

Abstract
Identifying cognitive and neural mechanisms involved in the development of schizophrenia requires longitudinal observation of individuals prior to onset. Here recent studies of prodromal individuals who progress to full psychosis are briefly reviewed in relation to models of schizophrenia pathophysiology. Together, this body of work suggests that disruption in brain connectivity, driven primarily by a progressive reduction in dendritic spines on cortical pyramidal neurons, may represent a key triggering mechanism. The earliest disruptions appear to be in circuits involved in referencing experiences according to time, place, and agency, which may result in a failure to recognize particular cognitions as self-generated or to constrain interpretations of the meaning of events based on prior experiences, providing the scaffolding for faulty reality testing.
Schizophrenia, psychosis, and the prodromal risk paradigm
Delusions, such as a belief that alien forces control one’s thoughts, and hallucinations, such as hearing voices, are core features of schizophrenia. Broadly, these symptoms imply an impairment of reality testing, or a disturbance in interpreting the sources and meaning of experience. Rather than representing a discrete module whereby experiences are tagged as “real” versus “unreal,” reality testing seems to reflect an emergent property of perceptual, attentional, mnemonic, and other systems operating in tandem to construct an interpretive/predictive framework for determining the “what,” “when,” “where,” and “by whom” of events reaching consciousness on a moment-to-moment basis [1]. As such, many have come to view schizophrenia as fundamentally a disorder of dysconnection within and between certain functional networks in the brain [2, 3]. Challenges remain, however, with respect to identifying which networks are critically affected and what levels or patterns of dysconnection are sufficient for psychotic symptom expression. Because schizophrenia is complexly determined, clinically heterogeneous, and (frequently) chronic and debilitating, neuroimaging studies comparing those with and without this condition can not by themselves differentiate which cognitive and neural changes are primary causes of particular symptoms, which are epiphenomena, and which are secondary to factors associated with chronicity of illness or antipsychotic drug treatment. Compounding this issue, different psychotic symptoms may involve different sets of network interactions, and certain symptoms may develop as secondary adaptations to (or explanations of) other symptoms.
A crucial aim is thus isolation of the changes in cognition and neural signaling immediately preceding the onset of psychosis that, by virtue of their temporal priority, may represent primary mechanisms in the cascades of events leading to the emergence of impaired reality testing. Identifying such changes requires a paradigm for ascertaining at risk individuals prior to psychosis onset and following them over time. Such a paradigm has been developed, based on the observation that the first psychotic episode is often preceded by the emergence of subtler changes in belief, thought, and/or perception that appear to represent attenuated forms of delusions, formal thought disorder, and hallucinations, respectively (see Box 1). Among individuals aged 12 to 35 years with a recent onset of such symptoms, termed “prodromal” or clinical high-risk (CHR) cases, approximately 20–35% develop fully psychotic symptoms over a 2-year period [4], with most of the conversions occurring during the first year following ascertainment and a decelerating conversion rate thereafter [5]. The vast majority of CHR cases who convert to psychosis develop a disorder in the schizophrenia spectrum, though some develop psychotic forms of depression or bipolar disorder (termed “affective psychoses”). In the material that follows, recent findings on cognitive and neural markers in CHR samples are evaluated with respect to their ability to inform and constrain models of the mechanisms involved in the onset of psychosis, with particular reference to schizophrenia. A special emphasis is placed on those markers that change dynamically (i.e., worsen) as one moves from a pre-onset, at risk, vulnerability state to a fully psychotic state, as such markers have the potential to hint at mechanisms representing proximal sufficient causes of psychosis (see Box 2). Discussion of this work is organized according to three prominent theories of schizophrenia onset – the neurodevelopmental model [6], the excitation-inhibition imbalance model [7], and the dopamine hypothesis [8]. Considered from the perspective of the CHR literature, rather than representing mutually exclusive explanations, the brain systems and processes implicated in these models are probably interconnected influences in the cascading changes in cognition and neural signaling underlying onset of psychosis. Though built on the foundation of these established models, many of the ideas advanced below with regard to the temporal sequencing and causal primacy of putative mechanisms are novel and in some cases necessarily speculative, given that the temporal sequencing of processes leading to onset of psychosis has not been an area of inquiry in the field until quite recently. Nevertheless, this theoretical framework provides a number of critical predictions that can be tested and potentially disconfirmed, leading to further model building and refinement.
Box 1. What is the prodrome to schizophrenia?
About 80–90% of patients with schizophrenia have a “prodrome” characterized by the emergence of attenuated or sub-threshold symptoms that appear to be on a continuum with delusions and hallucinations [93]. Common prodromal symptoms include perplexity, unusual and overvalued beliefs, guardedness, and hearing indistinct noises [48]. The dividing line between prodromal and psychotic intensity is based on the symptoms becoming more frequent, pervasive, and impairing, with loss of insight as to the non-reality basis of the beliefs and experiences. Although there is some variability, the prodromal phase on average lasts about 1 year [94]. Operational criteria have been developed to ascertain individuals in a prodromal or clinical high-risk (CHR) state, instantiated in two semi-structured interviews: the Comprehensive Assessment of At-Risk Mental States [95] and the Structured Interview for Prodromal Risk Syndromes [96]. It is important to note that CHR patients are generally distressed and seeking help, typically for mood/anxiety issues and/or school failure, and often keep their changing thoughts and perceptions to themselves until specifically asked about these experiences during screening. Given age at onset distributions for psychosis, screening is recommended for such presentations among individuals aged 12–35 years.
The CHR construct is a potent predictor of psychosis. According to a meta-analysis incorporating data from 27 studies comprising a total of 2,502 patients [4], 22% of such cases transitioned to a fully psychotic form of illness by 1-year and 36% by 3-years from initial ascertainment. (Conversion rates appear to vary according to referral pathways for CHR cases [97].) Because most studies employ follow-up periods of 3-years or less, the rate of conversions after this point remains unclear. Nevertheless, most of the conversions occur during the first year following ascertainment, and the conversion rate significantly decelerates thereafter, suggesting that the CHR criteria are sensitive to an imminent risk for onset of full psychosis [5]. Among those who convert, about 80% of the diagnostic outcomes are in the schizophrenia spectrum and the remaining 20% are in relation to mood-related and atypical forms of psychosis. Importantly, among the cases who do not convert, roughly half have been observed to remit the symptoms that indexed their initial risk status and improve functionally, while the remainder show continuing levels of attenuated psychotic-like symptoms and functional impairment [98, 99] (see Figure I).
Figure I.
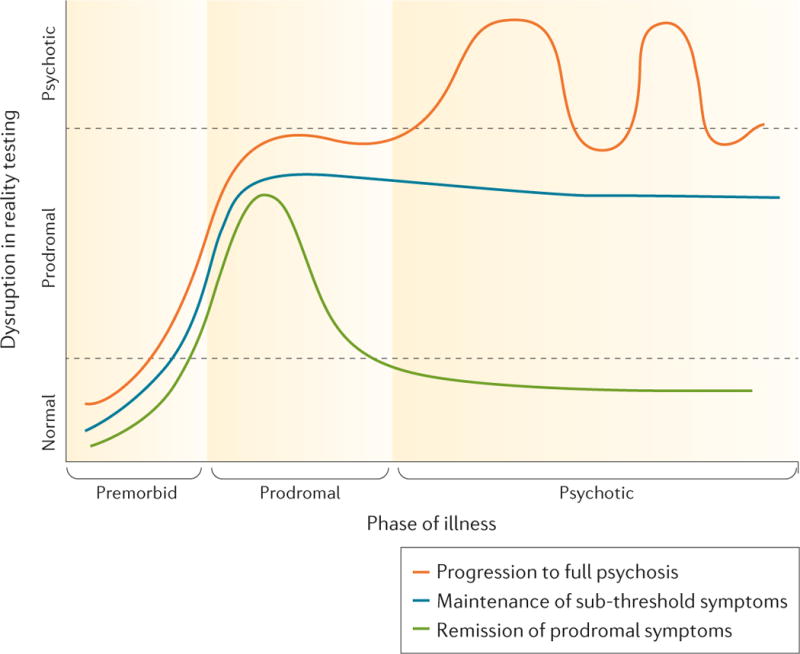
Descriptive model of the onset and course of psychotic symptoms among individuals who develop a prodromal risk syndrome. Approximately one-third of prodomal patients progress to full psychosis (red line), one-third maintain stable levels of sub-threshold symptoms (blue line), and one-third remit the prodromal symptoms (green line). Reprinted by permission from Macmillan Publishers Ltd: Nature Disease Primers [102], copyright 2015.
