

Abstract
Alpha-thalassemia (α-thal) is a disorder caused by the deletion of single or double α-globin genes, and/or point mutations in the α-globin genes. There are 2 common types of α-globin genes; HBA2 and HBA1. Recently, it has been discovered that the HBA2 gene is replaced by a unique HBA12 gene convert in 5.7% of the Saudi population. The α-globin genes have been emerging as a molecular target for the treatment of β-thalassemia (β-thal). Hence, it is essential to understand the molecular nature of α-globin genes to treat the most prevalent hemoglobin disorders, such as sickle cell disease, α-thal, and β-thal prevalent in the Kingdom of Saudi Arabia. Thirty-two different α-globin genotypes have been observed in the Saudi population. This review outlines the classification of the α-globin genes on the basis of their molecular nature and complex combinations of α-globin genes, and their variants predominant in Saudis.
Thalassemia (alpha [α] and beta [β]) and sickle cell disease (SCD) are the most prevalent hemoglobin disorders in the Kingdom of Saudi Arabia.1-11 Alpha-thalassemia (α-thal) is a disorder caused by the deletion of single or double α-globin genes, and/or point mutations in the α-globin genes.10 Alpha-thalassemia phenotype varies from very mild or microcytic hypochromic anemia to a lethal form of hemolytic anemia, depending on the type of molecular defects in α-globin genes.10 Alpha-globin genes are of 2 types; hemoglobin alpha 1 (HBA1) and hemoglobin alpha 2 (HBA2). The HBA1 and HBA2 genes are located in the p arm (short arm) of chromosome 16 at region one, band 3, and sub-band 3. Altogether there are 4 genes (α1α2/α1α2) in a person corresponding to 4 α-globin proteins. Out of the 4 α-globin genes, 2 are inherited from the father, and others from the mother. Loss of single or all of these genes results in different types of α-thal. The α-globin protein is a subunit of hemoglobin, which is a larger protein in red blood cells (RBC) that carries oxygen throughout the body. Alpha-globin proteins of HBA2 and HBA1 genes are nearly identical. Alpha-globin protein is subunit of fetal hemoglobin (HbF), which is active only in the human fetus and in the newborn period until roughly 6 months old. Exceptionally, in non-transfusion dependent β-thal cases, HbF is elevated and active throughout life. Reduced synthesis of α-globin protein ameliorates the clinical severity of β-thallassemia. Alpha-globin is an emerging molecular target for treatment of β-thal.12 Hence, it is essential to understand the different types of globin genes and its variants prevalent in Saudi population.
The α-globin genes in Saudis
A number of research reports were available on the analysis of mutations and deletion in the α-globin genes from Saudi population.1-11,13-20 Commonly, α-globin genes are of 2 types (HBA2 and HBA1), while in Saudis it is of 3 types namely HBA2 (α2), HBA1 (α1), and HBA12 (α12).8 The HBA12 is a new convert of the HBA2 gene, discovered in Saudis. The HBA2 has been replaced with HBA12 in 5.7% of Saudi population.8 The poly A mutation [AATAAA>AATAAG] (41%), and the α3.7 α+ heterozygous deletion are the most reported mutations and deletion in Saudi population. Co-inheritance of α-globin gene and β-globin gene mutations are prevalent in Saudis.7-9
The α-globin gene conversion
A recent study by Borgio et al8 using direct sequencing of HBA1 and HBA2 in Saudis revealed a new gene, which was very closely related to the common α-globin genes (HBA1 and HBA2). They named the new gene as 2. They clearly described that the formation the HBA12 was formed by the combination of HBA1 and HBA2 gene sequences through a process called gene conversion (Figure 1). Gene conversion is the process of the transfer of genetic material unidirectionally from a donor to an acceptor.21 Gene conversion between the 2 homologous α-globin genes is common.8 The HBA12 gene has the region starting -6bp until 581bp (3’ promoter, exon1, IVSI, exon2, and 5’IVSII) from HBA1 gene, and 774bp (3’enhancer) onwards from HBA2 gene.8 The region in-between 581bp and 774bp (3’ IVSII, exon 3’, and 5’ enhancer) were matching with HBA1 and HBA2, hence this region was considered as an indistinguishable region.8 The α-globin protein from HBA12 gene is not available in the literature, detailed studies are needed to confirm the similarity of HBA12 protein with the α-globin protein of HBA2 and HBA1 genes.
