

Abstract
SLiCE (Seamless Ligation Cloning Extract) is a novel cloning method that utilizes easy to generate bacterial cell extracts to assemble multiple DNA fragments into recombinant DNA molecules in a single in vitro recombination reaction. SLiCE overcomes the sequence limitations of traditional cloning methods, facilitates seamless cloning by recombining short end homologies (15–52 bp) with or without flanking heterologous sequences and provides an effective strategy for directional subcloning of DNA fragments from bacterial artificial chromosomes or other sources. SLiCE is highly cost-effective and demonstrates the versatility as a number of standard laboratory bacterial strains can serve as sources for SLiCE extract. We established a DH10B-derived E. coli strain expressing an optimized λ prophage Red recombination system, termed PPY, which facilitates SLiCE with very high efficiencies.
Keywords: SLiCE cloning, In vitro recombination, Seamless cloning, Bacterial cell extract, DNA cloning, Recombinant DNA
1 Introduction
The generation of recombinant DNA molecules is an essential tool in modern molecular biology. The conventional DNA cloning strategies that have been used for several decades typically involve the use of type II restriction enzymes to generate appropriate DNA fragments, the modification of DNA ends to generate blunt or sticky ends, and the ligation of the DNA fragments to generate plasmid or other type DNA vectors [1–3]. However, these procedures depend on the presence of appropriate restriction sites to generate both vector and insert molecules and often leave unwanted sequences at the junction sites. In addition, the restriction enzymes and modifying enzymes required for these manipulations are often expensive making these procedures costly especially in high throughput settings. To circumvent these limitations, we developed a new restriction site-independent cloning method that does not leave any unwanted sequences at the junction sites (seamless) and is based on in vitro recombination between short regions of homologies (15–52 bp) in bacterial cell extracts termed SLiCE (Seamless Ligation Cloning Extract) [4]. SLiCE allows for efficient restriction site-independent cloning of DNA fragments generated by restriction digestion or PCR amplification into linearized vectors. In addition, SLiCE does not require the use of enzymes for the modification of vector and insert end sequences (such as Klenow or T4 DNA polymerase) or ligases.
The SLiCE method [4] can be used for virtually any type of cloning approach such as the cloning of a single DNA fragment with or without flanking heterologous sequences (see Fig. 1), the cloning of multiple DNA fragments in one step (see Fig. 2), and the directional subcloning of large DNA fragments from bacterial artificial chromosomes (BACs) (see Fig. 3).
Fig. 1.

SLiCE cloning of a single DNA fragment. (a) SLiCE cloning without flanking heterologous sequences. (b) SLiCE cloning with flanking heterologous sequences (Reproduced from [4], with permission from Oxford University Press)
Fig. 2.

SLiCE cloning of multiple DNA fragments. (a ) Schematic illustrating multiple-way SLiCE cloning. A three-way cloning approach is shown. (b ) Schematic illustrating SLiCE batch cloning (Reproduced from [4], with permission from Oxford University Press)
Fig. 3.
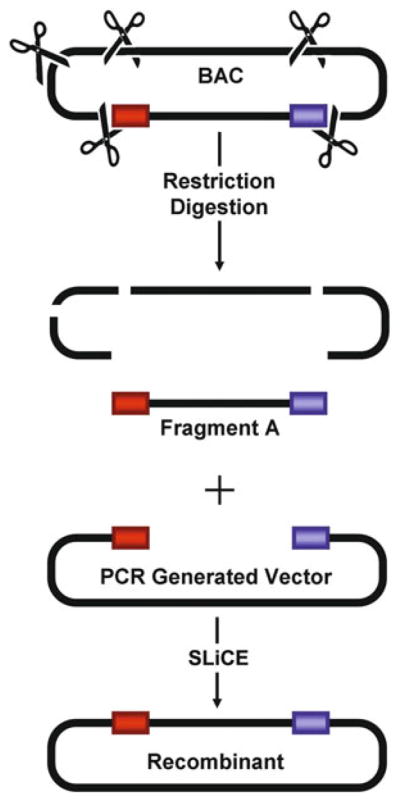
BAC SLiCE cloning (Reproduced from [4 ], with permission from Oxford University Press)
The SLiCE method is based on bacterial extracts that can be derived from a variety of common RecA− E. coli laboratory strains such as DH10B and JM109 [4]. In vivo homologous recombination in E. coli can be facilitated by three different recombination pathways: The RecA-dependent pathway, an RecA-independent pathway of unknown nature and an RecA-independent pathway that utilizes prophage Red/ET recombination systems [5–10]. Our studies indicate that an RecA-independent pathway catalyzes SLiCE. To improve SLiCE cloning efficiencies and capabilities, we established a DH10B-derived strain, termed PPY that was engineered to contain an optimized λ prophage Red recombination system [4–7]. This strain provides the highest cloning efficiencies thus far and can be used for all cloning approaches that are routinely used in the laboratory [4].