Box 2. Determining causes and mechanisms in studies of human psychopathology.
The causes of schizophrenia are complex, involving on the order of thousands of common and rare genetic variants [100], neurally-disruptive influences at both early and later stages of development [16], social stress [15], and other factors. Most theories postulate one or more final common pathways through which multiple different etiologic factors can influence disease risk. If psychosis reflects dyscoordination among systems governing source memory, prediction error signaling, attentional salience, etc., different aggregations of multiple etiological factors may be sufficient to perturb connectivity within and between these circuits to a degree that manifests in overt psychosis. The resulting circuit perturbations would thus be considered proximal sufficient causes of psychosis.
A crucial issue related to establishing whether a particular cognitive or neural trait reflects a sufficient condition for psychosis is whether it changes as one moves from a pre-onset, at risk, vulnerability state to a fully psychotic state. A marker that is stably deviant in those who develop psychosis from pre- to post-onset may be necessary, but clearly is not sufficient for psychotic symptom formation. Conversely, a marker that worsens as symptoms worsen in the ramp-up to full psychosis has the potential to represent a proximal sufficient cause of psychosis (see Figure II).
Most studies of the causes of psychosis are observational in nature, making it difficult to establish causation definitively. Exceptions include animal models and prevention studies that use experimental procedures, including random assignment of subjects to conditions. However, it is difficult to construct animal models that can adequately reflect the complexity of a phenomenon defined with reference to disruptions in the conscious interpretation of experience. Prevention studies could potentially be used to help prove causality through inverse deduction (i.e., by removing factor A, B is no longer observed), but it is critical to have well-informed intervention targets that are likely to be the correct ones before embarking on such studies. Although observational studies are not generally capable of proving causality in as definitive a way as an experiment, use of a prospective, longitudinal design permits analyses assessing for temporal precedence and meditational influences among numerous (potentially interacting) variables [90–92]. Such studies can significantly advance a field by helping to rule out competing theories whose critical predictions are refuted by patterns in the data [101]. In particular, if factor B is shown to occur temporally after factor A, B is unlikely to be a cause of factor A; this does not then prove that factor A is a cause of factor B, but it refutes models in which the reverse is held to be true.
Figure II.
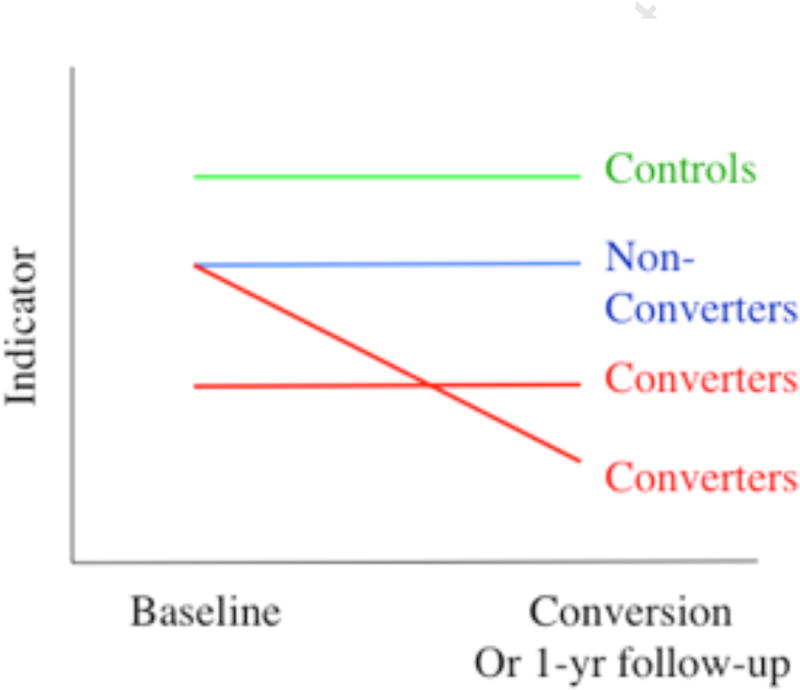
Hypothetical trajectories of indicators across illness phases. Different cognitive and neural mechanisms may be stably deviant in CHR cases from the pre- to post-onset phase (in which case they could be necessary, but not sufficient, for symptom onset) or may deteriorate during this period (in which case, they could represent a sufficient cause for symptom formation).
Neurodevelopment, medial prefrontal cortex, and errors in source monitoring
Brain dysconnectivity in schizophrenia is thought to derive primarily from deficits in dendritic spines that arise during development [3], though axonal pathology (e.g., disrupted myelination) may also play a role [9]. Such deficits are likely to be present at least in part in some cases from birth, resulting from an interplay of genetic factors and obstetric complications [6, 10], but may progress beyond a threshold critical for expression of psychotic symptoms as a function of normal neuromaturational events (i.e., apoptosis and synaptic pruning) during adolescence [11]. In other cases, dysconnectivity may emerge during late adolescence and early adulthood as a consequence of overly aggressive synaptic pruning [12, 13] and/or other aberrant processes, such as elevated cortisol leading to dendritic atrophy [14, 15]. The contributions of early (pre- and perinatal) and later (adolescent) brain developmental processes to psychosis risk are not mutually exclusive, and both sets of processes may be operative in some cases (Fig 1) [16].
Figure 1.
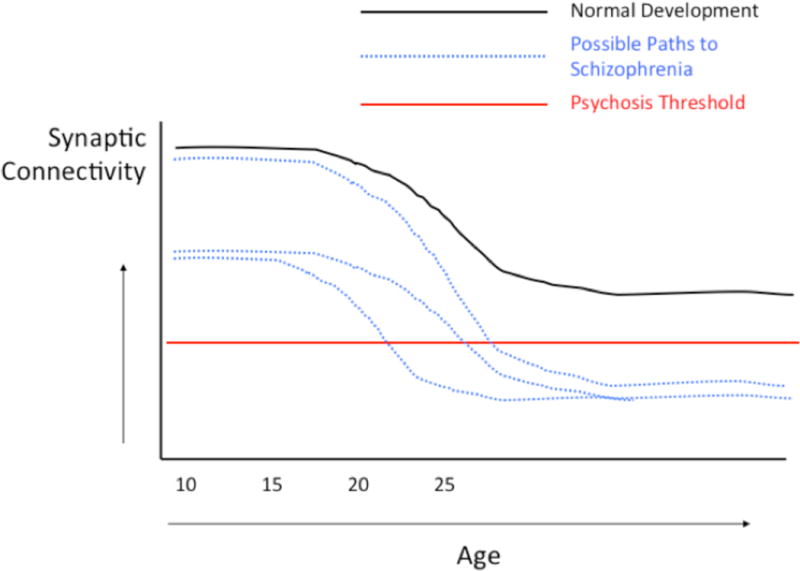
Developmental trajectories of cortical synaptic connectivity, including a number of trajectories (blue) in which connectivity is reduced below a hypothetical threshold sufficient for expression of psychotic symptoms.
Although the models just described have existed in some form or another for nearly 30 years, until recently there has been very little in the way of direct empirical tests bearing on the question of whether a progressive change in brain connectivity is associated with the emergence of psychosis. Several prospective longitudinal neuroimaging studies of CHR cases have now been published [17–24]. In the largest study, and the only one to employ a stringent control for multiple testing throughout the brain, CHR converters to psychosis were found to show a steeper rate of gray matter thinning in right superior and medial prefrontal cortex (PFC) (Fig 2a) [22]. Critically, converters who had not been exposed to antipsychotics during the interscan interval showed significantly greater thinning of PFC than CHR non-converters (regardless of medication status) and healthy controls. This finding represents a critical advance, as antipsychotics clearly complicate the interpretation of progressive brain changes among patients with established illness [25–28].
Figure 2.
