers Squibb (BMS) identified hit compound 1 (IC50=10 μM) as a small molecule non-peptidic Y2 receptor ligand by high-throughput screening (HTS).40 SAR studies were explored to improve the affinity and to eliminate the potential metabolically labile functionalities, cinnamide and sulfur moieties. (Table 1). The replacement of cinnamic acid moiety with trans-cyclopropyl phenyl (2) and benzothiophene (3) improved the binding affinity by 6- and 10-fold, respectively, while with benzofuran (4) did not improve the affinity. Substituting the 3-position of the thiophene with a chloro group (5) was detrimental. The exploration of length of basic amine side chain suggested the three carbon linker was optimal for the affinity (not shown). The meta- and para-substituted aniline analog had no affinity (not shown), indicating the ortho-substitution was favorable for the binding affinity. The replacement of sulfur at the ortho-position of the aniline moiety either with a methylene group (6) or amide bond (7) abolished the affinity. Interestingly, the benzyl group at the ortho-position (8) improved the affinity by 2-fold (IC50=450 nM). However, no functional activity as well as selectivity over the sub-family of Y receptors was reported.
Figure 1.

Representative structures of neuropeptide Y Y2 receptor antagonists.
Table 1.
SAR and binding affinity data of Y2 receptor ligands based on functionalized diamines.40

| |||
|---|---|---|---|
| Compound | R1 | R2 | IC50 (μM)a |
| 1 (hit) |
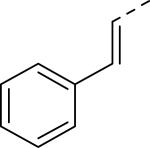
|
|
10 |
| 2 |
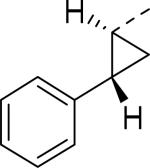
|
|
3.7 |
| 3 |
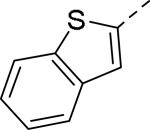
|
|
1.0 |
| 4 |
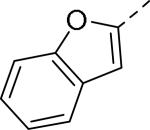
|
|
>10 |
| 5 |
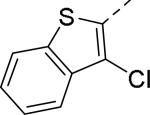
|
|
>10 |
| 6 |
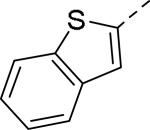
|
|
>10 |
| 7 |
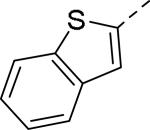
|
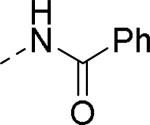
|
>10 |
| 8 |
|
|
0.45 |
50% inhibition of binding of radio ligand [125I]PYY.
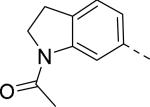
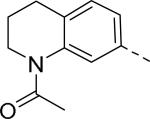
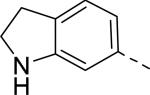
Researchers at Johnson and Johnson (JNJ) discovered a series of Y2 antagonists by HTS that had interestingly similar pharmacophore as Y2 ligands reported by BMS. The hit molecule 9 showed moderate affinity (IC50 = 4 μM, Table 2) in Y2 receptor binding assay.41 SAR studies were carried out systematically by modification of the indoline, cinnamide and piperidine moieties (Table 2-5). The modification of the N-acetyl group of the indoline moiety with formyl (10), ethyl glyoxylate (11) and methyl (12) were tolerable, but none of these analogs had better affinity. The un-substituted indoline (13), N-methyl sulfonyl (14) and N-trifluoromethyl acetyl (15) analog showed inferior affinity than the hit. The replacement of N-acetyl indoline by N-acetyl indole (16), N-acetyl tetrahydroquinoline (17) or 2-oxoindoline (18) decreased the affinity. (Table 2). Subsequently, efforts were focused on modification of the cinnamide moiety (Table 3). The olefin and carbonyl group of cinnamide were essential for the affinity as the saturated (19), non-carbonyl (20) and sulfonyl (21) analogs displayed weak affinity, while the trans- cyclopropyl analog (22) was nearly 3-fold less active. The Z-olefin isomer (23) and acetylene (24) analogs also showed lower affinity. Notably, the incorporation of an additional olefin (25) marginally improved the affinity. The removal of the phenyl of the cinnamide was detrimental for the affinity (26). The exploration of aromatic heterocycles such as pyridyl isomers (27, 28) and imidazole (29) considerably reduced the affinity, whereas thiophene (30) slightly increased the affinity. The investigation of substitutions on the phenyl ring of cinnamide revealed both electron-withdrawing and electron-donating groups were tolerable at the 3- and 4-position (31-39, Table 4), of which, substitution at the 3-position was slightly preferable with the exception of CF3 group. The presence of methyl (33), nitro (36) and cyano (34) groups at the 3-position of phenyl ring of the cinnamide improved the affinity, particularly the cyano by 4-fold. The substitution at both the 3- and 5-position (40-42) was not beneficial. Further modifications were made to N-benzyl group of the piperidine moiety of the hit molecule (43-48, Table 5). Replacing the benzyl group with benzoyl group (43) resulted in dramatic loss of the affinity, signifying the necessity of a basic amine group. Varying the carbon linker suggested the two carbon linker (44) was detrimental, whereas the three carbon linker (45) was slightly favorable. Remarkably, replacing the benzyl group with a cyclohexyl methyl (46) improved the affinity that was further increased by extending the linker by one methylene group (47). The cyclopentyl ethyl analog (48) showed comparable affinity to the cyclohexyl ethyl analog (47). Several modifications of the piperidine did not result in increase of the affinity (not shown). Combining the optimal modifications, cyano group at the 3-position of phenyl ring of the cinnamide moiety and cyclopentyl ethyl at N-1 of the piperidine, led to the discovery of JNJ-5207787 (Fig. 1) that displayed an IC50 of 100 nM in binding assay. JNJ-5207787 exhibited antagonist activity by inhibition of the PYY-stimulated [35 S]GTPγS binding with a pIC50 of 7.2 and displayed high selectivity (>100 fold) against human Y1, Y4 and Y5 sub-family receptors. It did not show binding affinity at concentrations up to 1 μM (< 50% inhibition) in a panel of 50 receptors, ion channels and transporters except for sodium channel 2.42 JNJ-5207787 was intraperitoneally (i.p.) bioavailable (33%), but not orally, with a plasma half-life of 2.03 h. JNJ-5207787 given i.p. (30 mg/kg, rats) showed brain/plasma ratio of 0.23 with a Cmax of 1351 ng/mL at 30 min. and occupied maximally 50% of Y2 receptor binding sites in the hypothalamic area. The doses higher than 30 mg/kg of JNJ-5207787 could not be administered due to the solubility and formulation difficulties. The use of JNJ 5207787 as a pharmacological tool is limited because of moderate antagonist activity and partial receptor occupancy.
Table 2.
SAR and binding affinity data of the modified indoline analogs.41

| |||||
|---|---|---|---|---|---|
| Compd | R | IC50 (μM)a | Compd | R | IC50 (μM)a |
| 9 (hit) |
|
4.0 | 14 |
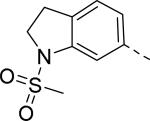
|
15 |
| 10 |
|
3.5 | 15 |
|
30 |
| 11 |
|
4.8 | 16 |
|
30 |
| 12 |
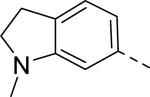
|
5.3 | 17 |
|
10 |
| 13 |
|
22 | 18 |
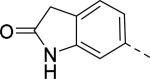
|
30 |
50% inhibition of binding of radio ligand [125I]PYY at human Y2 receptors.
Table 5.
Exploration of substitution on the piperidine nitrogen.41

| |||||
|---|---|---|---|---|---|
| Compd | R | IC50 (μM)a | Compd | R | IC50 (μM)a |
| 43 | C6H5CO | 29 | 46 | C6H11CH2 | 1.1 |
| 44 | C6H5(CH2)2 | 26 | 47 | C6H11(CH2)2 | 0.6 |
| 45 | C6H5(CH2)3 | 2.3 | 48 | C5H9(CH2)2 | 0.8 |
50% inhibition of binding of radio ligand [125I]PYY at human Y2 receptors.
Table 3.
SAR exploration of the cinnamide moiety.41