Figure 1.
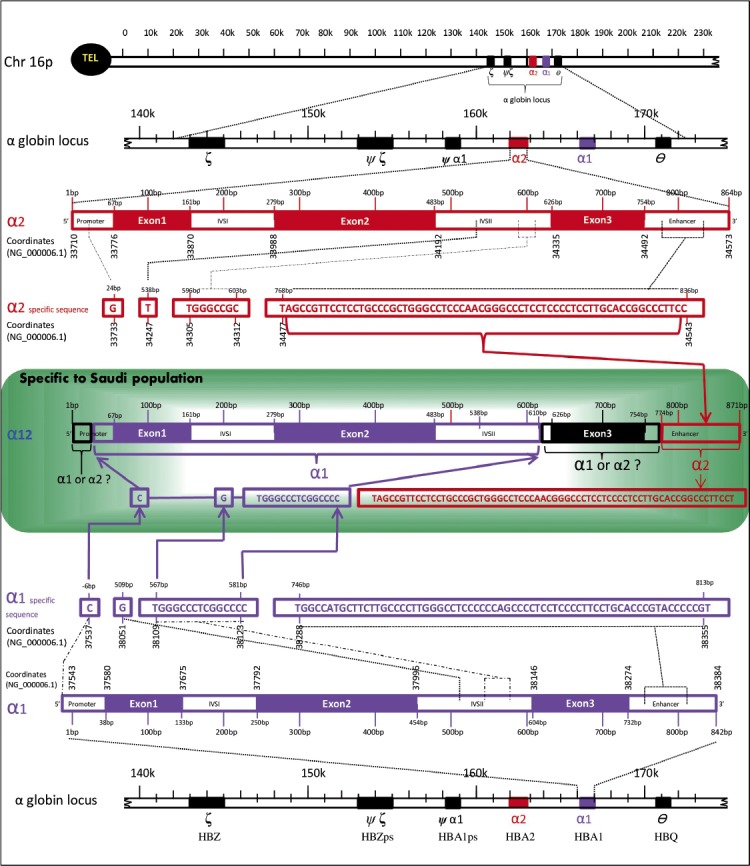
An image showing 3 types of globin genes prevalent in the Saudi population: HBA2 (α2), HBA1 (α1), and HBA12 (α12). The α2 gene is colored in nut brown and α1 gene is colored in violet. The undistinguished sequences (α1 or α2 ?) are colored in black. Reproduced and modified from: Borgio JF, AbdulAzeez S, Al-Nafie AN, Naserullah ZA, Al-Jarrash S, Al-Madan MS, et al. A novel HBA2 gene conversion in cis or trans: “α12 allele” in a Saudi population. Blood Cells Mol Dis 2014; 53: 199-203.8 With permission from Elsevier.
A total of 5.7% of the study population including sickle cell trait, hemophilia-A patient, SCD patients, and β-thal major patients were reported to have the new gene convert, α12 gene. The inheritance of the HBA12 gene was proven on an elaborated family study, any one of the parent of individual with HBA12 was a carrier for the HBA12 gene. The HBA12 gene was reported to be co-inherited with any one of the common α-globin gene defects like α3.7 deletion, ααα3.7 triplications, and α4.2 deletion, but not always. The reported HBA12 gene from Saudis was distinguishably different from the α-globin patch works, such as α212 and α121.8 Except the nullizygous, all the other 3 types (hemizygous α1-/α1α12, heterozygous α1α2/α1α12, and homozygous α1α12/α1α12) of zygosities were observed for the α12 gene from Saudi population. α-globin gene convert was highly prevalent in the Saudis due to the high percentage of consanguinity. Slight increase in mean corpuscular volume, elevated HbF (α2γ2), and reduced HbA2 (α2δ2) were noted on the subjects with α12 gene convert.8
Alpha12 and HbA2
Deep analysis by Borgio et al8 revealed the influence of the α12 gene on the level of hemoglobin A2 (HbA2). Subgrouping the population with the α12 gene into 6 groups (HbScarrier; β-thalcarrier; β-thalmajor/α-thalcarrier; SCD+ve, and α-thalcarrier; HbScarrier α-thalcarrier; and NormalNo α-thal&β-thal) by the authors was able to identify the reduced level of HbA2 in the first 5 groups with α12 gene.8 Thorough investigation on the large-scale micromapping of phenomics for this α12 gene is mandatory to uncover the hematologic effects of the new α12 gene.