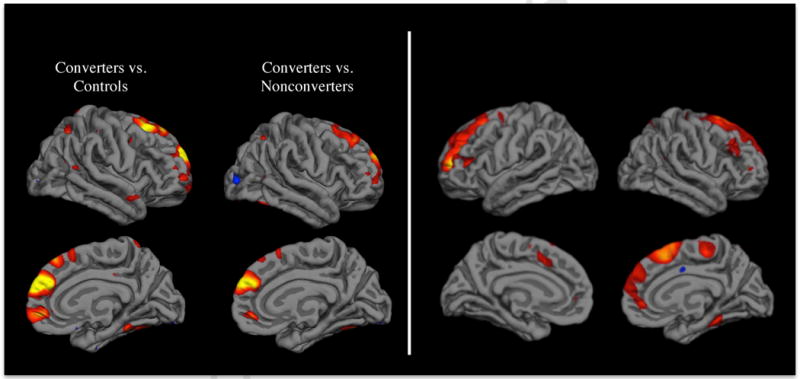
(Left) Cortical surface maps showing regions in which converters to psychosis had significantly greater progressive loss of gray matter thickness compared with non-converters and controls [22]; (Right) Cortical surface maps showing regions in which higher levels of unusual thought content at baseline predicted a steeper rate of gray matter thinning among converters [47].
Given that the accelerated gray matter loss associated with psychosis onset does not appear to be secondary to exposure to antipsychotic medications, it could be related to factors that participate in the pathophysiology of schizophrenia and related disorders, such as neuroinflammation [29, 30]. Neuroinflammatory markers are elevated in postmortem neural tissue from patients with schizophrenia [31], and these same markers are associated with microglial-mediated synaptic pruning and dendritic retraction in animal models [32, 33], providing a potential mechanistic basis for the reduced synaptic density seen in patients [3]. In the recent longitudinal imaging study of CHR subjects, higher levels of proinflammatory cytokines at baseline were strongly predictive of steeper rates of gray matter reduction in superior and medial PFC among those who converted to psychosis [22]. The fact that physiological triggers of microglial activation predict the rate of gray matter thinning on MRI supports the view that progressive gray matter change in this context is likely to reflect dendritic retraction and synaptic pruning at the cellular level, a hypothesis also supported by evidence on positron emission tomography (PET) scan of greater activated microglia in patients with schizophrenia in some [34, 35] but not all [36] studies.
The topography of regions showing accelerated change in converters to psychosis may provide important clues as to the role of these changes in symptom formation. In the recent study of cortical thickness [22], though there were medium to large effect sizes across most of bilateral prefrontal cortex as well as parahippocampal gyrus, superior temporal gyrus, and some circumscribed regions of parietal cortex, the effects were maximal in superior and medial aspects of PFC. Networks involving the medial PFC play critical roles in source monitoring during memory acquisition and retrieval, with particular relevance to whether items were actually perceived versus imagined and whether they were perceived or imagined by oneself or another person [37–40]. For example, while lateral prefrontal regions have a general role in memory retrieval, medial prefrontal regions exhibit differentially greater activity when remembering performed rather than imagined actions [37]. Further, structural variations in the paracingulate sulcus, located in the medial PFC, correlate with individual differences in performance on reality monitoring tasks [38]. These findings converge with evidence of differential impairment of episodic memory in schizophrenia, such that there is a deficit in deep or relational encoding, reducing contextual referencing of memories and making patients relatively more reliant on familiarity-based processes [41–44]. A similar deficit in episodic memory has been observed in CHR subjects [41], and those who later convert to psychosis have been found to show learning and memory deficits on clinical neuropsychological tests [45, 46]. The effect sizes on these measures in CHR cases who later convert are somewhat smaller than those seen in first-episode schizophrenia [41, 46], suggesting progression of episodic memory deficits from the prodromal to fully psychotic phases. However, whether there is progressive deterioration in episodic memory within CHR subjects who develop psychosis has not yet been examined.
Given that experience has a major impact on the formation of belief and in a way that depends critically on episodic memory, delusions may emerge as a consequence of a progressive loss of connectivity of regions involved in source monitoring during memory encoding and retrieval. That is, disruptions in source monitoring during learning and memory may lay the groundwork for the subsequent development of disruptions in belief evaluation. This hypothesis is supported by evidence that more severe unusual ideas and beliefs at baseline predict the steeper rate of gray matter loss in superior and medial PFC bilaterally among CHR cases progressing to full psychosis [47] (Fig 2b). As such, disruptions in source memory may underlie the earliest appearing prodromal symptoms, which include perplexity or confusion about what is real versus imaginary, changes in the perception of time, and changes in the interpretation of events and experiences such that the familiar begins to feel strange, confusing, ominous, threatening, or otherwise to have special meaning [48]. Over time, more elaborate beliefs – such as alien control of thought, ideas of reference, and systemized persecutory delusions – may be built up as explanations of the faulty source attributions and generalized sense of foreboding, and skepticism in these explanations may erode as an increasing proportion of experience salient in memory is subject to source confusion. The relationship of source memory to the pathogenesis of hallucinations is unclear, though hearing voices (the most common hallucinatory experience in schizophrenia) likely involves a disruption in source monitoring of internally generated ideation (or subvocal speech) [49, 50], and accelerated gray matter loss among converters to psychosis has been observed in regions relevant to both source monitoring and auditory verbal processing [20, 22]. A focus on deficits in network interactions supporting source memory may also help to explain the appearance of psychotic symptoms in patients with frontotemporal dementia [51], as well as the observation of relatively greater deficits in episodic memory among cases of mood disorders with psychotic features than among cases without psychotic features [52].
Whether deficits in source memory per se appear early and progress during the psychosis prodrome remains to be evaluated empirically. At this point, the link between source monitoring deficits and onset of psychosis in CHR cases is circumstantial, based primarily on the observations that brain regions active during source memory evaluations overlap with regions undergoing progressive cortical thinning during the development of psychosis. Relying on structural brain changes that can be measured using MRI to discern the networks involved in psychosis onset is problematic because, given overlap on the distributions of measures of cortical gray matter between patients with schizophrenia and healthy controls, it does not appear that there is a critical threshold on such measures below which psychosis develops, at least not in any one region. Although better discrimination may be achieved through multivariate pattern analysis [53], it seems likely that structural MRI measures are at some distance away from the levels of observation at which the sufficient conditions for psychosis can be ascertained. A challenge for this area going forward is thus to bridge from the relatively gross anatomical perspective afforded by MRI (which has nevertheless helped to identify a progressive marker with some localizing significance) to events at the molecular and physiological levels that presumably drive disrupted connectivity of regions supporting source monitoring and eventual psychosis. Initial findings of altered functional connectivity of multiple networks that include superior, medial, and lateral frontal nodes (e.g., default mode, salience, and executive) in CHR cases are promising [54–58], but thus far no studies have appeared examining whether these changes are linked specifically to disruptions in source monitoring or are progressive in the ramp up to full psychosis.
NMDA receptors, excitation-inhibition balance, and prediction error
Abusers of phencyclidine and ketamine have been observed to manifest a psychotic state reminiscent of many aspects of schizophrenia, including depersonalization, derealization, ideas of reference, paranoia, and perceptual abnormalities [59–61]. In parallel, low doses of ketamine exacerbate psychotic symptoms in patients with schizophrenia [62]. The psychotogenic effects of these compounds are driven by their antagonism of NMDA receptors, which are activated by the excitatory amino acid transmitter glutamate and several other molecules working collaboratively (i.e., co-agonists) [59–61]. Given that glutamatergic pyramidal cells synapse on a population of interneurons expressing the inhibitory transmitter gamma-aminobutryic acid (GABA) that in turn project back to the pyramidal cells, completing a set of local circuits [63], NMDA receptor hypofunction may result in an imbalance of excitation and inhibition that could contribute to the pathophysiology of schizophrenia [7].
Additional evidence of NMDA receptor involvement in schizophrenia comes from studies showing associations of the disorder with polymorphisms in genes that participate in the glutamate cascade [20, 64]. Magnetic resonance spectroscopy (MRS) studies have found evidence of increased glutamate levels in PFC among antipsychotic-naïve schizophrenia patients in their first psychotic episode [65] and increased glutamine in the anterior cingulate cortex in CHR cases [66], a pattern that appears consistent with disinhibition of pyramidal cells due to NMDA hypofunction at synapses on GABAergic interneurons; however, reduced glutamate/glutamine levels on MRS have also been observed in prefrontal cortex among patients with chronic schizophrenia [67] and in thalamus among CHR subjects [66]. Indirect evidence of NMDA involvement is supported by evidence of reduced amplitudes of the mismatch negativity (MMN) waveform in patients with schizophrenia [68] as well as in CHR cases who later convert to psychosis [69]. MMN is an electrophysiological evoked potential generated automatically when a deviant stimulus occurs during a series of standard stimuli. Given that MMN is dependent on NMDA receptors [70], and given that NMDA receptors in turn are critical for long-term potentiation (LTP), the cellular basis of learning and memory [71], reduced MMN in the prodromal and fully psychotic phases of schizophrenia is consistent with a disruption in NMDA-mediated synaptic plasticity.