| |||||
|---|---|---|---|---|---|
| Compd | R | IC50 (μM)a | Compd | R | IC50 (μM)a |
| 19 |
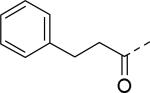
|
18 | 25 |
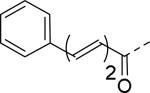
|
2.8 |
| 20 |
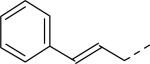
|
30 | 26 |
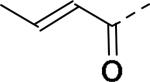
|
30 |
| 21 |
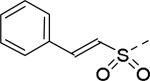
|
30 | 27 |
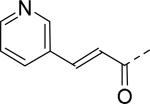
|
30 |
| 22 |
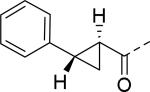
|
11 | 28 |
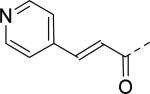
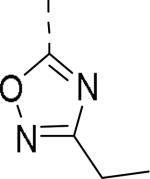
|
12 |
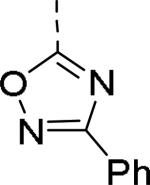
| 23 |
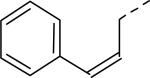
|
22 | 29 |
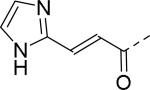
|
17 |
| 24 |
|
9.2 | 30 |
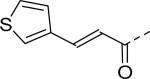
|
3.2 |
50% inhibition of binding of radio ligand [125I]PYY at human Y2 receptors.
Table 4.
Exploration of substitution on the phenyl ring of the cinnamide moiety.41

| |||||
|---|---|---|---|---|---|
| Compd | R | IC50 (μM)a | Compd | R | IC50 (μM)a |
| 31 | 3-F | 3.6 | 37 | 3-CF3 | 8.9 |
| 32 | 3-Cl | 3.0 | 38 | 4-Cl | 3.6 |
| 33 | 3-CH3 | 1.4 | 39 | 4-CF3 | 2.8 |
| 34 | 3-CN | 1.0 | 40 | 3,5-diF | 2.5 |
| 35 | 3-Br | 3.3 | 41 | 3,5-diCH3 | 3.9 |
| 36 | 3-NO2 | 1.9 | 42 | 3,5-diCl | 30 |
50% inhibition of binding of radio ligand [125I]PYY at human Y2 receptors.
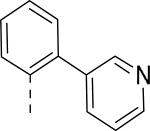
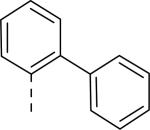
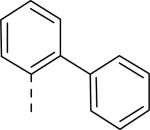
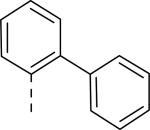
JNJ discovered a different series of novel and potent NPY Y2 antagonists by pharmacophore-directed virtual screening of the compound collection based on known Y2 ligands, including JNJ-5207787 and a neuropeptide Y analog. The lead (49, Table 6, IC50 = 240 nM) identified from the screening was originally synthesized as a part of microsomal triglyceride transfer protein (MTTP) inhibitors program.43,44 SAR studies were extensively carried out to improve the potency and to eliminate potentially the liable functionalities, methyl ester (metabolic stability), biaryl amide (solubility) and the 4-pyridine (CYP450 inhibitor). The exploration of different alkyl amides as a replacement of the metabolically labile methyl ester group suggested N, N-diethyl carboxamide was optimal for the Y2 binding affinity that was improved by 3- to 7- fold. Replacing the central phenyl ring with heteroaryl groups such as pyridyl, pyrimidyl and pyridazinyl drastically reduced the affinity (not shown).44 Substituting the central phenyl ring, ortho to the piperazine ring, significantly improved the affinity in the order of CN>F>CH3>Br>>H, whereas the OCH3 and the substitution at the meta-position were detrimental for the affinity, suggesting the role of both steric and electronic effects (52-54, Table 6).44 The 3-pyridylphenyl amide and biphenyl analogs (50, 51) showed comparable affinities to the 4-pyridylphenyl analog (49). The fluoro group was chosen as a substituent at the ortho-position of the central phenyl ring. A variety of alkyl and heteroaryl amides as exemplified in Table 7 (57-70) were explored to replace the biaryl amide moiety, primarily to reduce the molecular weight and lipophilicity. Particularly, 3,5-dimethylisoxazole-4-carboxamide (57), N(1)-ethyl-4-methylpyrazole-5-carboxamide (58), 3,5-dimethylisoxazole-4-acetamide (60), α,α-dialkylated phenyl acetamide (63) and 2-ethylbutyramide (64) analogs showed good binding affinity.44 The potency seemed to depend on the size and orientation of the lipophilic group present at this position. These groups showed better affinity when the cyano group was present at the ortho-position of the central phenyl ring. The urea analogs as shown by 61 displayed similar affinities to the amide analogs, while the sulfonamide analogs had poor affinities (not shown). Methylation of the anilide NH decreased the affinity (not shown). The affinities of reverse amide analogs depended on the type of alkyl or heteroaryl groups present; for instance, 3, 5-dimethylisoxazol-4-yl methyl analog had comparable affinity, while 2-ethylbutyl analog significantly lost the affinity (not shown). The introduction of a methylene group (benzyl amine analogs) was unfavorable for the affinity. Further modifications were made preserving the fluoro at the ortho position of the central phenyl ring, 2-ethylbutyl anilide and diethyl amide of the phenyl glycine moiety. Both electron-donating and electron-withdrawing groups were tolerable at the 3- and 4-positions of the phenyl ring of the phenyl glycine moiety (Table 8, 71-82). Notably, OCH3 (77) and OCF3 (79) groups at the 4-position were beneficial, increasing the affinity by 3- to 5-fold. The replacement of the phenyl ring with 2-pyridyl was also tolerable. The modification of the piperazine ring with 2-methyl piperazines and bridged piperazine were not beneficial, whereas the piperidine analogs maintained the affinity with a slight improvement depending on the anilide substituent (Table 9, 83-88), signifying the basic amine was not essential. In the piperidine series, the anilide substituent, 3,5-dimethylisoxazole (86 and 88) displayed greater selectivity over MTTP than the biaryl substituent (83).44,45 Both the piperazine and piperidine series of compounds exhibited poor microsomal stability. Consequently, in vivo pharmacokinetics were performed via subcutaneous (s.c.) administration. The 3,5-dimethylisoxazole-4-carboxamide analog (86) was brain-penetrant and displayed approximately 50% of Y2 receptors occupancy in the brain (10 mg/kg, rats), while 3,5-dimethylisoxazole urea analog (88) showed no occupancy. The piperazine 56 (JNJ-31020028, Table 6) was selected to further characterize in vivo and in vitro.46 JNJ-31020028 showed high Y2 affinity with an IC50 of 6 nM and > 100 fold selectivity against Y1, Y4 and Y5 sub-family receptors. The enantiomers of JNJ-31020028 had similar binding affinities. In the functional assay, JNJ-31020038 was demonstrated to be antagonist with a pKB value of 8.04. JNJ-31020038 had no significant binding up to 10 μM in a panel of 50 receptors, ion channels and transporters and did not inhibit a panel of 65 kinases. JNJ-31020028 had poor oral bioavailability (6%) in rats (10 mg/kg), but 100% subcutaneous bioavailability with a good Cmax (4.35 μM) and a shorter half-life of 0.83 h. JNJ-31020028 was brain-penetrant and occupied maximally 90% of the Y2 receptors in rats at a dose of 10 mg/kg (s.c.) with an ED50 of 1.6 mg/kg for occupancy.
Table 6.
SAR and binding affinity data of arylpiperazine analogs.43