SLiCE is a simple and efficient procedure with the entire process consisting of three steps: (1) The preparation of linear vector and insert fragments containing short end homologies introduced by PCR with primers having appropriate 5′ extension sequences; (2) the SLiCE in vitro reaction; and (3) the standard transformation (electroporation or chemical transformation) of recombination products into suitable host bacteria [4].
In this chapter, we describe the preparation of the PPY SLiCE extract, the maintenance of the PPY strain, and the SLiCE cloning procedure.
2 Materials
2.1 Preparation of PPY SLiCE Extract
PPY strain (available from Dr. Yongwei Zhang, yongwei.zhang@einstein.yu.edu) (seeNote 1).
2XYT medium: Dissolve 16 g Bacto-tryptone, 10 g Bacto-yeast extract, and 5 g NaCl in 900 mL ddH2O. Adjust pH to 7.2 with NaOH. Adjust to 1 L with ddH2O. Autoclave to sterilize and store at room temperature.
Antibiotics: Streptomycin and chloramphenicol.
L-(+)-Arabinose (Sigma, A3256).
CelLytic™ B Cell Lysis Reagent (Sigma, B7435).
100 % Glycerol.
Protein LoBind Tube 1.5 mL (Eppendorf).
Protein LoBind Tube 0.5 mL (Eppendorf).
50-mL centrifuge tubes.
250-mL Nalgene Lab Quality Wide-Mouth Bottles.
37 °C shaker.
37 °C incubator.
Centrifuge.
Spectrophotometer.
−20 and −80 °C freezer.
2.2 Maintenance of PPY strain
PPY strain.
Lysogeny Broth (LB) medium: Dissolve 10 g Bacto tryptone, 5 g Bacto-yeast extract, and 10 g NaCl in 900 mL ddH2O. Adjust pH to 7.2 with NaOH. Adjust to 1 L with ddH2O. Autoclave to sterilize and store at room temperature.
Antibiotics: Streptomycin and chloramphenicol.
20 % Glycerol.
37 °C shaker.
−80 °C freezer.
2.3 SLiCE Cloning
PPY SLiCE extract (prepared as described in Subheading 3.1).
10× SLiCE buffer: 500 mM Tris–Cl (pH 7.5 at 25 °C), 100 mM MgCl2, 10 mM ATP, 10 mM DTT, store at −20 °C.
Materials and equipment for restriction cutting, PCR, DNA purification and transformation.
3 Methods
3.1 Preparation of PPY SLiCE Extract
Streak PPY glycerol stock or fresh culture on an LB agar plate (10 μg/mL streptomycin and 12.5 μg/mL chloramphenicol) and incubate at 37 °C overnight.
Inoculate one single colony into a 50-mL centrifuge tube containing 25 mL 2XYT (10 μg/mL streptomycin) and shake at 37 °C and 330 rpm overnight.
The next day, measure the OD600 (seeNote 2).
Dilute the o/n culture to 0.03 OD600, i.e., inoculate appropriate volume of the o/n culture into a 250-mL Nalgene Lab Quality Wide-Mouth Bottle containing 50 mL 2XYT medium (10 μg/mL streptomycin) (see Note 3).
Shake at 37 °C and 330 rpm until the culture reaches an OD600 of 5.0–5.5 (see Note 4).