If NMDA receptor hypofunction participates in the development of psychosis, we would expect markers of this disruption to worsen during the ramp up to full psychosis. Thus far, no longitudinal studies of markers of NMDA function in CHR subjects have appeared. However, the reductions in dendritic spines and synapse density noted in postmortem studies of patients with schizophrenia are localized primarily to glutamatergic pyramidal cells [3], and LTP and other processes dependent on NMDA receptors are critically involved in the synaptic pruning that characterizes normal adolescent brain development [72], thus linking the neurodevelopmental and excitation-inhibition imbalance models (Fig 3). In fact, experiencedependent plasticity of NMDA synapses is critical in this regard, as inactive synapses are targeted for pruning by activated microglia during this sculpting process [73]. Thus, deficits in NMDA-dependent synaptic plasticity may drive the accelerated gray matter loss observed in CHR cases who convert to psychosis.
Figure 3.
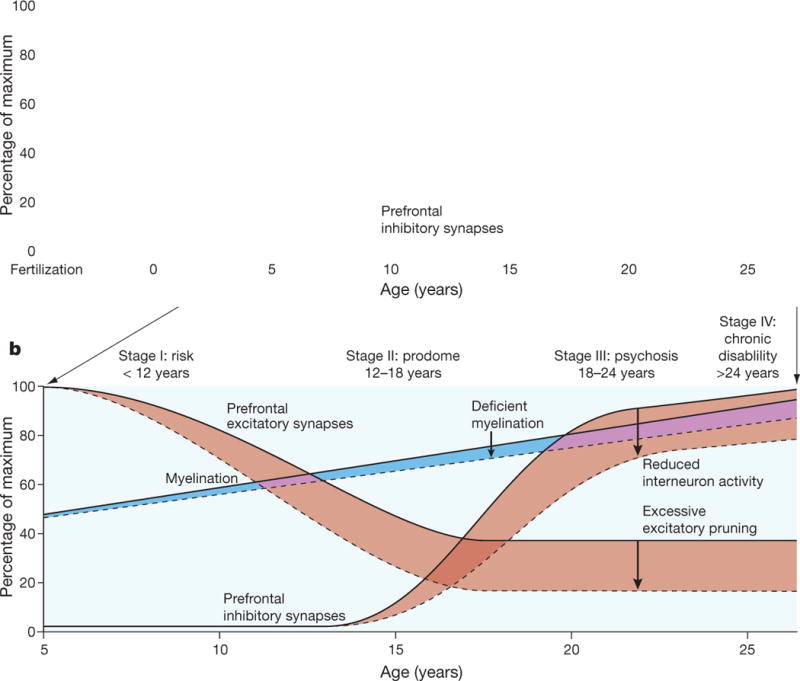
Schematic of neurodevelopmental changes resulting in disruptions in excitation-inhibition balance. Reprinted by permission from Macmillan Publishers Ltd: Nature [103], copyright 2010.
An emerging deficit in NMDA signaling may help to account for the formation of delusions by interrupting prediction error signals that mediate between beliefs and experiences [74]. In the initial phases of this process, the network of previously learned associations that guide reality construction and interpretation of on-going experience are challenged by new experiences inconsistent with these predictive codings, setting off a cacophony of prediction error signals, manifest phenomenologically as perplexity and confusion (the earliest precursors of delusions). As the winnowing away of NMDA receptors continues, the constraints of previously learned contingencies on the interpretation of new experiences are loosened, and prediction error signaling drops off. This process creates a permissive environment for the development of new explanations of experience (indeed, new constructions of reality). For example, the experience of ideation that is not perceived as self-generated appearing suddenly in consciousness (e.g., due to a disruption of the medial PFC networks coordinating self monitoring) may be explained as having been transmitted from an external source. Without the constraints of explanatory models based on previous contingencies (learned when LTP mechanisms were intact), such explanations are not rejected as improbable, as there is no longer a basis for generating the model-inconsistent error signals that would normally provide reality constraint.
A challenge for this theoretical perspective is explaining the fixity of delusional beliefs. One explanation is that there is a paradoxical strengthening of the associations (i.e., delusional elaborations) formed during states of NMDA hypofunction due to an absence of prediction error signals during memory reconsolidation [75].
Dopamine signaling and abnormal salience attributions
For many years, the central hypothesis of the field concerning the pathogenesis of schizophrenia focused on dopamine. This was based primarily on the fact that all drugs with antipsychotic potency block dopamine D2 receptors; conversely, drugs that stimulate dopamine release, including cocaine and amphetamine, produce short-lived psychotic states [8]. However, although there is an inherited polymorphism in the D2 receptor gene associating with the disorder [64], a functional receptor-based dopaminergic abnormality in schizophrenia has not been identified. Although some in vivo studies using PET have found evidence of increased density of dopamine D2/D3 type receptors in schizophrenia, the finding is not consistent and appears modest at best [76]. More recently, PET has been used to show that dopamine synthesis capacity, measured using radiolabelled L-DOPA (the precursor molecule for dopamine), is elevated in patients with schizophrenia, particularly while psychotic [76]. Further, amphetamine challenge is associated with an exaggerated increase in dopamine release and exacerbation of psychotic symptoms in these patients [77]. There is also evidence of increased dopamine synthesis capacity during the prodromal phase [78], with greater synthesis capacity in CHR patients who transition to psychosis compared with those who do not [79] and with greater increases in synthesis capacity over time among the converters [80]. These CHR studies, while performed by the same lab and based on small numbers of cases, are nevertheless provocative because they show increases in DA signaling prior to psychosis onset that are progressive and predictive of full psychosis.
Dopamine plays a central role in reward-based learning, such that in a particular context, dopamine release is associated with initiation of behaviors that predict reward [81]. Notably, in both prodromal and first-episode patients, the increases in dopamine synthesis are particularly pronounced in parts of the striatum that project to association cortex, including PFC [78]. If increased dopamine synthesis and release began to occur in contexts without prior reward-based contingencies, this could result in a heightened sense of salience of otherwise innocuous stimuli, which could plausibly play a role in the development of ideas of reference and paranoia [82].
Although the findings just summarized strongly implicate heightened dopamine synthesis and release as playing a role in psychotic symptoms, it is less clear where these abnormalities belong in the complex cascades of influences underlying onset of psychosis. With respect to the antipsychotic effects of D2 receptor antagonism, it is important to note that while both first-and second-generation antipsychotic drugs reduce the severity of psychotic symptoms in many patients, they are not curative, and long-term maintenance is required [83]. Further, antipsychotic drug treatment in the prodromal phase blunts symptom severity during active treatment, but short-term trials with these agents do not prevent the initial psychotic episode [84, 85]. In considering the relevance of dopamine to models of psychosis onset, we require a mechanism to explain increases in dopamine synthesis capacity and release during the late teens and early twenties, as symptoms progress from attenuated to fully psychotic levels of intensity. Stress could play a role, but thus far, there is no direct evidence linking stress or hypothalamic pituitary adrenal (HPA) axis activation to emergence of DA-related pathology in CHR cases. On the other hand, based on the cellular interactions of glutamate, GABA, and dopamine [63], the initiator may in fact be changes in NMDA receptor activity (Fig 4). Consistent with this view, NMDA hypofunction induced by PCP or ketamine results in changes in dopamine signaling that resemble the patterns observed in schizophrenia in terms of both the striatal (hyperdopaminergic) and cortical (hypodopaminergic) systems [86–88]. In view of these considerations, it seems reasonable to hypothesize that altered dopamine signaling is a downstream consequence of NMDA hypofunction in schizophrenia.
Figure 4.