| ||||
|---|---|---|---|---|
| Compd | R1 | R2 | R3 | IC50 (μM)a |
| 49 | COOCH3 | H |
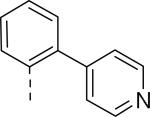
|
0.24 |
| 50 | COOCH3 | H |
|
0.22 |
| 51 | COOCH3 | H |
|
0.53 |
| 52 | COOCH3 | CN |
|
0.025 |
| 53 | COOCH3 | CH3 |
|
0.100 |
| 54 | COOCH3 | F |
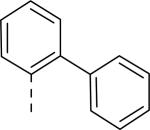
|
0.052 |
| 55 | CONHC2H5 | F |
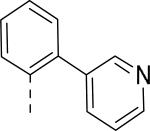
|
0.042 |
| 56 (JNJ 31020028) | CON(C2H5)2 | F |
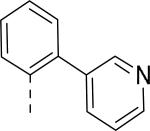
|
0.006 |
50% inhibition of binding of radio ligand [125I]PYY at human Y2 receptors.
Table 7.
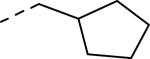
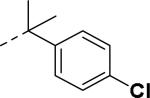
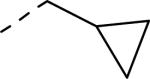
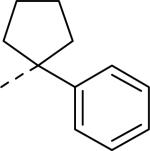
SAR and binding affinity data of anilide analogs.44

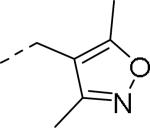
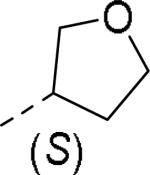
| |||||
|---|---|---|---|---|---|
| Compd | R3 | IC50 (μM)a | Compd | R3 | IC50 (μM)a |
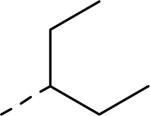
| 57 |
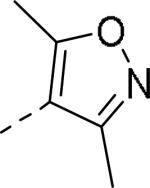
|
0.026 | 64 |
|
0.050 |
| 58 |
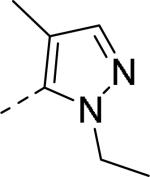
|
0.035 | 65 |
|
0.07 |
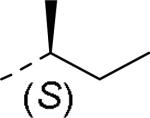
| 59 |
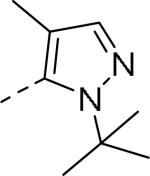
|
0.3 | 66 |
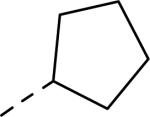
|
0.08 |
| 60 |
|
0.02 | 67 |
|
0.065 |
| 61 |
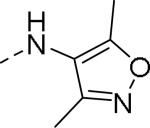
|
0.016 | 68 |
|
3.0 |
| 62 |
|
0.072 | 69 |
|
0.3 |
| 63 |
|
0.020 | 70 |
|
0.07 |
50% inhibition of binding of radio ligand [125I]PYY at human Y2 receptors.
Table 8.
Exploration of substitution on the phenyl ring of the phenyl glycine moiety.44

| |||||
|---|---|---|---|---|---|
| Compd | R | IC50 (μM)a | Compd | R | IC50 (μM)a |
| 71 | 3-F | 0.033 | 77 | 4-OCH3 | 0.018 |
| 72 | 3-Cl | 0.05 | 78 | 4-OC2H5 | 0.044 |
| 73 | 3-OCH3 | 0.04 | 79 | 4-OCF3 | 0.026 |
| 74 | 4-F | 0.2 | 80 | 4-OH | 0.1 |
| 75 | 4-Cl | 0.1 | 81 | 3,4-diF | 0.33 |
| 76 | 4-CN | 0.044 | 82 | 2,4-diF | 0.15 |
50% inhibition of binding of radio ligand [125I]PYY at human Y2 receptors.
Table 9.

| ||
|---|---|---|
| R = | ||
|
|
|
| 83 | 84 | 85 |
| IC50a = 7 nM (<300 nM) | IC50 = 12 nM | IC50 = 25 nM |
|
|
|
| 86 | 87 | 88 |
| IC50 = 26 nM (>10,000 nM) | IC50 = 14 nM | IC50 = 10 nM (>10,000 nM) |
50% inhibition of binding of radio ligand [125I]PYY at human Y2 receptors. The values in the brackets are IC50 value at MTTP.
Further modifications were re-investigated perhaps to improve the metabolic stability of the compounds. It appeared efforts were focused on modification of both the anilide and diethyl amide of the phenyl glycine moieties.47, 48 A variety of heteroaryls such as oxadiazoles, oxazoles, isoxazoles, triazoles and pyrazoles were explored to replace the anilide substituent (Table 10, 89-108). Remarkably, 5-substituted-1,2,4-oxadiazoles showed good affinity. The alkyl substitution particularly isopropyl at the 3-position of the 1,2,4-oxadiazoles were beneficial for the affinity, while electron withdrawing groups such as CF3, CN, ester and amide were detrimental (not shown). Similarly, a number of heterocycles were investigated to replace the diethyl amide of the phenyl glycine moiety (Table 11, 109-118). The affinity data was not disclosed for all the compounds. It seemed 5-substituted-1,2,4-oxadiazole, 2-oxazole, 2-pyrimidine and 5-isoxazole conserved good affinity. In 5-substituted 1,2,4-oxadiazole series, the affinity was decreased as the size of alkyl chain increased, suggesting perhaps a limited hydrophobic space was available. The representative examples of the series were shown in Figure 2 (114, 119). These molecules had better physicochemical properties such as molecular weight (<500 Da) and lipophilicity (cLogP < 5) than JNJ-31020028 (565 Da, 5.5).
Table 10.
Exploration of heteroaryl groups on phenyl ring of aniline moiety.47

| |||||
|---|---|---|---|---|---|
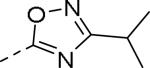
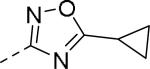
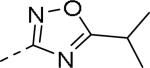
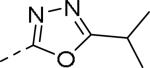
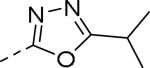
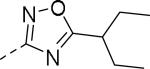
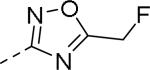
| Compd | Ar | IC50 (μM)a | Compd | Ar | IC50 (μM)a |
| 89 |
|
0.085 | 99 |
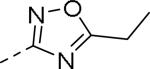
|
0.24 |
| 90 |
|
0.041 | 100 |
|
0.27 |
| 91 |
|
0.13 | 101 |
|
0.6 |
| 92 |
|
0.22 | 102 |
|
0.48 |
| 93 |
|
0.83 | 103 |
|
0.16 |
| 94 |
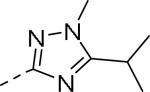
|
1.95 | 104 |
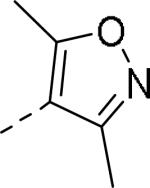
|
10 |
| 95 |
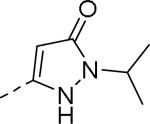
|
0.34 | 105 |
|
0.9 |
| 96 |
|
0.46 | 106 |
|
0.74 |
| 97 |
|
0.81 | 107 |
|
0.42 |
| 98 |
|
0.11 | 108 |
|
0.078 |
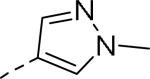
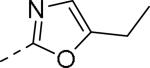
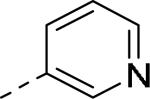
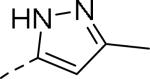
50% inhibition of binding of radio ligand [125I]PYY at human Y2 receptors.
Table 11.
Modifications of diethyl amide of phenyl glycine moiety with various heteroaromatics.48

| |||||
|---|---|---|---|---|---|
| Compd | Ar | IC50 (μM)a | Compd | Ar | IC50 (μM)a |
| 109 |

|
0.24 | 114 |

|
0.29 |
| 110 |

|
0.027 (7.4)b | 115 |
|
0.15 |
| 111 |
|
0.027 (7.45) | 116 |

|
0.038 (7.5) |
| 112 |

|
0.17 | 117 |

|
0.034 (7.2) |
| 113 |
|
0.72 | 118 |

|
0.033 |
50% inhibition of binding of radio ligand [125I]PYY at human Y2 receptors.
Figure 2.