Add 0.2 % L-(+)-arabinose into the culture and continue shaking at 330 rpm for 2 h at 37 °C, to induce expression of λ prophage protein Red. Remove 500 μL from the culture to measure the actual OD600.
Transfer 48 mL of the bacterial culture into two 50-mL centrifuge tubes (24 mL each).
Pellet the cells by centrifugation at 5,000 × g for 20 min at 4 °C.
Wash the cell pellet from the 24 mL original culture with 50 mL ddH2O once.
Pellet the cells by centrifugation at 5,000 × g for 20 min at 4 °C.
Measure the wet weight.
Resuspend the cell pellet of about 0.23 g of wet weight or from 24 mL of original culture at OD600 ≈ 6 in 300 μL CelLytic™ B Cell Lysis Reagent (Sigma, B7435).
Transfer the resuspended cells into a low binding 1.5 mL tube (Protein LoBind Tube 1.5 mL, Eppendorf) and incubate at room temperature for 10 min to allow lysis to occur.
Centrifuge the cell lysate at 20,000 × g for 2 min at room temperature to pellet any insoluble material.
Remove the resulting supernatant from the cell debris into a low binding 1.5 mL tube (Protein LoBind Tube 1.5 mL, Eppendorf).
Mix the cell extracts with equal volume of 100 % glycerol and aliquot into 40–60 μL portions in low binding 0.5 mL tubes (Protein LoBind Tube 0.5 mL, Eppendorf), labeled as “PPY SLiCE extract.”
Store the PPY SLiCE extract at −20 °C for about 2 months or at −80 °C for long-term storage, which can be thawed on wet ice and refrozen up to ten times without significant loss of activity.
3.2 Maintenance of PPY strain
Inoculate 1 single colony of PPY strain from LB agar plate (10 μg/mL streptomycin and 12.5 μg/mL chloramphenicol) into 5 mL LB medium (10 μg/mL streptomycin and 12.5 μg/mL chloramphenicol) and shake at 37 °C and 225–338 rpm overnight.
In a sterile tube mix an equal volume of PPY culture with 20 % autoclaved glycerol.
Store at −80 °C.
3.3 SLiCE Cloning
-
Preparation of vector and insert DNA.
PCR primer design and synthesis. The primers for SLiCE have the following characteristics: (1) The 5′-end of the primer contains 15–52 bases that are homologous to the sequence at one end of a DNA fragment for co-assembly or to other appropriate positions within a DNA fragment for co-assembly (i.e., the vector or another insert) (see Note 5); (2) The 3′-end of the primer contains 15–24 bases that are specific to the DNA fragment for amplification; (3) The primers do not need any further special treatment or modification except for desalting.
Preparation of vector DNA. Linearize vector DNA used for SLiCE by restriction digestion (see Note 6) or PCR amplification (see Notes 5 and 7). For BAC SLiCE cloning, PCR amplify the vectors using primers containing 5′-end homologies to the target fragments described as above (see Notes 5 and 7).
Preparation of insert DNA. Amplify the cloning inserts using PCR with primers containing 5′-end homologies to the vector or to other inserts for co-assembly described as above (see Notes 5 and 7). For BAC SLiCE cloning, digest the BAC DNA with restriction enzymes.
DNA purification. Purify the vector and insert DNAs by gel extraction, column-based purification methods, phenol/chloroform extraction, or other DNA purification methods and elute in EB (10 mM Tris-Cl, pH 8.5) or ddH2O (see Note 8). For BAC SLiCE cloning, purify the restriction-digested BAC DNA fragments by phenol/chloroform extraction.
Alternatively, the DNA purification step can be omitted, especially in simple cloning approaches (see Note 9).
-
SLiCE Reaction.
-
Transformation (see Note 12).
Electroporate 1 μL SLiCE reaction mix into 20 μL ElectroMAX DH10B™ Cells (Invitrogen) or chemically transform 1 μL SLiCE reaction mix into 100 μL MAX Efficiency® DH10B™ Competent Cells (Invitrogen) following the manufacturer’s instruction. Other competent cells used for DNA cloning can also be used.