Alpha-globin genotypes
The term “α-globin genotype” refers to the genetic makeup of an individual’s complete set of α-genes. Two alleles (α/α) at each α-globin gene position is called diploid. In general, 2 pairs of alleles from 2 α-globin genes, α1/α1 (HBA1) and α2/α2 (HBA2) represents the genotype (α1α2/α1α2) of an α-globin gene. In terms of Saudi population, the HBA2 gene has been replaced with HBA12 gene convert, a pair of alleles α12/α12 of an α-globin gene convert, HBA12 specific to Saudis represents the genotype, α1α12/α1α12. Hence there are 3 genotypes, α1α2/α1α2, α1α2/α1α12, and α1α12/α1α12 are the possible normal genotypes in Saudi population (Table 1). There were 32 different genotypes reported from Saudi population (Table 1). The α-globin genotypes, -α3.7/αα, and -α3.7/-α3.7 are the most prevalent in Saudis.7,15 Alpha-globin genotype of each individual contributes to its α-thal phenotype. On the basis of genotypes, α-thal can be classified into 4 types, group one: deletion of 4 α-globin genes, termed Hb Bart’s; group 2: deletion of 3 α- globin genes, called HbH disease; group 3: deletion of 2 α- globin genes, named α-thal trait; group 4: deletion of one α-globin gene, designated α-thal Silent (Table 1, Figure 2). Two particular identical alleles are described as homozygous (for example, α3.7 homozygous deletion -α3.7/-α3.7), and if the 2 alleles differ, it is termed as heterozygous (for example, α3.7 heterozygous deletion -α3.7/αα). Hemizygous (for example, α1-4.2/α1α12) form of α-globin genotypes were also reported from Saudis.8 Severity of the α-thal disorder is indirectly proportional to the number of functional