Representative high affinity Y2 antagonists with heteroaryl groups that substituted the anilide amide and diethyl amide of the phenyl glycine moieties of JNJ-31020028 series.47, 48
Novartis disclosed a new series of Y2 antagonists based on imidazoline-2,5-dione and 4,5-dihydro-2H-imidazol-5-one in two patent applications.49, 50 The antagonist activity was only revealed in % of inhibition at 10 μM, obtained from [35S]GTPγS binding assay, ranging from 28% to 100%. The salient features seemed to be the presence of the aryl ketone and 4,4′-diphenyl-imidazol-5-one or 4-phenyl-4′-npropyl-imidazol-5-one (Fig. 3). It appeared to be the preferable anilide substitutions were 2-ethylbutanamide, 3,5-dimethylisoxaxol-4-yl acetamide and 3,5-dimethylisoxaxol-4-yl urea similar to JNJ series. Novartis also published a patent on Y2 antagonists that were similarly to the JNJ-31020028 series.51 These compounds primarily had aryl or heteroaryl groups instead of the diethyl amide of the phenyl glycine moiety of the JNJ-31020028 series (not shown).
Figure 3.

Representative structure of imidazoline-2,5-dione and 4,5-dihydro-2H-imidazol-5-one based NPY Y2 antagonists.49, 50
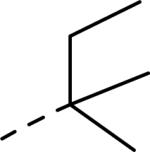
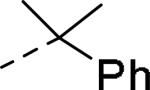
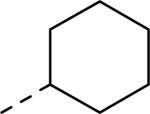
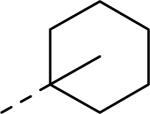
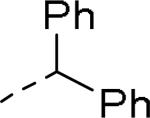
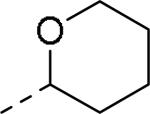
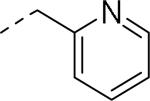
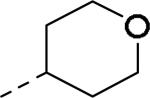
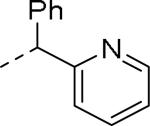
GlaxoSmithKline (GSK) reported a series of di-amide compounds structurally related to the JNJ-31020028 series as selective Y2 antagonists.52, 53 The functional HTS of GSK compound collection identified di-amide compound 121 (Table 12) as one of the hits that displayed moderate Y2 antagonist activity and selectivity against Y1 and Y5 receptors. The SAR of anilide substituents and ortho-substitution of the central phenyl ring was similar to JNJ-31020028 series. GSK focused mainly on the aryl acetamide substituents on the right-hand side of the molecule and preferred chloro substituent, more potent than the fluoro, on the central phenyl ring. Increasing the bulkiness of t-butyl group resulted in increased potency as exemplified by 121-132 (Table 12). Particularly, the 1-methyl cyclohex-1-yl analog (124) showed more than 20-fold higher potency than the hit, but had poor solubility. Introducing 2- and 4-tetrahydropyran moieties (125 and 126) to increase the polarity of the molecule were not beneficial for the potency. Further modifications were focused on replacing the t-butyl amide with a variety of substituted phenyl acetamides. The phenyl acetamide analog 127 had better potency than the t-butyl analog. The substitution of the benzylic position with two methyl groups (128) or one phenyl group (130) improved the potency by nearly 10-fold. Introducing a methyl at the meta-position of the phenyl group (129) resulted in small increase in the potency. Though potent, substituted phenyl acetamide analogs had poor solubility, high intrinsic clearance and high non-specific rat brain tissue binding (Table 14). Switching one of the phenyl rings of benzhydryl analog with 2-pyridyl moiety (132) was tolerable but did not improve either the solubility or the metabolic stability (Table 14). Further efforts were focused on modification of benzamide of the piperazine ring. Both electron-donating and -withdrawing groups were tolerable, of which CF3 group at the ortho-position of the phenyl ring moderately improved the potency (Table 13, 133-136). The incorporation of a heteroatom into the phenyl ring though reduced the potency, maintained good potency and improved the solubility as well as brain free fraction (Table 14, 136). Compound 136 displayed moderate brain/plasma ratio (0.6) and reasonable brain free fraction of 2.9%, but had poor exposure (Table 17). The lead optimization was embarked to achieve sufficient exposure levels to maximally occupy the receptor by balancing the potency, exposure and brain free fraction. The corresponding benzyl analogs of benzamides showed comparable potencies and provided opportunities to explore a variety of amines. The 4-amino piperidine analogs were found to display good potency, low intrinsic clearance and aqueous solubility (Table 14 and 15, 137-146). The size of the alkyl chain on 4-amino group of the piperidine had impact on the potency that was increased in the order of ethyl>isopropyl>n-propyl>cyclopropylmethyl>iso-butyl. Notably, cyclopropylmethyl amine analog (141) had reduced lipophilicity and higher solubility. The addition of methyl or CF3 at the meta-position of the phenyl ring of phenyl acetamide (142, 143) increased the potency and interestingly the solubility, but, also increased the lipophilicity. Notably, the CF3 group also improved the metabolic stability. Incorporating heteroatom(s) in the phenyl ring of phenyl acetamide moiety to decrease the lipophilicity significantly reduced the potency (144-146). Reinvestigation of the basic amine moiety to have optimal potency, solubility and lipophilicity identified spirocyclic amine as exemplified by 147 with decent potency. The substitution of the spirocyclic amine with cyclopropylmethyl (148) remarkably improved the potency but also the lipophilicity. The pyrimidyl analog 149 with cyclopropylmethyl spirocyclic amine showed good potency, solubility, moderate intrinsic clearance and adequate brain free fraction (Table 14 and 16). Compound 149 displayed comparable potencies at rat and human Y2 receptor, slightly higher for rat Y2 receptor, as well as selectivity against Y1 and Y5 receptors. Compound 149 had IC50 of > 10 μM against major CYP isoforms. In comparison to compound 136, compound 149 exhibited a higher brain/plasma ratio (2.3) and a higher brain exposure (Cmax of 73 ng/g) in the rat at 1 h after a single s.c. administration of 2 mg/kg. The later GSK series of compounds were significantly different at the left side of the molecule compared to JNJ-31020028 series. No further in vivo studies of GSK compound 149 were reported. Compound 149 is a valuable pharmacological tool to investigate the therapeutic potential of Y2 receptor in animal models.
Table 12.
SAR data of anilide analogs.52

| |||||||
|---|---|---|---|---|---|---|---|
| Compd | R | fpKia | Solubility (μg/mL) | Compd | R | fpKi | Solubility (μg/mL) |
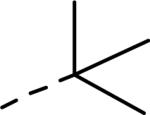
| 121(hit) |
|
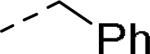
5.6 | 113 | 127 |
|
7.1 | 3 |
| 122 |
|
7.3 | 22 | 128 |
|
8.0 | 0 |
| 123 |
|
6.8 | 4 | 129 |
|
8.3 | 0 |
| 124 |
|
7.7 | 4 | 130 |
|
8.2 | 7 |
| 125 |
|
6.5 | nt | 131 |
|
6.5 | 70 |
| 126 |
|
5.2 | nt | (±)132 |
|
7.9 | 8 |
Functional activity against human Y2 receptors using [35S]GTPγS binding assay.
Table 14.
| Compd | fpKia | cLogP | Solubility (μg/mL) | Clint rat (mL/min/g) | Clint human (mL/min/g) | BTB rat (%) |
|---|---|---|---|---|---|---|
| 127 | 7.1 | 4.34 | 3 | 13.8 | 0.5 | 99.0 |
| 128 | 8.0 | 5.05 | 0 | 7.4 | 0.6 | 99.5 |
| (±)132 | 7.9 | 4.19 | 8 | 15.3 | <0.5 | 99.7 |
| 136 | 7.6 | 4.32 | 56 | 0.7 | 2.7 | 97.1 |
| 141 | 6.9 | 4.4 | 121 | 5.7 | 0.8 | 97.6 |
| 142 | 7.6 | 4.9 | 268 | 16.0 | 4.8 | 99.1 |
| 143 | 7.7 | 5.28 | 219 | 1.4 | <0.5 | 99.6 |
| 146 | 6.1 | 2.44 | 248 | 1.9 | <0.5 | 88.2 |
| 149 | 6.8 | 4.03 | 225 | 1.8 | 0.8 | 97.2 |
Functional activity against human Y2 receptors using [35S]GTPγS binding assay.
Table 13.
SAR data of benzamide analogs.52