Plate the transformed cells onto agar plates containing appropriate antibiotics.
Table 1.
SLiCE reaction conditions
| Components | Standard cloning | Rush cloning | BAC cloning |
|---|---|---|---|
| Vector DNA | Purified, 50–200 ng | Unpurified, 1 μLa | Purified, 50–200 ng |
| Insert DNA | Purified, 1:1–10:1b | Unpurified, 1 μLa | Phenol/chloroform-purified, 5–10 μg restriction enzyme digested BAC fragments |
| 10× SLiCE buffer | 1 μL | 1 μL | 1 μL |
| PPY SLiCE extract | 1 μL | 1 μL | 1 μL |
| ddH2O | to 10 μL | to 10 μL | to 10 μL |
| Reaction conditions | |||
| Incubation temperature | 37 °C | 37 °C | 37 °C |
| Incubation time | 15 minc–1 h | 15 minc | 1 h |
The volume of vector and insert DNAs can be adjusted. Ensure that the volume of unpurified DNA does not exceed 1/5 of total reaction volume
Molar ratio of insert to vector
15 min-incubation is enough for most SLiCE cloning tasks with a slightly reduced efficiency compared with 1 h
Acknowledgments
This work was supported by National Institute of Health [1R01CA76329 to W.E., 1R01CA93484 to W.E].
Footnotes
The genotype of the PPY strain is as follows: F− endA1 recA1 galE15 galK16 nupG rpsLΔlacX74 Φ80lacZΔM15 araD139Δ(ara,leu)7697 mcrA Δ(mrr-hsdRMS-mcrBC) cynX:: [araC pBAD- redα EM7-redβ Tn5-gam] λ−. The PPY strain is recommended as a source of SLiCE extract for its highest efficiency and fidelity in our experiments. Other Rec A and restriction system-deficient strains such as DH10B or JM109 can also be used. For these strains, (1) the cloning efficiency is lower than PPY-derived SLiCE [4]; (2) the preparation of DH10B, JM109, or other bacterial strains cloning extracts follows the same protocol mentioned in Subheading 3.1, except the arabinose induction step (step 6) is omitted and appropriate antibiotics need to be used.
Centrifuge 500–1,000 μL bacterial cultures at top speed for 2 min and aspirate off all of the medium. Use ddH2O to resuspend the bacterial pellet and as a black control to measure the absorbance at wavelength = 600 nm using a spectrophotometer. Note that high OD600 readings must be calculated by diluting the sample in ddH2O to enable photometric measurement in the linear range between 0.1 and 0.5 OD600.
For preparation of PPY SLiCE extract, avoid using chloramphenicol for drug selection because it inhibits the growth of PPY strain significantly.
It typically requires 6–10 h to reach the ideal cell density at 330 rpm and 37 °C. Addition of 0.1 % glycerol to the culture can increase the growth rate. Avoid growing the PPY cells longer than 14 h.
In cases where DNA fragments for co-assembly (i.e., vectors linearized with restriction enzymes) have sticky ends, count the end homology length from the 3′-end of DNA fragments. The cloning efficiency of SLiCE increases with increasing end homology length in a range from 15 to 52 bp, but drops significantly when the end homologies are further increased [4].
The nature of vector and insert ends such as blunt ends or 3′ or 5′ sequence overhangs does not influence SLiCE efficiency or accuracy. However, the use of vectors with complementary 5′ or 3′ overhanging ends for SLiCE increases the formation of empty vector background colonies, which is probably due to annealing of the single stranded ends and vector recircularization in the bacterial extracts or in the transformed host cells. It is recommended to avoid linearizing vectors with a single sticky end restriction enzyme or pairs of restriction enzymes generating complementary 5′ or 3′ overhangs. If this cannot be avoided, blunt the vector ends before SLiCE reaction. In addition, the PCR generated vectors yield a 50–100-fold lower cloning efficiency than the restriction linearized vectors isolated from regular bacterial host strains such as DH10B or DH5α. A reason for the lower cloning efficiencies of PCR amplified vectors may be the lack of DNA modifications (such as methylation) in the vector backbone that are introduced during replication in the host bacteria and that might enhance the cloning efficiencies.