α-globin genes. The severities of α-thal tend to be more in group one results from the loss of all 4 α-globin genes, while signs and symptoms are almost nil in group 4 (Figure 2). Techniques for the updated genotyping (the process of determining a genotype) of α-globin genes in Saudis for the proper diagnosis should be given to health professional.
Table 1.
Alpha-globin genotypes prevalent in Saudi population according to various studies in Saudi Arabia.
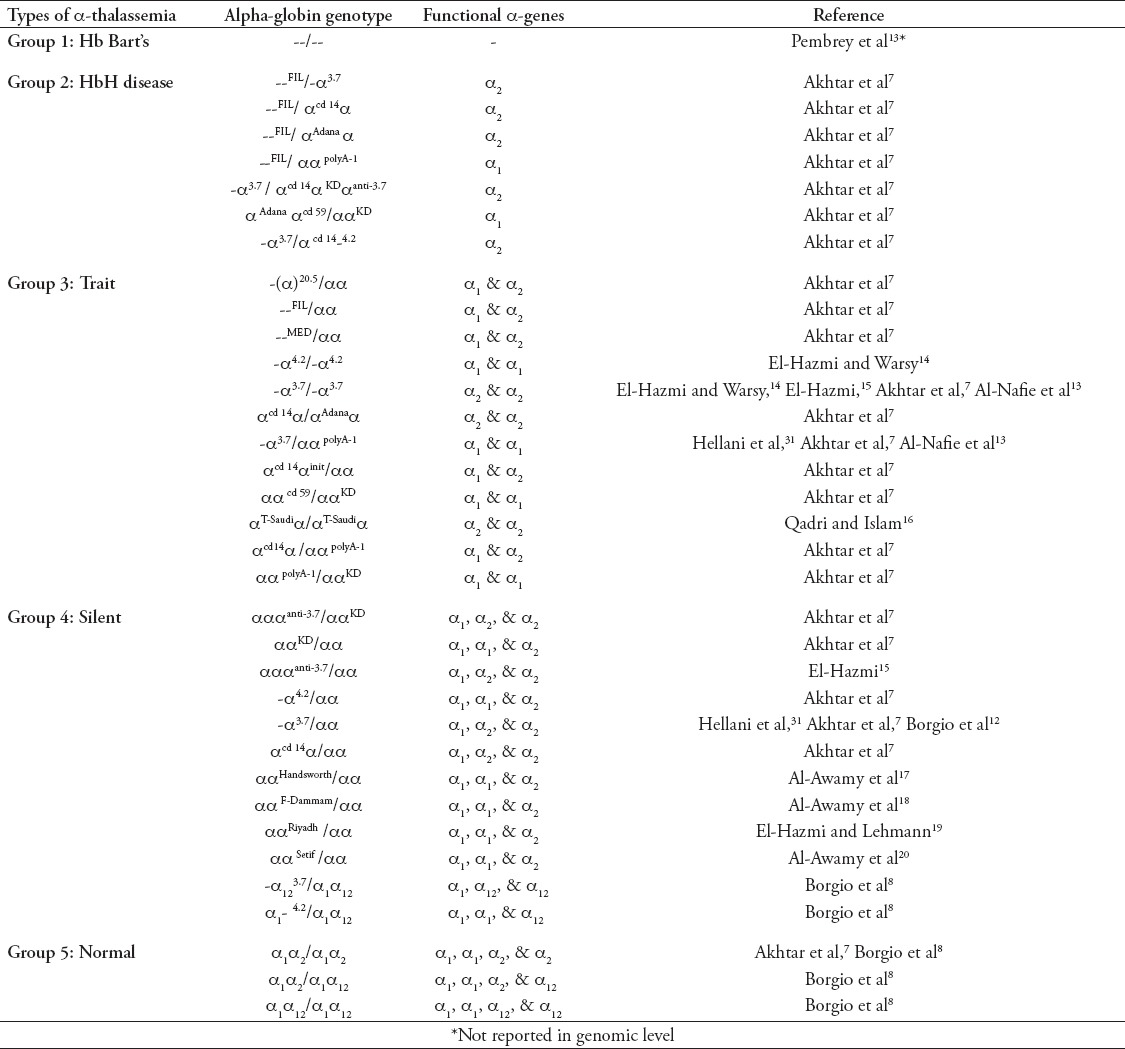
Figure 2.
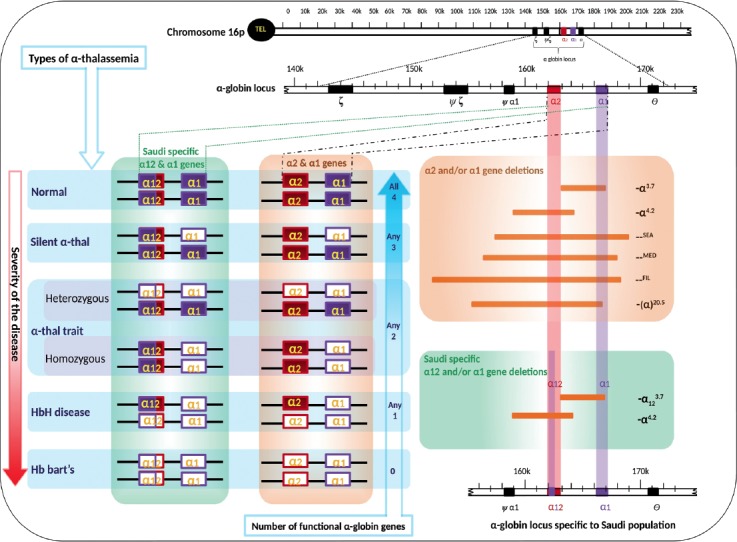
Molecular types of thalassemia and types of globin gene deletions prevalent in the Saudi population. Filled boxes of genes α1, α2, and α12 indicates normal genes, while empty boxes of genes α1, α2, and α12 indicate the deleted genes.
Down regulation factors of the α-globin gene expression
In general, down regulation of the expression of the α-globin genes due to mutations in ATRX (α-thal x-linked mental retardation) gene, usually lead to α-thal like phenotype.22,23 Very recently, there were 4 novel mutations (IVS I-5(G→C), Cd39(C→T), c.623delA, and c.848T>C) on ATRX gene reported in Saudi population. The 2 exonic mutations (c.623delA and c.848T>C) were reported in patients co-inherited with α-globin genes mutations.9 The study is a clear alarm that the α-thal-like phenotype in Saudi population may be due to mutations in α-globin genes, or in ATRX gene. It seems reasonable to suggest that screening for the presence of mutations in the ATRX gene along with mutations in the HBA2, HBA1, and HBA12 genes are essential for proper identification of the disease burden in this population.