| |||||
|---|---|---|---|---|---|
| Compd | X | R | fpKia | cLogP | Solubility (μg/mL) |
| 133 | C | CF3 | 8.4 | 6.07 | 1 |
| 134 | C | OCH3 | 8.6 | 5.27 | 2 |
| 135 | N | CF3 | 7.9 | 4.9 | 11 |
| 136b | N | OCH3 | 7.6 (7.3)c | 4.32 | 56 |
Functional activity against human Y2 receptors using [35S]GTPγS binding assay
tested as the HCl salt.
The functional activity against rat Y2 receptors.
Table 17.
| Compd | Dose (mg/kg) | Sample time (h) | Brain/Blood | Cmax (Br) (ng/g) | Cmax (Bl) (ng/mL) |
|---|---|---|---|---|---|
| 136 | 3 | 1.0 | 0.6 | 6 | 13 |
| 149 | 2 | 1.0 | 2.3 | 73 | 32 |
Table 15.
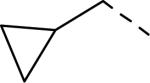
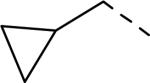
SAR data of 4-aminopiperidine analogs.53

| |||||
|---|---|---|---|---|---|
| Compd | R | Ar | fpKia | cLogP | Solubility(μg/mL) |
| 137 | Et | Ph | 5.7 | 3.96 | nt |
| 138 | iPr | Ph | 6.0 | 4.26 | nt |
| 139 | n-Pr | Ph | 6.6 | 4.48 | nt |
| 140 | i-Bu | Ph | 7.1 | 4.88 | nt |
| 141 |
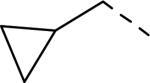
|
Ph | 6.9 | 4.4 | 121 |
| 142 |
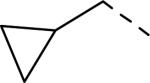
|
(m-Me)Ph | 7.6 | 4.9 | 268 |
| 143 |
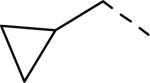
|
(m-CF3)Ph | 7.7 | 5.28 | 219 |
| 144 |
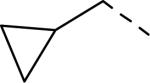
|
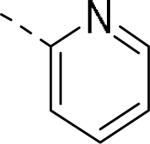
|
5.9 | 2.9 | nt |
| 145 |
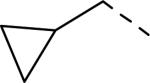
|
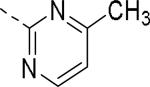
|
6.4 | 2.9 | 112 |
| 146 |
|
|
6.1 | 2.44 | 248 |
Functional activity against human Y2 receptors using [35S]GTPγS binding assay.
Table 16.
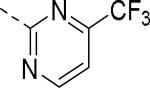
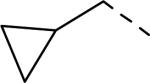
SAR data spirocyclic amine analogs.53

| |||||
|---|---|---|---|---|---|
| Compd | R | X | fpKia | cLogP | Solubility (μg/mL) |
| 147 | H | C | 7.8 | 5.16 | 96 |
| 148 |
|
C | 8.7 | 6.41 | 53 |
| 149 |
|
N | 6.8 (7.2)b | 4.03 | 225 |
Functional activity against human Y2 receptors using [35S]GTPγS binding assay
The functional activity against rat Y2 receptors.
Brothers et al. of The Scripps Research Institute (TSRI), Florida performed whole-cell based high throughput screening of a library of small molecules available through the auspices of the National Institutes of Health with an aim to identify selective and novel non-peptidic NPY Y2 antagonists.39 The HTS campaign led to the discovery of four distinct new chemotypes that displayed binding Ki values ranging from 1.55 and 60 nM in a radio ligand binding assay and functional IC50 values varying from 0.19 to 4.4 μM in the cAMP biosensor assay (Fig.4). Of the four hit molecules tested against Y1 receptor and 40 off-target receptors, ion channel and transporters related to CNS only SF-22 displayed significant binding with a Ki value of 255 nM at 5-HT2B receptor. We explored SAR studies around the SF-11 scaffold to improve the potency and drug-likeness (Table 18 and 19).54 The removal of ethoxy group (150) of the hit molecule SF-11 resulted in a complete loss of activity, suggesting a substitution on the aryl ring was essential. Replacing the ethoxy group with a methoxy (151), isopropoxy (152) or trifluoromethoxy (153) decreased the potency, indicating the presence of a hydrophobic pocket with steric-constraints at the binding site. Interestingly, the n-propyl analog (154) was less potent than SF-11, suggesting the presence of both a hydrogen-bond acceptor and hydrophobic group at this position were essential for the antagonist activity. Of the hydrogen-bond acceptor containing groups, such as dialkylamines, esters, carboxamides, and sulfonamides, diethyl amine, diethyl carboxamide and dimethyl sulfonamide groups were advantageous (Table 18). Notably, compound 159 (CYM 9484) with dimethyl sulfonamide was the most potent with an IC50 of 19 nM. The exploration of 3-substitution (161 and 162) or 2,4- or 3,4-disubstitution (163-166) was either detrimental or not beneficial. The removal of the hydroxyl group or methylation of hydroxyl group of the diphenylcarbinol led to complete loss of the activity, signifying the essentiality of a hydrogen-bond donor (Table 19, 168 and 169). Interestingly, the benzhydryl piperazine analog (170) retained the activity in contrast to the 4-benzhydrylpiperidine analog (168), supporting the requirement of a hydrogen-bond donor. The replacement of one of the phenyl rings with a cyclohexyl group or deletion of one of the phenyl rings resulted in a complete loss of activity (167 and 171), indicating perhaps steric interactions between the phenyl rings reserved them in a specific conformation that was favorable for the activity. Switching one of the phenyl rings of diphenylcarbinol with 2-pyridyl and 3-thienyl were tolerable, whereas with the 3-furyl, 3-pyridyl and 3, 5-dimethylisoxazol-4-yl were detrimental (172-176). The substitution of a fluorine at the 4-position of one of the phenyl rings of diphenylcarbinol (177) was also tolerable, but not beneficial. Further efforts were focused on the replacement of the thiourea functionality which is associated with potential toxicity. While the exploration of urea functionality to replace the thiourea resulted in a complete loss of activity, the carbamate was very-well tolerated. The carbamate analogs displayed a similar SAR to the thiourea series, of which compound 180 with a diethyl sulfonamide at the 4-position of the phenyl ring showed highest activity with an IC50 of 12 nM (Table 20, 178-185). However, the carbamate analogs were not stable in the rat plasma due to the hydrolysis of the carbamate group (unpublished results) that makes them unsuitable for in vivo studies.
Figure 4.