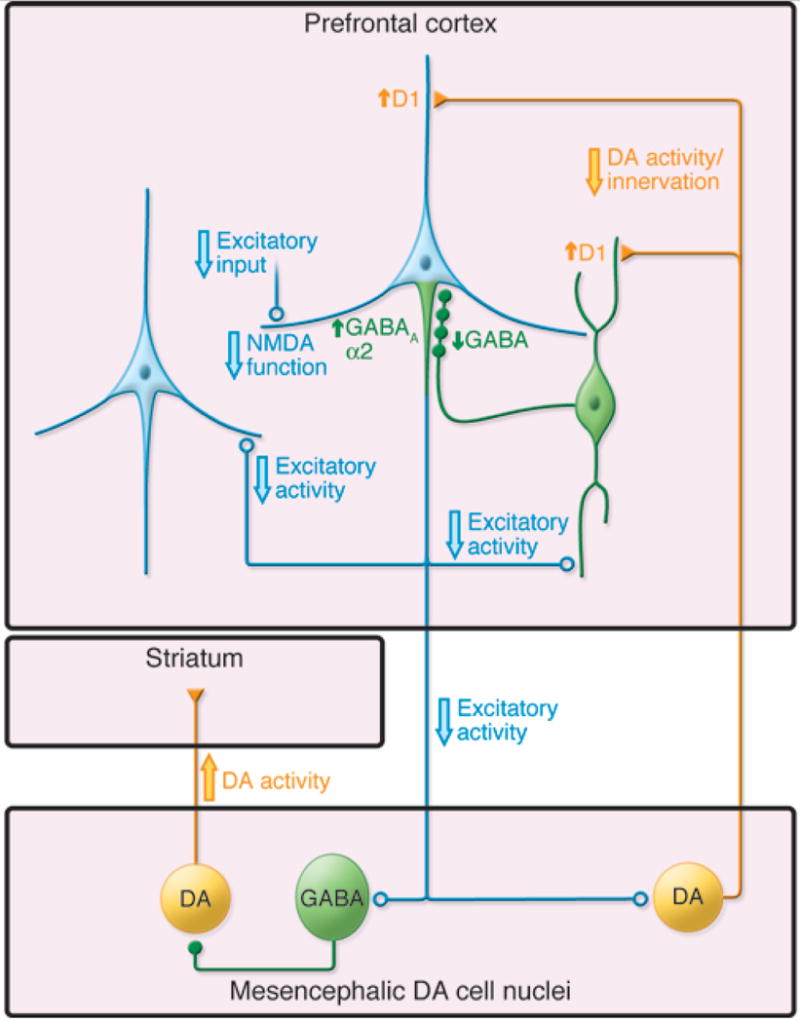
Circuit model of neuronal changes relevant to models of schizophrenia. Excitatory activity of cortical pyramidal neurons (light blue) is thought to be reduced in schizophrenia, most likely due to NMDA receptor hypofunction. Because of interactions with inhibitory, GABAergic interneurons (green), decreased excitatory input to neurons in the mesencephalon would lead to increased dopamine activity in the striatum and decreased dopamine activity in the cortex (yellow). Reprinted by permission from the American Society of Clinical Investigation: Journal of Clinical Investigation [63], copyright 2009.
Concluding remarks
It is hoped that knowledge of the mechanisms involved in the onset of psychosis will yield interventions that could prevent or mitigate the changes in cognition and neural signaling underlying symptom formation [89]. Understanding the sequencing of changes is crucial in this regard. Though the temporal sequencing of causal factors could be to some extent obfuscated by the emergence of prodromal symptoms themselves, at present there is no efficient way to study these processes longitudinally prior to onset of full psychosis except in CHR cases. Based on the foregoing, a disruption in brain connectivity, driven primarily by a reduction in dendritic spines expressing NMDA synapses that accelerates during late adolescence and early adulthood, is likely to represent a key triggering mechanism in the onset of psychosis. What factors cause this process to begin? A (non-exhaustive) list includes disrupted neurite outgrowth, deficient synaptic plasticity, overly aggressive pruning, excessive immune activation, and elevated cortisol. In principle, each of these processes could be mediated by microglial activation that preferentially impacts the balance in glutamate/GABA signaling in relation to cortical pyramidal cells. If it could be established that increasing neuroinflammation precedes and predicts changes in brain connectivity in pathways relevant to reality testing prior to onset of psychosis, targeting microglial activation in at risk individuals may be a promising preventive strategy. Alternatively, if dysregulated NMDA-dependent synaptic plasticity, reflected in an overabundance of weak synapses that are then pruned as a natural consequence of adolescent brain development, is the key driver, microglial activation would then represent a secondary signal (i.e., as in a clean-up operation), and strengthening of NMDA-related plasticity mechanisms would appear a more promising strategy. Parsing these alternatives will require research designs involving multiple points of observation prior to onset of psychosis and application of time-lagged and growth curve analytic approaches [90, 91] that can help to establish temporal precedence and mediation among multiple cascading influences over time [92]. Of course, it is also possible that the dysconnectivity underlying the development of psychosis results from different initiating causes in different sets of patients, in which case, biomarkers may be helpful for selecting the appropriate intervention for a given patient. Whatever the origins of the progressive changes in connectivity among CHR cases, if we are to be successful in preventing onset of full psychosis, restoration of source monitoring, prediction error signaling, and/or memory-driven salience attributions during the interpretation of experience may represent key rate limiting steps.
Outstanding Questions Box.
Does progressive gray matter reduction in clinical high risk (CHR) cases who convert to psychosis drive changes in functional connectivity in networks critical for source monitoring, prediction error signaling, and/or attentional salience, and are these changes in functional connectivity progressive in the ramp up to full psychosis?
Does altered functional connectivity of networks involved in source monitoring, prediction error signaling, and/or attentional salience define a state of the brain sufficient for psychotic symptom formation? That is, is there a level or pattern of activity and connectivity within and between these networks that perfectly discriminates between those who do and do not manifest psychotic symptoms (and between periods of active psychosis versus remission within the same case)?
Does increasing microglial activation precede and predicts changes in brain connectivity in pathways relevant to reality testing prior to onset of psychosis, and if so, can intervention with anti-inflammatory agents prevent the progressive changes in brain structure and function and the emergence of psychotic symptoms?
Does dysregulated NMDA-dependent synaptic plasticity result in an overabundance of weak synapses that are then pruned as a natural consequence of adolescent brain development in the ramp up to full psychosis? If so, can interventions that enhance synaptic activity and plasticity prevent the progressive changes in brain structure and function and the emergence of psychotic symptoms?
Are different initiating mechanisms (e.g., neuroinflammation, plasticity deficits) differentially relevant in different cases, and if so, could biomarkers be used to select interventions specifically targeting the mechanisms relevant to particular cases?
Trends Box.
Young people who are distressed and treatment seeking and presenting with attenuated forms of delusions and hallucinations have a 20–35% risk for developing full psychosis within 2-years; such individuals have a clinical high-risk (CHR) syndrome.
CHR cases who convert to psychosis show a steeper rate of gray matter reduction, most pronounced in superior and medial prefrontal cortex (PFC), and manifest deficits in source monitoring during encoding and retrieval of information in memory, functions critically dependent on networks involving medial PFC. Abnormal source attributions in turn may a play a role in the development of delusions and hallucinations.
CHR cases who convert to psychosis have deficits in a physiological marker of synaptic plasticity, which may help to explain changes in prediction error signaling thought to underlie disrupted reality testing.
CHR cases who convert to psychosis show increases in dopamine synthesis capacity, which may underlie the aberrant salience attributions associated with paranoia and ideas of reference.
Acknowledgments
Tyrone Cannon receives funding from the NIH and is a consultant to the Los Angeles County Department of Mental Health and Boehringer Ingelheim Pharmaceuticals. The author wishes to thank multiple collaborators and colleagues for discussions over the past several years that contributed to many of the ideas in this manuscript, including Diana Perkins, Daniel Mathalon, Marcia Johnson, Phillip Corlett, Elaine Walker, Scott Woods, Alan Anticevic, Amy Arnsten, Robert Heinssen, Thomas McGlashan, John Krystal, Jean Addington, Larry Seidman, Kristin Cadenhead, Barbara Cornblatt, Carrie Bearden, Ming Tsuang, and Marvin Chun.
Glossary
- Apoptosis
a normative developmental process of programmed death of cells (e.g., neurons) that are no longer needed.