The main limitation of this review is that, some of the articles dealing with α-thal within the Saudi population were not publicly accessible full text scholarly articles. Health sector professional should keep themselves updated with the genotyping techniques for α-globin and ATRX genes in Saudis. The very high frequency of α-thal, SCD, and β-thal in the Kingdom and their co-inheritance obliges the addition of sequence based testing system for HBA2, HBA1, HBA12, and ATRX genes along with the existing pre-marital testing program, to detect the risk for the offspring of affected individuals.
In conclusion, large-scale screening is mandatory to identify the influence of point mutations and deletion on the phenotype of Saudi population. In-depth, the studies on the prevalence of the sequence variations in α12 and their influence on the phenotypes are needed. Comparative studies on the protein structure and molecular modeling of α1, α2, and α12 have to be initiated. Specific diagnostic kits for Saudi population should be developed to identify the sequence defects in α1, α2, and α12 genes. The clinical effects due to the changes in α-globin gene expression from the α12 gene should be studied at large-scale. Cellular studies are needed to understand the process of down regulation of α-globin gene expression due to ATRX gene mutation in Saudis.
Acknowledgment
The author thanks Dr. A. K. Al-Ali, Director, Center for Research and Medical Consultation, University of Dammam, Dammam, Kingdom of Saudi Arabia for his continuous support and encouragement. The author wishes to express his appreciation to Mrs. B. Jesvin Bency for her critical comments. The author also acknowledgs Dr. S. AbdulAzeez, Dr. S. Chathoth, Dr. S. A. Baikady, Dr. C. Cyrus, Dr. C. B. Vatte, Dr. K. Alkharsah, Dr. K. A. Ahmed, Dr. N. B. Almandil, and Dr. M. Farhat for their encouragement. The author would like to extend his appreciation to Mr. Mohammed H. Al-Shamlan for Arabic translation of the abstract.
Footnotes
Copyright.
Whenever a manuscript contains material (tables, figures, etc.) which is protected by copyright (previously published), it is the obligation of the author to obtain written permission from the holder of the copyright (usually the publisher) to reproduce the material in Saudi Medical Journal. This also applies if the material is the authors own work. Please submit copies of the material from the source in which it was first published.
References
- 1.Jastaniah W. Epidemiology of sickle cell disease in Saudi Arabia. Ann Saudi Med. 2011;31:289–293. doi: 10.4103/0256-4947.81540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mehdi SR, Al Dahmash BA. A comparative study of hematological parameters of α and β thalassemias in a high prevalence zone: Saudi Arabia. Indian J Hum Genet. 2011;17:207–211. doi: 10.4103/0971-6866.92106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alsultan A, Alabdulaali MK, Griffin PJ, Alsuliman AM, Ghabbour HA, Sebastiani P, et al. Sickle cell disease in Saudi Arabia: the phenotype in adults with the Arab-Indian haplotype is not benign. Br J Haematol. 2014;164:597–604. doi: 10.1111/bjh.12650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Al-Owain M, Al-Zaidan H, Al-Hassnan Z. Map of autosomal recessive genetic disorders in Saudi Arabia: concepts and future directions. Am J Med Genet A. 2012;158A:2629–2640. doi: 10.1002/ajmg.a.35551. [DOI] [PubMed] [Google Scholar]
- 5.Al-Shahrani M. Steps toward the prevention of hemoglobinopathies in the kingdom of Saudi Arabia. Hemoglobin. 2009;33(Suppl 1):S21, S24. doi: 10.3109/03630260903346437. [DOI] [PubMed] [Google Scholar]
- 6.Al-Sultan A, Phanasgaonkar S, Suliman A, Al-Baqushi M, Nasrullah Z, Al-Ali A. Spectrum of β-thalassemia mutations in the eastern province of Saudi Arabia. Hemoglobin. 2011;35:125–134. doi: 10.3109/03630269.2011.553567. [DOI] [PubMed] [Google Scholar]