Structures of four different chemotypes identified from HTS.39
Table 18.
SAR and functional activity data of the thiourea analogs.54

| |||||
|---|---|---|---|---|---|
| Compd | R | IC50a (μM) | Compd | R | IC50a (μM) |
| SF-11 | 4-OC2H5 | 0.199 | 158 | 4-CON(C2H5)2 | 0.078 |
| 150 | -H | NAc | 159 | 4-SO2N(CH3)2 | 0.019 |
| 151 | 4-OCH3 | 1.1 | 160 | 4-SO2N(C2H5)2 | 0.136 |
| 152 | 4-OiPr | 1.77 | 161 | 3-OCH3 | 8.162b |
| 153 | 4-OCF3 | 1.047 | 162 | 3- COOC2H5 | 0.238b |
| 154 | 4-nC3H7 | 0.777 | 163 | 3-Cl, 4-N(C2H5)2 | 0.085 |
| 155 | 4-N(C2H5)2 | 0.072b | 164 | 3-CN, 4-OC2H5 | 0.323 |
| 156 | 4-COOC2H5 | 0.207 | 165 | 2-CH3, 4- OC2H5 | NA |
| 157 | 4-CON(CH3)2 | 0.301 | 166 | 2-F, 4- OC2H5 | 1.35 |
Functional activity using cAMP biosensor assy. All compounds were inactive at the NPY Y1 receptor
exhibited partial antagonism
not active (NA) at the highest concentration tested (10 μM).
Table 19.
SAR exploration of the diphenylcarbinol moiety.54

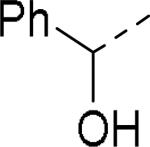
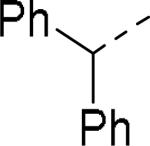
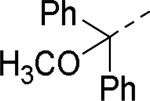
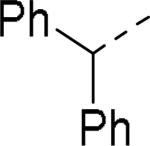





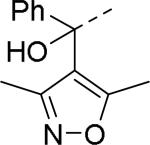
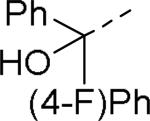
Table 20.
SAR and functional activity data of the carbamate analogs.54