Digest PCR products using plasmid DNA as templates with DpnI prior to purification or SLiCE reaction to remove residual plasmid template DNA (add 1–2 μL DpnI into a standard 30 μL PCR, at 37 °C for 15–60 min). DpnI works fine in PCR buffers. DpnI treatment can decrease cloning background efficiently in some cases. If unpurified DpnI-treated PCR products are subjected to SLiCE reaction directly, inactivate it at 60 °C for 20 min.
For gel purification, it is recommended to use new prepared agarose gels and unused TAE or TBE for electrophoresis to avoid DNA damage caused by inappropriate pH and other factors. Gel purification can be replaced by other DNA purification methods such as column-based purification methods or phenol/chloroform extraction. Note that these methods sometimes lead to higher background caused by uncut vector plasmid or unspecific PCR products.
Limited volume of unpurified vector or insert DNA prepared from general PCR or restriction system leads to a significantly reduced but still acceptable SLiCE cloning efficiency, especially for simple cloning. Note that heat inactivation is required when the unpurified DNA contains the restriction enzymes that recognize the restriction sites of recombinant DNA molecules.
Before use, thaw 10× SLiCE buffer at 37 °C and vortex vigorously to dissolve any precipitated material. Thaw SLiCE extract stored at −80 °C on ice and vortex.
We provide three SLiCE reaction systems (see Table 1). Standard cloning is for general cloning of one or multiple DNA fragments into vectors [4]. Rush cloning is an alternative approach mainly for simple cloning. BAC cloning is for directional subcloning of DNA fragments from BAC vectors [4].
Standard transformation methods such as electroporation and chemical transformation are all compatible with SLiCE cloning. For cloning of large or complex DNA fragments, electroporation is recommended (yielding 10–100-fold higher transformation efficiencies than that of chemical transformation).
References
- 1.Smith HO, Wilcox KW. A restriction enzyme from Hemophilus influenzae. I. Purification and general properties. J Mol Biol. 1970;51:379–391. doi: 10.1016/0022-2836(70)90149-x. [DOI] [PubMed] [Google Scholar]
- 2.Danna K, Nathans D. Specific cleavage of simian virus 40 DNA by restriction endonuclease of Hemophilus influenzae. Proc Natl Acad Sci U S A. 1971;68:2913–2917. doi: 10.1073/pnas.68.12.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen SN, Chang AC, Boyer HW, et al. Construction of biologically functional bacterial plasmids in vitro. Proc Natl Acad Sci U S A. 1973;70:3240–3244. doi: 10.1073/pnas.70.11.3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y, Werling U, Edelmann W. SLiCE: a novel bacterial cell extract-based DNA cloning method. Nucleic Acids Res. 2012;40(8):e55. doi: 10.1093/nar/gkr1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Little JW. An exonuclease induced by bacteriophage lambda. II. Nature of the enzymatic reaction. J Biol Chem. 1967;242(4):679–686. [PubMed] [Google Scholar]
- 6.Radding CM, Carter DM. The role of exonuclease and beta protein of phage lambda in genetic recombination. 3. Binding to deoxyribonucleic acid. J Biol Chem. 1971;246(8):2513–2518. [PubMed] [Google Scholar]
- 7.Carter DM, Radding CM. The role of exonuclease and beta protein of phage lambda in genetic recombination. II. Substrate specificity and the mode of action of lambda exonuclease. J Biol Chem. 1971;246(8):2502–2512. [PubMed] [Google Scholar]
- 8.Lovett ST, Hurley RL, Sutera VA, et al. Crossing over between regions of limited homology in Escherichia coli. RecA-dependent and RecA-independent pathways. Genetics. 2002;160:851–859. doi: 10.1093/genetics/160.3.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dutra BE, Sutera VA, Lovett ST. RecA-independent recombination is efficient but limited by exonucleases. Proc Natl Acad Sci U S A. 2007;104:216–221. doi: 10.1073/pnas.0608293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuzminov A. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol Mol Biol Rev. 2002;63:751–813. doi: 10.1128/mmbr.63.4.751-813.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]