- 7.Akhtar MS, Qaw F, Borgio JF, Albuali W, Suliman A, Nasserullah A, et al. Spectrum of α-thalassemia mutations in transfusion-dependent β-thalassemia patients from the Eastern Province of Saudi Arabia. Hemoglobin. 2013;37:65–73. doi: 10.3109/03630269.2012.753510. [DOI] [PubMed] [Google Scholar]
- 8.Borgio JF, AbdulAzeez S, Al-Nafie AN, Naserullah ZA, Al-Jarrash S, Al-Madan MS, et al. A novel HBA2 gene conversion in cis or trans: “α12 allele” in a Saudi population. Blood Cells Mol Dis. 2014;53:199–203. doi: 10.1016/j.bcmd.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 9.Al-Nafie AN, Borgio JF, AbdulAzeez S, Al-Suliman AM, Qaw FS, Naserullah ZA, et al. Co-inheritance of novel ATRX gene mutation and globin (α & β) gene mutations in transfusion dependent beta-thalassemia patients. Blood Cells Mol Dis. 2015;55:27–29. doi: 10.1016/j.bcmd.2015.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Harteveld CL, Higgs DR. Alpha-thalassaemia. Orphanet J Rare Dis. 2010;5:13. doi: 10.1186/1750-1172-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hellani A, Fadel E, El-Sadadi S, El-Sweilam H, El-Dawood A, Abu-Amero KK. Molecular spectrum of alpha-thalassemia mutations in microcytic hypochromic anemia patients from Saudi Arabia. Genet Test Mol Biomarkers. 2009;13:219–221. doi: 10.1089/gtmb.2008.0123. [DOI] [PubMed] [Google Scholar]
- 12.Mettananda S, Gibbons RJ, Higgs DR. α-Globin as a molecular target in the treatment of β-thalassemia. Blood. 2015;125:3694–3701. doi: 10.1182/blood-2015-03-633594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pembrey ME, Weatherall DJ, Clegg JB, Bunch C, Perrine RP. Haemoglobin Bart’s in Saudi Arabia. Br J Haematol. 1975;29:221–234. doi: 10.1111/j.1365-2141.1975.tb01816.x. [DOI] [PubMed] [Google Scholar]
- 14.El-Hazmi MA, Warsy AS. Double heterozygote leftward/rightward deletion type alpha-thalassaemia in Saudi Arabs. Hum Hered. 1986;36:222–226. doi: 10.1159/000153630. [DOI] [PubMed] [Google Scholar]
- 15.El-Hazmi MA. Alpha-thalassemia in Saudi Arabia: deletion pattern. Hum Genet. 1987;76:196–198. doi: 10.1007/BF00284921. [DOI] [PubMed] [Google Scholar]
- 16.Qadri MI, Islam SA. Hemoglobin H disease in the eastern region of Saudi Arabia. Saudi Med J. 2000;21:666–671. [PubMed] [Google Scholar]
- 17.Al-Awamy BH, Niazi GA, Naeem MA, Wilson JB, Huisman TH. Hemoglobin Handsworth or alpha2 18(A16)Gly----Arg beta2 in a Saudi newborn. Hemoglobin. 1985;9:183–186. doi: 10.3109/03630268508997001. [DOI] [PubMed] [Google Scholar]
- 18.Al-Awamy BH, Niazi GA, Al-Mouzan MI, Wilson JB, Chen SS, Webber BB, et al. Hb F-Dammam or alpha 2A gamma 2(79) (EF3) Asp----Asn. Hemoglobin. 1985;9:171–173. doi: 10.3109/03630268508996998. [DOI] [PubMed] [Google Scholar]
- 19.El-Hazmi MA, Lehmann H. Hemoglobin Riyadh--alpha2beta2 (120(GH3)Lys replaced by Asn). A new variant found in association with alpha-thalassemia and iron deficiency. Hemoglobin. 1976-1977;1:59–74. doi: 10.3109/03630267609031022. [DOI] [PubMed] [Google Scholar]
- 20.Al-Awamy B, Niazi GA, Wilson JB, Huisman TH. Hb Setif or alpha 2 94(G1)Asp----Tyr beta 2 observed in a Saudi Arabian family. Hemoglobin. 1985;9:87–90. doi: 10.3109/03630268508996987. [DOI] [PubMed] [Google Scholar]
- 21.Lachance J, Tishkoff SA. Biased gene conversion skews allele frequencies in human populations, increasing the disease burden of recessive alleles. Am J Hum Genet. 2014;95:408–420. doi: 10.1016/j.ajhg.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Viprakasit V. α-Thalassaemia: a genotype-phenotype correlation and management. ISBT Science Series. 2015;10:295–304. [Google Scholar]
- 23.Gibbons RJ. α-Thalassemia, mental retardation, and myelodysplastic syndrome. Cold Spring Harb Perspect Med. 2012;2:a011759. doi: 10.1101/cshperspect.a011759. [DOI] [PMC free article] [PubMed] [Google Scholar]