Of the identified non-peptidic Y2 antagonists, JNJ-31020028 was examined in animal models of alcohol consumption, anxiety and depression. JNJ-31020028 administered s.c. or i.c.v did not reduce alcohol-intake in alcohol preferring rats or operant self-administration of alcohol by wistar rats.55 On the other hand, peptidomimetic Y2 antagonist BIIE0246 decreased operant self-administration of alcohol by rats and the effect was thought to be because of sedation at the administered dose.26 JNJ-31020028 was not effective in a variety of anxiety animal models, although it reduced the alcohol-withdrawal induced anxiety.55 The authors concluded from these results Y2 antagonists may not be useful for alcoholism, but may be useful for the treatment of negative affective states associated with alcohol withdrawal. Systemically administered JNJ-31020028 also reduced nicotine withdrawal induced social anxiety56. Chronic administration (i.c.v) of JNJ-31020028 displayed antidepressant-like behavior in an animal model of depression similar to BIIE0246.57
NPY Y2 receptor agonists
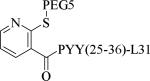
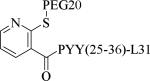
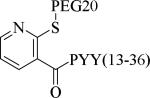
Small molecule, non-peptidic Y2 agonists are not yet identified. The endogenous peptide PYY(3-36) is a potent and Y2 preferring agonist, but it also activates Y1 and Y5 receptor subtypes with a significant binding affinity. The inconsistent results of PYY(3-36) in animal models of obesity was partly thought to be due to the activation of Y1 and Y5 receptors that stimulate food-intake, and poor pharmacokinetics. Consequently, several potent, selective and systemically active peptidic agonists have been discovered. The N-terminal truncated PYY(3-36) analogs displayed greater Y2 selectivity but also decreased affinity (Table 21, 187-188).58 The shortest truncated analog PYY(25-36) showed moderate potency for Y2 and minimal affinity for Y1 and Y5. The acetylation of N-terminal -amino group of PYY(25-36) significantly improved the Y2 potency as well maintained the selectivity (191). Further N-terminal modification of PYY(25-36) with benzoic acid analogs resulted in potent and selective Y2 agonists as exemplified by 192. The N-terminal 2-amino benzoic acid was modified to derivatize with varying lengths of monomethoxypolyethylene glycols (PEGs) to improve the pharmacokinetics (Table 21, 193-195).59 The PEG5 and PEG20-ylated peptides showed 2- and 8-fold less efficacy than the peptide 192. The use of longer sequence of PYY (13-36) restored the Y2 efficacy60 (196). The PEGylated peptide Y2 agonist 196, administered s.c., reduced dose-dependently the food intake in lean mice and wistar rats and body weight in diet-induced obese (DIO) mice following repeated dosing for 14 days with superior efficacy and a longer duration of action in contrast to PYY(3-36).59, 60 7-TM pharma disclosed several modified analogs of PYY (3-36) as potent and selective Y2 agonists in two patent applications.61, 62 Of these, TM-30335 reduced the body weight in DIO mice model on daily s.c. dosing for 40 days63 but not superior to PYY (3-36). Bulaj et al. recently identified another systemically active Y2 peptidic agonist NPY-BBB2 by introducing lipidization–cationization motif at the N-terminal of the truncated NPY analog (Ahx5-24)NPY.64 The modified peptide NPY-BBB2 (198) administered i.p. potently suppressed seizures in the 6 Hz mouse model of epilepsy with an ED50 of 1.1 mg/kg. The clinical efficacy of selective and systemically active Y2 agonists is remain to be demonstrated.
Table 21.
| Compd | Peptidea | Y2 EC50 (nM) | Y2 Ki (nM) | Y1 Ki (nM) | Y5 Ki (nM) |
|---|---|---|---|---|---|
| 186 | PYY (3-36) | 0.3 | 0.4 | 21 | 20 |
| 187 | PYY (22-36) | 11 | 3 | 390 | >1000 |
| 188 | PYY (25-36) | 240 | 270 | >1000 | >1000 |
| 189 | AcPYY (22-36) | 13 | 9 | 118 | >1000 |
| 190 | AcPYY (24-36)-L31 | 6 | 5 | 210 | >1000 |
| 191 | AcPYY (25-36) | 27 | 30 | >1000 | >1000 |
| 192 |
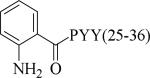
|
3 | 4 | >1000 | >1000 |
| 193 |
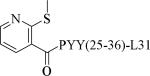
|
4 | 11 | >1000 | >1000 |
| 194 |
|
6 | 7 | >1000 | >1000 |
| 195 |
|
25 | 41 | 4000 | 1600 |
| 196 |
|
6.5 | 9.1 | 760 | 630 |
| 197 | (Ahx5-24)NPYb | 9 | 5 | 100 | >1000 |
| 198 | Ac-YKK(Kp)(Ahx5-24)ANPYc | nd | 26.1 | nd | nd |
All peptides are amidated at the C-terminal. PYY(25-36)-L31 or PYY(24-36)-L31 are peptides in which Val 31 of corresponding PYY peptide is replaced with Leu.
5-24 amino acid residues of NPY are replaced with 6-aminohexanoic acid.
Kp is Nε-palmitoyl-L-lysine.
nd: not determined.
Conclusions
In recent years, the Y2 receptor has attracted considerable interest because of its role in various physiological and pathological processes such as food-intake, affective disorders, alcohol and drugs of abuse, bone formation and epilepsy. The pharmacological role of Y2 receptor was mostly investigated by knock-out experiments and using peptidic antagonist BIIE0246 due to the lack of nonpeptidic modulators. Some of the results obtained with BIIE0246 might be misleading because of the off-target, sedative effects and the i.c.v route of administration. Several non-peptidic, selective and brain-penetrant Y2 antagonists are currently available. Though the clinical utility of Y2 receptor modulators is not clear, the identified non-peptidic Y2 antagonists particularly JNJ-31020028 and GSK compound 141 are valuable as in vivo tools to elucidate the precise pharmacological role of Y2 receptor and to validate Y2 receptor as a therapeutic target. JNJ-31020028 has been investigated in animal models of anxiety and alcoholism. JNJ-31020028 reduced alcohol- and nicotine-withdrawal induced anxiety, demonstrating the potential therapeutic utility of Y2 antagonists for the treatment of affective disorders. The studies with JNJ-31020028 do not support the role of Y2 in alcoholism in contrast to BIIE0246. The oral bioavailability, brain-penetration and/or metabolic stability are the key issues of most of the currently available non-peptidic Y2 antagonists. Selective and systemically active Y2 peptidic agonists such as NPYBBB2 and 196 displayed efficacy in animal models of epilepsy and obesity, respectively. However, small molecule non-peptidic Y2 agonists are lacking. The future work should focus on the discovery of non-peptidic Y2 agonists, Y2 antagonists that have good oral bioavailability, brain-penetration and good plasma half-life, and characterization of their in vivo efficacy and side effect profile associated with the pharmacological (in)activation of Y2 receptor.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Michel MC, Beck-Sickinger A, Cox H, Doods HN, Herzog H, Larhammar D, Quirion R, Schwartz T, Westfall T. Pharmacological reviews. 1998;50:143. [PubMed] [Google Scholar]
- 2.Blomqvist AG, Herzog H. Trends in neurosciences. 1997;20:294. doi: 10.1016/s0166-2236(96)01057-0. [DOI] [PubMed] [Google Scholar]
- 3.Lin S, Boey D, Couzens M, Lee N, Sainsbury A, Herzog H. Neuropeptides. 2005;39:21. doi: 10.1016/j.npep.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Parker RM, Herzog H. The European journal of neuroscience. 1999;11:1431. doi: 10.1046/j.1460-9568.1999.00553.x. [DOI] [PubMed] [Google Scholar]
- 5.Fetissov SO, Byrne LC, Hassani H, Ernfors P, Hokfelt T. The Journal of comparative neurology. 2004;470:256. doi: 10.1002/cne.11047. [DOI] [PubMed] [Google Scholar]
- 6.Shi YC, Lin S, Castillo L, Aljanova A, Enriquez RF, Nguyen AD, Baldock PA, Zhang L, Bijker MS, Macia L, Yulyaningsih E, Zhang H, Lau J, Sainsbury A, Herzog H. Obesity (Silver Spring, Md.) 2011;19:2137. doi: 10.1038/oby.2011.99. [DOI] [PubMed] [Google Scholar]
- 7.Greber S, Schwarzer C, Sperk G. British journal of pharmacology. 1994;113:737. doi: 10.1111/j.1476-5381.1994.tb17055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen X, DiMaggio DA, Han SP, Westfall TC. The American journal of physiology. 1997;273:H1737. doi: 10.1152/ajpheart.1997.273.4.H1737. [DOI] [PubMed] [Google Scholar]
- 9.Brothers SP, Wahlestedt C. EMBO molecular medicine. 2010;2:429. doi: 10.1002/emmm.201000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parker SL, Balasubramaniam A. British journal of pharmacology. 2008;153:420. doi: 10.1038/sj.bjp.0707445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamiji MM, Inui A. Current topics in medicinal chemistry. 2007;7:1734. doi: 10.2174/156802607782340957. [DOI] [PubMed] [Google Scholar]
- 12.Shi YC, Baldock PA. Bone. 2012;50:430. doi: 10.1016/j.bone.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Wu G, Feder A, Wegener G, Bailey C, Saxena S, Charney D, Mathe AA. Expert opinion on therapeutic targets. 2011;15:1317. doi: 10.1517/14728222.2011.628314. [DOI] [PubMed] [Google Scholar]
- 14.Meurs A, Clinckers R, Ebinger G, Michotte Y, Smolders I. Current topics in medicinal chemistry. 2007;7:1660. doi: 10.2174/156802607782340975. [DOI] [PubMed] [Google Scholar]