- Cytokines
signaling molecules in the immune system that control inflammatory as well as cellular regeneration and repair processes.
- Dendritic atrophy
degeneration of the branching components of neurons that help to support inter-neuronal communication.
- Derealization
a state in which the external world is perceived as unreal, often associated with perplexity.
- Depersonalization
a state in which one’s thoughts and feelings seem unreal or not to belong to oneself.
- Dopamine
a neurotransmitter critical for reward-based learning processes that participates in signaling the incentive value (attentional salience) of stimuli.
- Frontotemporal dementia
a degenerative brain syndrome characterized by progressive loss of tissue in the frontal and temporal lobes.
- Ideas of reference
the experience of innocuous or coincidental events as having strong personal significance.
- Microglia
resident immune cells in the brain that when activated, help to sculpt cortical circuits by selectively eliminating inactive synapses.
- Myelination
the development of an insulating fatty sheath around the axons of neurons that increases the conduction velocity of electrical signals.
- Neurite
any projection from the cell body of a neuron, as in a dendrite or axon, that supports inter-neuronal communication.
- NMDA receptors
key receptor molecules for the excitatory neurotransmitter glutamate, their activation is critical for long-term potentiation, the cellular basis of learning and memory, and for prediction error signaling; when blocked by certain drugs (phencyclidine, ketamine), associated with psychotic symptoms.
- Prodromal or clinical high-risk syndrome
a syndrome in a distressed and treatment seeking young person presenting with attenuated forms of delusions and hallucinations, associated with a heightened risk for conversion to full psychosis within 2–3 years.
- Prediction error
neural signals that indicate when a particular observation appears inconsistent with prediction, based on an internal schema or model explaining the meaning of such observations given prior learning.
- Psychosis
a clinical state in which reality testing is disrupted, manifest as delusions (false beliefs) and hallucinations (perceptions in the absence of objective sensation).
- Salience
the incentive value of stimuli, as encoded neurally and expressed in effects on attention and other cognitive processes.
- Schizophrenia
a psychiatric syndrome in which psychotic symptoms such as delusions and hallucinations are prominent.
- Source memory
memory for the context of learning, with particular relevance to whether the item, event, experience, etc., was real or imagined and by what agent (oneself or someone else).
- Synaptic pruning
a process of normal adolescent brain development by which weak or inactive synapses are eliminated, resulting in increased efficiency of cortical networks.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Johnson MK. Memory and reality. Am Psychol. 2006;61:760–771. doi: 10.1037/0003-066X.61.8.760. [DOI] [PubMed] [Google Scholar]
- 2.Stephan KE, et al. Dysconnection in schizophrenia: from abnormal synaptic plasticity to failures of self-monitoring. Schizophr Bull. 2009;35:509–527. doi: 10.1093/schbul/sbn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glausier JR, Lewis DA. Dendritic spine pathology in schizophrenia. Neuroscience. 2013;251:90–107. doi: 10.1016/j.neuroscience.2012.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fusar-Poli P, et al. Predicting psychosis: meta-analysis of transition outcomes in individuals at high clinical risk. Arch Gen Psychiatry. 2012;69:220–229. doi: 10.1001/archgenpsychiatry.2011.1472. [DOI] [PubMed] [Google Scholar]
- 5.Cannon TD, et al. Prediction of psychosis in youth at high clinical risk: a multisite longitudinal study in North America. Arch Gen Psychiatry. 2008;65:28–37. doi: 10.1001/archgenpsychiatry.2007.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fatemi SH, Folsom TD. The neurodevelopmental hypothesis of schizophrenia, revisited. Schizophr Bull. 2009;35:528–548. doi: 10.1093/schbul/sbn187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gonzalez-Burgos G, Lewis DA. NMDA receptor hypofunction, parvalbumin-positive neurons, and cortical gamma oscillations in schizophrenia. Schizophr Bull. 2012;38:950–957. doi: 10.1093/schbul/sbs010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III–the final common pathway. Schizophr Bull. 2009;35:549–562. doi: 10.1093/schbul/sbp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karlsgodt KH, et al. Structural and Functional Brain Abnormalities in Schizophrenia. Curr Dir Psychol Sci. 2010;19:226–231. doi: 10.1177/0963721410377601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karlsgodt KH, et al. The neurodevelopmental hypothesis of schizophrenia. In: David AS, et al., editors. Schizophrenia: The Final Frontier. Psychology Press; 2011. [Google Scholar]
- 11.Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry. 1987;44:660–669. doi: 10.1001/archpsyc.1987.01800190080012. [DOI] [PubMed] [Google Scholar]
- 12.Feinberg I. Schizophrenia: caused by a fault in programmed synaptic elimination during adolescence? J Psychiatr Res. 1982;17:319–334. doi: 10.1016/0022-3956(82)90038-3. [DOI] [PubMed] [Google Scholar]
- 13.McGlashan TH, Hoffman RE. Schizophrenia as a disorder of developmentally reduced synaptic connectivity. Arch Gen Psychiatry. 2000;57:637–648. doi: 10.1001/archpsyc.57.7.637. [DOI] [PubMed] [Google Scholar]
- 14.Wellman CL. Dendritic reorganization in pyramidal neurons in medial prefrontal cortex after chronic corticosterone administration. J Neurobiol. 2001;49:245–253. doi: 10.1002/neu.1079. [DOI] [PubMed] [Google Scholar]
- 15.Walker E, et al. Stress and the hypothalamic pituitary adrenal axis in the developmental course of schizophrenia. Annu Rev Clin Psychol. 2008;4:189–216. doi: 10.1146/annurev.clinpsy.4.022007.141248. [DOI] [PubMed] [Google Scholar]
- 16.Cannon TD, et al. Early and late neurodevelopmental influences in the prodrome to schizophrenia: contributions of genes, environment, and their interactions. Schizophr Bull. 2003;29:653–669. doi: 10.1093/oxfordjournals.schbul.a007037. [DOI] [PubMed] [Google Scholar]
- 17.Borgwardt SJ, et al. Reductions in frontal, temporal and parietal volume associated with the onset of psychosis. Schizophr Res. 2008;106:108–114. doi: 10.1016/j.schres.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 18.Pantelis C, et al. Neuroanatomical abnormalities before and after onset of psychosis: a crosssectional and longitudinal MRI comparison. Lancet. 2003;361:281–288. doi: 10.1016/S0140-6736(03)12323-9. [DOI] [PubMed] [Google Scholar]
- 19.Sun D, et al. Progressive brain structural changes mapped as psychosis develops in ‘at risk’ individuals. Schizophr Res. 2009;108:85–92. doi: 10.1016/j.schres.2008.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takahashi T, et al. Progressive gray matter reduction of the superior temporal gyrus during transition to psychosis. Arch Gen Psychiatry. 2009;66:366–376. doi: 10.1001/archgenpsychiatry.2009.12. [DOI] [PubMed] [Google Scholar]
- 21.Ziermans TB, et al. Progressive structural brain changes during development of psychosis. Schizophr Bull. 2012;38:519–530. doi: 10.1093/schbul/sbq113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cannon TD, et al. Progressive reduction in cortical thickness as psychosis develops: a multisite longitudinal neuroimaging study of youth at elevated clinical risk. Biol Psychiatry. 2015;77:147–157. doi: 10.1016/j.biopsych.2014.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takahashi T, et al. Insular cortex gray matter changes in individuals at ultra-high-risk of developing psychosis. Schizophr Res. 2009;111:94–102. doi: 10.1016/j.schres.2009.03.024. [DOI] [PubMed] [Google Scholar]
- 24.Walter A, et al. Hippocampal volume in subjects at high risk of psychosis: a longitudinal MRI study. Schizophr Res. 2012;142:217–222. doi: 10.1016/j.schres.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 25.Dorph-Petersen KA, et al. The influence of chronic exposure to antipsychotic medications on brain size before and after tissue fixation: a comparison of haloperidol and olanzapine in macaque monkeys. Neuropsychopharmacology. 2005;30:1649–1661. doi: 10.1038/sj.npp.1300710. [DOI] [PubMed] [Google Scholar]