- 15.Thorsell A. Peptides. 2007;28:480. doi: 10.1016/j.peptides.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 16.Zhang L, Bijker MS, Herzog H. Pharmacology & therapeutics. 2011;131:91. doi: 10.1016/j.pharmthera.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 17.Boggiano MM, Chandler PC, Oswald KD, Rodgers RJ, Blundell JE, Ishii Y, Beattie AH, Holch P, Allison DB, Schindler M, Arndt K, Rudolf K, Mark M, Schoelch C, Joost HG, Klaus S, Thone-Reineke C, Benoit SC, Seeley RJ, Beck-Sickinger AG, Koglin N, Raun K, Madsen K, Wulff BS, Stidsen CE, Birringer M, Kreuzer OJ, Deng XY, Whitcomb DC, Halem H, Taylor J, Dong J, Datta R, Culler M, Ortmann S, Castaneda TR, Tschop M. Obesity reviews : an official journal of the International Association for the Study of Obesity. 2005;6:307. doi: 10.1111/j.1467-789X.2005.00218.x. [DOI] [PubMed] [Google Scholar]
- 18.Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, Wren AM, Brynes AE, Low MJ, Ghatei MA, Cone RD, Bloom SR. Nature. 2002;418:650. doi: 10.1038/nature00887. [DOI] [PubMed] [Google Scholar]
- 19.Abbott CR, Small CJ, Kennedy AR, Neary NM, Sajedi A, Ghatei MA, Bloom SR. Brain research. 2005;1043:139. doi: 10.1016/j.brainres.2005.02.065. [DOI] [PubMed] [Google Scholar]
- 20.El Bahh B, Balosso S, Hamilton T, Herzog H, Beck-Sickinger AG, Sperk G, Gehlert DR, Vezzani A, Colmers WF. The European journal of neuroscience. 2005;22:1417. doi: 10.1111/j.1460-9568.2005.04338.x. [DOI] [PubMed] [Google Scholar]
- 21.Stroud LM, O'Brien TJ, Jupp B, Wallengren C, Morris MJ. Brain research. 2005;1033:151. doi: 10.1016/j.brainres.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 22.El Bahh B, Cao JQ, Beck-Sickinger AG, Colmers WF. British journal of pharmacology. 2002;136:502. doi: 10.1038/sj.bjp.0704751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Redrobe JP, Dumont Y, Herzog H, Quirion R. Behavioural brain research. 2003;141:251. doi: 10.1016/s0166-4328(02)00374-1. [DOI] [PubMed] [Google Scholar]
- 24.Tasan RO, Nguyen NK, Weger S, Sartori SB, Singewald N, Heilbronn R, Herzog H, Sperk G. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:6282. doi: 10.1523/JNEUROSCI.0430-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Redrobe JP, Dumont Y, Fournier A, Quirion R. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2002;26:615. doi: 10.1016/S0893-133X(01)00403-1. [DOI] [PubMed] [Google Scholar]
- 26.Bacchi F, Mathe AA, Jimenez P, Stasi L, Arban R, Gerrard P, Caberlotto L. Peptides. 2006;27:3202. doi: 10.1016/j.peptides.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 27.Thorsell A, Slawecki CJ, El Khoury A, Mathe AA, Ehlers CL. Pharmacology, biochemistry, and behavior. 2006;83:28. doi: 10.1016/j.pbb.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 28.Thiele TE, Naveilhan P, Ernfors P. Peptides. 2004;25:975. doi: 10.1016/j.peptides.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 29.Thorsell A, Rimondini R, Heilig M. Neuroscience letters. 2002;332:1. doi: 10.1016/s0304-3940(02)00904-7. [DOI] [PubMed] [Google Scholar]
- 30.Rimondini R, Thorsell A, Heilig M. Neuroscience letters. 2005;375:129. doi: 10.1016/j.neulet.2004.10.084. [DOI] [PubMed] [Google Scholar]
- 31.Baldock PA, Sainsbury A, Couzens M, Enriquez RF, Thomas GP, Gardiner EM, Herzog H. The Journal of clinical investigation. 2002;109:915. doi: 10.1172/JCI14588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kitlinska J, Abe K, Kuo L, Pons J, Yu M, Li L, Tilan J, Everhart L, Lee EW, Zukowska Z, Toretsky JA. Cancer research. 2005;65:1719. doi: 10.1158/0008-5472.CAN-04-2192. [DOI] [PubMed] [Google Scholar]
- 33.Lu C, Everhart L, Tilan J, Kuo L, Sun CC, Munivenkatappa RB, Jonsson-Rylander AC, Sun J, Kuan-Celarier A, Li L, Abe K, Zukowska Z, Toretsky JA, Kitlinska J. Oncogene. 2010;29:5630. doi: 10.1038/onc.2010.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dumont Y, Cadieux A, Doods H, Pheng LH, Abounader R, Hamel E, Jacques D, Regoli D, Quirion R. British journal of pharmacology. 2000;129:1075. doi: 10.1038/sj.bjp.0703162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dautzenberg FM, Neysari S. Pharmacology. 2005;75:21. doi: 10.1159/000085897. [DOI] [PubMed] [Google Scholar]
- 36.Pluym N, Baumeister P, Keller M, Bernhardt G, Buschauer A. ChemMedChem. 2013;8:587. doi: 10.1002/cmdc.201200566. [DOI] [PubMed] [Google Scholar]
- 37.Pluym N, Brennauer A, Keller M, Ziemek R, Pop N, Bernhardt G, Buschauer A. ChemMedChem. 2011;6:1727. doi: 10.1002/cmdc.201100241. [DOI] [PubMed] [Google Scholar]
- 38.Doods H, Gaida W, Wieland HA, Dollinger H, Schnorrenberg G, Esser F, Engel W, Eberlein W, Rudolf K. European journal of pharmacology. 1999;384:R3. doi: 10.1016/s0014-2999(99)00650-0. [DOI] [PubMed] [Google Scholar]
- 39.Brothers SP, Saldanha SA, Spicer TP, Cameron M, Mercer BA, Chase P, McDonald P, Wahlestedt C, Hodder PS. Molecular pharmacology. 2010;77:46. doi: 10.1124/mol.109.058677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andres CJ, Antal Zimanyi I, Deshpande MS, Iben LG, Grant-Young K, Mattson GK, Zhai W. Bioorganic & medicinal chemistry letters. 2003;13:2883. doi: 10.1016/s0960-894x(03)00554-7. [DOI] [PubMed] [Google Scholar]
- 41.Jablonowski JA, Chai W, Li X, Rudolph DA, Murray WV, Youngman MA, Dax SL, Nepomuceno D, Bonaventure P, Lovenberg TW, Carruthers NI. Bioorganic & medicinal chemistry letters. 2004;14:1239. doi: 10.1016/j.bmcl.2003.12.057. [DOI] [PubMed] [Google Scholar]
- 42.Bonaventure P, Nepomuceno D, Mazur C, Lord B, Rudolph DA, Jablonowski JA, Carruthers NI, Lovenberg TW. The Journal of pharmacology and experimental therapeutics. 2004;308:1130. doi: 10.1124/jpet.103.060459. [DOI] [PubMed] [Google Scholar]
- 43.Swanson DM, Wong VD, Jablonowski JA, Shah C, Rudolph DA, Dvorak CA, Seierstad M, Dvorak LK, Morton K, Nepomuceno D, Atack JR, Bonaventure P, Lovenberg TW, Carruthers NI. Bioorganic & medicinal chemistry letters. 2011;21:5552. doi: 10.1016/j.bmcl.2011.06.136. [DOI] [PubMed] [Google Scholar]
- 44.Bonaventure P, Carruthers NI, Chai W, Dvorak CA, Jablonowski JA, Rudolph DA, Seierstad M, Shah CR, Swanson DM, Wong VD. US 20070100141 A1.
- 45.Chai W, Wong VD, Nepomuceno D, Bonaventure P, Lovenberg TW, Carruthers NI. Bioorganic & medicinal chemistry letters. 2013;23:4141. doi: 10.1016/j.bmcl.2013.05.038. [DOI] [PubMed] [Google Scholar]
- 46.Shoblock JR, Welty N, Nepomuceno D, Lord B, Aluisio L, Fraser I, Motley ST, Sutton SW, Morton K, Galici R, Atack JR, Dvorak L, Swanson DM, Carruthers NI, Dvorak C, Lovenberg TW, Bonaventure P. Psychopharmacology. 2010;208:265. doi: 10.1007/s00213-009-1726-x. [DOI] [PubMed] [Google Scholar]
- 47.Chai W, Dvorak CA, Jablonowski JA, Rudolph DA, Shah CR, Wong V. WO 2009079593 A1.
- 48.Chai W, Dvorak CA, Jablonowski JA, Rudolph DA, Shah CR, Wong V. WO 2009079597 A1.
- 49.Nozulak J, Orain D. US 20090099243 A1.
- 50.Nozulak J, Orain D. US 20090099244 A1.
- 51.Nozulak J, Orain D, Cotesta S. US 20090099199 A1.
- 52.Lunniss GE, Barnes AA, Barton N, Biagetti M, Bianchi F, Blowers SM, Caberlotto L, Emmons A, Holmes IP, Montanari D, Norris R, Walters DJ, Watson SP. Bioorganic & medicinal chemistry letters. 2009;19:4022. doi: 10.1016/j.bmcl.2009.06.035. [DOI] [PubMed] [Google Scholar]
- 53.Lunniss GE, Barnes AA, Barton N, Biagetti M, Bianchi F, Blowers SM, Caberlotto LL, Emmons A, Holmes IP, Montanari D, Norris R, Puckey GV, Walters DJ, Watson SP, Willis J. Bioorganic & medicinal chemistry letters. 2010;20:7341. doi: 10.1016/j.bmcl.2010.10.065. [DOI] [PubMed] [Google Scholar]
- 54.Mittapalli GK, Vellucci D, Yang J, Toussaint M, Brothers SP, Wahlestedt C, Roberts E. Bioorganic & medicinal chemistry letters. 2012;22:3916. doi: 10.1016/j.bmcl.2012.04.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cippitelli A, Rezvani AH, Robinson JE, Eisenberg L, Levin ED, Bonaventure P, Motley ST, Lovenberg TW, Heilig M, Thorsell A. Alcohol (Fayetteville, N.Y.) 2011;45:567. doi: 10.1016/j.alcohol.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 56.Aydin C, Oztan O, Isgor C. Behavioural brain research. 2011;222:332. doi: 10.1016/j.bbr.2011.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Morales-Medina JC, Dumont Y, Bonaventure P, Quirion R. Neuropeptides. 2012;46:329. doi: 10.1016/j.npep.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 58.DeCarr LB, Buckholz TM, Coish PD, Fathi Z, Fisk SE, Mays MR, O'Connor SJ, Lumb KJ. Bioorganic & medicinal chemistry letters. 2007;17:538. doi: 10.1016/j.bmcl.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 59.Lumb KJ, DeCarr LB, Milardo LF, Mays MR, Buckholz TM, Fisk SE, Pellegrino CM, Ortiz AA, Mahle CD. Journal of medicinal chemistry. 2007;50:2264. doi: 10.1021/jm061454v. [DOI] [PubMed] [Google Scholar]
- 60.Ortiz AA, Milardo LF, DeCarr LB, Buckholz TM, Mays MR, Claus TH, Livingston JN, Mahle CD, Lumb KJ. The Journal of pharmacology and experimental therapeutics. 2007;323:692. doi: 10.1124/jpet.107.125211. [DOI] [PubMed] [Google Scholar]
- 61.Schwartz T. WO 2005089789 A2.
- 62.Schwartz T, Wang F. WO 2007038943 A1.
- 63.Heal DJ, Gosden J, Smith SL. Neuropharmacology. 2012;63:132. doi: 10.1016/j.neuropharm.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 64.Green BR, White KL, McDougle DR, Zhang L, Klein B, Scholl EA, Pruess TH, White HS, Bulaj G. Journal of peptide science : an official publication of the European Peptide Society. 2010;16:486. doi: 10.1002/psc.1266. [DOI] [PubMed] [Google Scholar]