- 26.Fusar-Poli P, et al. Progressive brain changes in schizophrenia related to antipsychotic treatment? A meta-analysis of longitudinal MRI studies. Neurosci Biobehav Rev. 2013;37:1680–1691. doi: 10.1016/j.neubiorev.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ho BC, et al. Long-term antipsychotic treatment and brain volumes: a longitudinal study of first-episode schizophrenia. Arch Gen Psychiatry. 2011;68:128–137. doi: 10.1001/archgenpsychiatry.2010.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Navari S, Dazzan P. Do antipsychotic drugs affect brain structure? A systematic and critical review of MRI findings. Psychol Med. 2009;39:1763–1777. doi: 10.1017/S0033291709005315. [DOI] [PubMed] [Google Scholar]
- 29.Frick LR, et al. Microglial dysregulation in psychiatric disease. Clin Dev Immunol. 2013;2013:608654. doi: 10.1155/2013/608654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Monji A, et al. Cytokines and schizophrenia: Microglia hypothesis of schizophrenia. Psychiatry Clin Neurosci. 2009;63:257–265. doi: 10.1111/j.1440-1819.2009.01945.x. [DOI] [PubMed] [Google Scholar]
- 31.Rao JS, et al. Increased neuroinflammatory and arachidonic acid cascade markers, and reduced synaptic proteins, in the postmortem frontal cortex from schizophrenia patients. Schizophr Res. 2013;147:24–31. doi: 10.1016/j.schres.2013.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Milner R, Campbell IL. The extracellular matrix and cytokines regulate microglial integrin expression and activation. J Immunol. 2003;170:3850–3858. doi: 10.4049/jimmunol.170.7.3850. [DOI] [PubMed] [Google Scholar]
- 33.Walker FR, et al. Dynamic structural remodelling of microglia in health and disease: A review of the models, the signals and the mechanisms. Brain Behav Immun. 2014 doi: 10.1016/j.bbi.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 34.Doorduin J, et al. Neuroinflammation in schizophrenia-related psychosis: a PET study. J Nucl Med. 2009;50:1801–1807. doi: 10.2967/jnumed.109.066647. [DOI] [PubMed] [Google Scholar]
- 35.van Berckel BN, et al. Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11C]PK11195 positron emission tomography study. Biol Psychiatry. 2008;64:820–822. doi: 10.1016/j.biopsych.2008.04.025. [DOI] [PubMed] [Google Scholar]
- 36.Kenk M, et al. Imaging neuroinflammation in gray and white matter in schizophrenia: an in-vivo PET study with [18F]-FEPPA. Schizophr Bull. 2015;41:85–93. doi: 10.1093/schbul/sbu157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brandt VC, et al. Did I turn off the gas? Reality monitoring of everyday actions. Cogn Affect Behav Neurosci. 2014;14:209–219. doi: 10.3758/s13415-013-0189-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buda M, et al. A specific brain structural basis for individual differences in reality monitoring. J Neurosci. 2011;31:14308–14313. doi: 10.1523/JNEUROSCI.3595-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simons JS, et al. Separable forms of reality monitoring supported by anterior prefrontal cortex. J Cogn Neurosci. 2008;20:447–457. doi: 10.1162/jocn.2008.20036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sugimori E, et al. Brain mechanisms underlying reality monitoring for heard and imagined words. Psychol Sci. 2014;25:403–413. doi: 10.1177/0956797613505776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haut KM, et al. Contributions of Feature Binding During Encoding and Functional Connectivity of the Medial Temporal Lobe Structures to Episodic Memory Deficits Across the Prodromal and First-Episode Phases of Schizophrenia. Clin Psychol Sci. 2015;3:159–174. doi: 10.1177/2167702614533949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ragland JD, et al. Neural correlates of relational and item-specific encoding during working and long-term memory in schizophrenia. Neuroimage. 2012;59:1719–1726. doi: 10.1016/j.neuroimage.2011.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ranganath C, et al. The cognitive neuroscience of memory function and dysfunction in schizophrenia. Biol Psychiatry. 2008;64:18–25. doi: 10.1016/j.biopsych.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Erp TG, et al. Remember and know judgments during recognition in chronic schizophrenia. Schizophr Res. 2008;100:181–190. doi: 10.1016/j.schres.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fusar-Poli P, et al. Cognitive functioning in prodromal psychosis: a meta-analysis. Arch Gen Psychiatry. 2012;69:562–571. doi: 10.1001/archgenpsychiatry.2011.1592. [DOI] [PubMed] [Google Scholar]
- 46.Giuliano AJ, et al. Neurocognition in the psychosis risk syndrome: a quantitative and qualitative review. Curr Pharm Des. 2012;18:399–415. doi: 10.2174/138161212799316019. [DOI] [PubMed] [Google Scholar]
- 47.Chung Y, et al. Prodromal Symptom Severity Predicts Accelerated Gray Matter Reduction and Third Ventricle Expansion Among Clinically High Risk Youth Developing Psychotic Disorders. Mol Neuropsychiatry. 2015;1:13–22. doi: 10.1159/000371887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marshall C, et al. The content of attenuated psychotic symptoms in those at clinical high risk for psychosis. Psychiatry Res. 2014;219:506–512. doi: 10.1016/j.psychres.2014.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Allen PP, et al. Misattribution of external speech in patients with hallucinations and delusions. Schizophr Res. 2004;69:277–287. doi: 10.1016/j.schres.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 50.Woodward TS, Menon M. Misattribution models (II): Source monitoring in hallucinating schizophrenia subjects. In: Jardi R, et al., editors. The Neuroscience of Hallucinations. Springer; 2013. [Google Scholar]
- 51.Galimberti D, et al. Psychiatric Symptoms in Frontotemporal Dementia: Epidemiology, Phenotypes, and Differential Diagnosis. Biol Psychiatry. 2015 doi: 10.1016/j.biopsych.2015.03.028. [DOI] [PubMed] [Google Scholar]
- 52.Barch DM, Sheffield JM. Cognitive impairments in psychotic disorders: common mechanisms and measurement. World Psychiatry. 2014;13:224–232. doi: 10.1002/wps.20145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun D, et al. Elucidating a magnetic resonance imaging-based neuroanatomic biomarker for psychosis: classification analysis using probabilistic brain atlas and machine learning algorithms. Biol Psychiatry. 2009;66:1055–1060. doi: 10.1016/j.biopsych.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dandash O, et al. Altered striatal functional connectivity in subjects with an at-risk mental state for psychosis. Schizophr Bull. 2014;40:904–913. doi: 10.1093/schbul/sbt093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Heinze K, et al. Discrete alternations of brain network structural covariance in individuals at ultra-high risk for psychosis. Biol Psychiatry. 2015 doi: 10.1016/j.biopsych.2014.10.023. [DOI] [PubMed] [Google Scholar]
- 56.Lord LD, et al. Characterization of the anterior cingulate’s role in the at-risk mental state using graph theory. Neuroimage. 2011;56:1531–1539. doi: 10.1016/j.neuroimage.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 57.Wotruba D, et al. Aberrant coupling within and across the default mode, task-positive, and salience network in subjects at risk for psychosis. Schizophr Bull. 2014;40:1095–1104. doi: 10.1093/schbul/sbt161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anticevic A, et al. Association of thalamic dysconnectivity and conversion to psychosis in youth and young adults at elevated clinical risk. JAMA Psychiatry. doi: 10.1001/jamapsychiatry.2015.0566. (In press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- 60.Krystal JH, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- 61.Olney JW, Farber NB. Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry. 1995;52:998–1007. doi: 10.1001/archpsyc.1995.03950240016004. [DOI] [PubMed] [Google Scholar]
- 62.Lahti AC, et al. Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology. 2001;25:455–467. doi: 10.1016/S0893-133X(01)00243-3. [DOI] [PubMed] [Google Scholar]
- 63.Lewis DA, Sweet RA. Schizophrenia from a neural circuitry perspective: advancing toward rational pharmacological therapies. J Clin Invest. 2009;119:706–716. doi: 10.1172/JCI37335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dempster K, et al. Glutamatergic metabolite correlations with neuropsychological tests in first episode schizophrenia. Psychiatry Res. 2015 doi: 10.1016/j.pscychresns.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 66.Stone JM, et al. Glutamate dysfunction in people with prodromal symptoms of psychosis: relationship to gray matter volume. Biol Psychiatry. 2009;66:533–539. doi: 10.1016/j.biopsych.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 67.Lutkenhoff ES, et al. Proton MRS in twin pairs discordant for schizophrenia. Mol Psychiatry. 2010;15:308–318. doi: 10.1038/mp.2008.87. [DOI] [PubMed] [Google Scholar]
- 68.Umbricht D, Krljes S. Mismatch negativity in schizophrenia: a meta-analysis. Schizophr Res. 2005;76:1–23. doi: 10.1016/j.schres.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 69.Bodatsch M, et al. Prediction of psychosis by mismatch negativity. Biol Psychiatry. 2011;69:959–966. doi: 10.1016/j.biopsych.2010.09.057. [DOI] [PubMed] [Google Scholar]
- 70.Javitt DC, et al. Role of cortical N-methyl-D-aspartate receptors in auditory sensory memory and mismatch negativity generation: implications for schizophrenia. Proc Natl Acad Sci U S A. 1996;93:11962–11967. doi: 10.1073/pnas.93.21.11962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Feldman DE. Synaptic mechanisms for plasticity in neocortex. Annu Rev Neurosci. 2009;32:33–55. doi: 10.1146/annurev.neuro.051508.135516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tessier CR, Broadie K. Activity-dependent modulation of neural circuit synaptic connectivity. Front Mol Neurosci. 2009;2:8. doi: 10.3389/neuro.02.008.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schafer DP, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Corlett PR, et al. Disrupted prediction-error signal in psychosis: evidence for an associative account of delusions. Brain. 2007;130:2387–2400. doi: 10.1093/brain/awm173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Corlett PR, et al. Ketamine effects on memory reconsolidation favor a learning model of delusions. PLoS One. 2013;8:e65088. doi: 10.1371/journal.pone.0065088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Howes OD, et al. Mechanisms underlying psychosis and antipsychotic treatment response in schizophrenia: insights from PET and SPECT imaging. Curr Pharm Des. 2009;15:2550–2559. doi: 10.2174/138161209788957528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Abi-Dargham A, et al. Increased striatal dopamine transmission in schizophrenia: confirmation in a second cohort. Am J Psychiatry. 1998;155:761–767. doi: 10.1176/ajp.155.6.761. [DOI] [PubMed] [Google Scholar]
- 78.Howes OD, et al. Elevated striatal dopamine function linked to prodromal signs of schizophrenia. Arch Gen Psychiatry. 2009;66:13–20. doi: 10.1001/archgenpsychiatry.2008.514. [DOI] [PubMed] [Google Scholar]
- 79.Egerton A, et al. Presynaptic striatal dopamine dysfunction in people at ultra-high risk for psychosis: findings in a second cohort. Biol Psychiatry. 2013;74:106–112. doi: 10.1016/j.biopsych.2012.11.017. [DOI] [PubMed] [Google Scholar]
- 80.Howes O, et al. Progressive increase in striatal dopamine synthesis capacity as patients develop psychosis: a PET study. Mol Psychiatry. 2011;16:885–886. doi: 10.1038/mp.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kapur S. Psychosis as a state of aberrant salience: a framework linking biology, phenomenology, and pharmacology in schizophrenia. Am J Psychiatry. 2003;160:13–23. doi: 10.1176/appi.ajp.160.1.13. [DOI] [PubMed] [Google Scholar]
- 82.Roiser JP, et al. Neural and behavioral correlates of aberrant salience in individuals at risk for psychosis. Schizophr Bull. 2013;39:1328–1336. doi: 10.1093/schbul/sbs147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tandon R, et al. Schizophrenia, “just the facts” 5. Treatment and prevention. Past, present, and future. Schizophr Res. 2010;122:1–23. doi: 10.1016/j.schres.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 84.McGorry PD, et al. Randomized controlled trial of interventions designed to reduce the risk of progression to first-episode psychosis in a clinical sample with subthreshold symptoms. Arch Gen Psychiatry. 2002;59:921–928. doi: 10.1001/archpsyc.59.10.921. [DOI] [PubMed] [Google Scholar]
- 85.McGlashan TH, et al. Randomized, double-blind trial of olanzapine versus placebo in patients prodromally symptomatic for psychosis. Am J Psychiatry. 2006;163:790–799. doi: 10.1176/ajp.2006.163.5.790. [DOI] [PubMed] [Google Scholar]
- 86.Jentsch JD, et al. Enduring cognitive deficits and cortical dopamine dysfunction in monkeys after long-term administration of phencyclidine. Science. 1997;277:953–955. doi: 10.1126/science.277.5328.953. [DOI] [PubMed] [Google Scholar]
- 87.Javitt DC. Glutamate and schizophrenia: phencyclidine, N-methyl-D-aspartate receptors, and dopamine-glutamate interactions. Int Rev Neurobiol. 2007;78:69–108. doi: 10.1016/S0074-7742(06)78003-5. [DOI] [PubMed] [Google Scholar]
- 88.Kegeles LS, et al. Modulation of amphetamine-induced striatal dopamine release by ketamine in humans: implications for schizophrenia. Biol Psychiatry. 2000;48:627–640. doi: 10.1016/s0006-3223(00)00976-8. [DOI] [PubMed] [Google Scholar]
- 89.Insel TR, Scolnick EM. Cure therapeutics and strategic prevention: raising the bar for mental health research. Mol Psychiatry. 2006;11:11–17. doi: 10.1038/sj.mp.4001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Finkel D, et al. Latent growth curve analyses of accelerating decline in cognitive abilities in late adulthood. Dev Psychol. 2003;39:535–550. doi: 10.1037/0012-1649.39.3.535. [DOI] [PubMed] [Google Scholar]
- 91.McArdle JJ, et al. Structural modeling of dynamic changes in memory and brain structure using longitudinal data from the normative aging study. J Gerontol B Psychol Sci Soc Sci. 2004;59:P294–304. doi: 10.1093/geronb/59.6.p294. [DOI] [PubMed] [Google Scholar]
- 92.Kumsta R, et al. IX. Risk, causation, mediation, and moderation. Monogr Soc Res Child Dev. 2010;75:187–211. doi: 10.1111/j.1540-5834.2010.00556.x. [DOI] [PubMed] [Google Scholar]
- 93.Yung AR, McGorry PD. The prodromal phase of first-episode psychosis: past and current conceptualizations. Schizophr Bull. 1996;22:353–370. doi: 10.1093/schbul/22.2.353. [DOI] [PubMed] [Google Scholar]
- 94.Beiser M, et al. Establishing the onset of psychotic illness. Am J Psychiatry. 1993;150:1349–1354. doi: 10.1176/ajp.150.9.1349. [DOI] [PubMed] [Google Scholar]
- 95.Yung AR, et al. Mapping the onset of psychosis: the Comprehensive Assessment of At-Risk Mental States. Aust N Z J Psychiatry. 2005;39:964–971. doi: 10.1080/j.1440-1614.2005.01714.x. [DOI] [PubMed] [Google Scholar]
- 96.Miller TJ, et al. Prodromal assessment with the structured interview for prodromal syndromes and the scale of prodromal symptoms: predictive validity, interrater reliability, and training to reliability. Schizophr Bull. 2003;29:703–715. doi: 10.1093/oxfordjournals.schbul.a007040. [DOI] [PubMed] [Google Scholar]
- 97.Wiltink S, et al. Declining transition rates to psychosis: the contribution of potential changes in referral pathways to an ultra-high-risk service. Early Interv Psychiatry. 2015;9:200–206. doi: 10.1111/eip.12105. [DOI] [PubMed] [Google Scholar]
- 98.Addington J, et al. At clinical high risk for psychosis: Outcome for non-converters. American Journal of Psychiatry. 2011 doi: 10.1176/appi.ajp.2011.10081191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schlosser DA, et al. Recovery from an at-risk state: clinical and functional outcomes of putatively prodromal youth who do not develop psychosis. Schizophr Bull. 2012;38:1225–1233. doi: 10.1093/schbul/sbr098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.International Schizophrenia C et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cannon TD. What is the role of theories in the study of schizophrenia? Schizophr Bull. 2009;35:563–567. doi: 10.1093/schbul/sbp008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kahn RS, et al. Schizophrenia. Nature Disease Primers (In press) [Google Scholar]
- 103.Insel TR. Rethinking schizophrenia. Nature. 2010;468:187–193. doi: 10.1038/nature09552. [DOI] [PubMed] [Google Scholar]