

Abstract
DNA methylation is a well-characterized epigenetic modification that plays central roles in mammalian development, genomic imprinting, X-chromosome inactivation and silencing of retrotransposon elements. Aberrant DNA methylation pattern is a characteristic feature of cancers and associated with abnormal expression of oncogenes, tumor suppressor genes or repair genes. Ten-eleven-translocation (TET) proteins are recently characterized dioxygenases that catalyze progressive oxidation of 5-methylcytosine to produce 5-hydroxymethylcytosine and further oxidized derivatives. These oxidized methylcytosines not only potentiate DNA demethylation but also behave as independent epigenetic modifications per se. The expression or activity of TET proteins and DNA hydroxymethylation are highly dysregulated in a wide range of cancers including hematologic and non-hematologic malignancies, and accumulating evidence points TET proteins as a novel tumor suppressor in cancers. Here we review DNA demethylation-dependent and -independent functions of TET proteins. We also describe diverse TET loss-of-function mutations that are recurrently found in myeloid and lymphoid malignancies and their potential roles in hematopoietic transformation. We discuss consequences of the deficiency of individual Tet genes and potential compensation between different Tet members in mice. Possible mechanisms underlying facilitated oncogenic transformation of TET-deficient hematopoietic cells are also described. Lastly, we address non-mutational mechanisms that lead to suppression or inactivation of TET proteins in cancers. Strategies to restore normal 5mC oxidation status in cancers by targeting TET proteins may provide new avenues to expedite the development of promising anti-cancer agents.
Keywords: 5-methylcytosine oxidation, hematopoiesis, hematologic malignancies, TET protein, tumor suppression
INTRODUCTION
In mammals, the DNA methylation and hydroxymethylation status is dynamically regulated by the interplay between DNA methyltransferase (DNMT) and Ten-eleven-translocation (TET) family enzymes (Pastor et al., 2013; Shen et al., 2014; Suzuki and Bird, 2008). DNMTs carry out DNA methylation by transferring a methyl group (CH3) from S-adenosyl methionine (SAM) to the 5-carbon of cytosine in a CpG dinucleotide to generate 5-methylcytosine (5mC), which takes place predominantly in a symmetrical manner (Fig. 1A) (Ooi et al., 2009). DNMT3A and DNMT3B are called de novo DNA methyltransferases because they create the initial methylation marks on unmethylated substrates during the early stage of embryonic development. Replication of the DNA bearing the symmetrically methylated CpG dinucleotides results in hemimethylated DNA strands because new strands are synthesized in the absence of methylation marks. However, DNMT1/UHRF1 complex is preferably targeted to these hemimethylated regions and restores normal symmetrical methylation patterns, a process termed maintenance methylation (Fig. 1A) (Arita et al., 2008; Avvakumov et al., 2008; Bostick et al., 2007; Hashimoto et al., 2008; Sharif et al., 2007). Under conditions where maintenance methylation is blocked by aberrant targeting, expression, or function of the DNMT1/UHRF1 complex, 5mC undergoes progressive, passive dilution following each round of replication (Pastor et al., 2013; Shen et al., 2014).
Fig. 1.
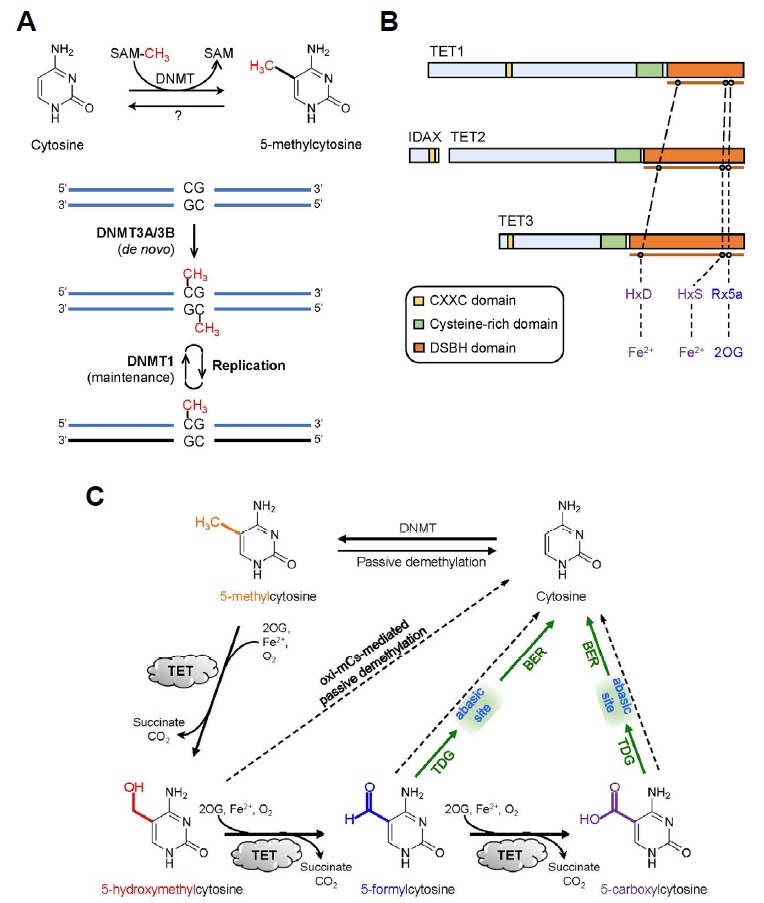
DNA methylation and demethylation processes mediated by DNMTs and TET proteins. (A) DNA methylation refers to the transfer of the methyl group from S-adenosyl methoinine (SAM) to the 5-carbon of cytosine to yield 5mC. De novo DNA methyltransferases DNMT3A and DNMT3B originally generate 5mC marks on unmethylated CpG dinucleotides. Replication leads to hemimethylated CpG sites that are remethylated by maintenance methyltransferase DNMT1. (B) Domain structure of the TET family proteins. TET proteins contain the catalytic core region that consists of cysteine-rich and double stranded β-helix (DSBH) domain. Within the DSBH domain, key catalytic residues that bind Fe2+ and 2-oxoglutarate (2OG) are present. (C) Oxidative reversal of DNA methylation marks in mammals. TET proteins promote both passive and active DNA demethylation. TET proteins oxidize 5mCs to produce oxi-mCs that antagonize DNA binding of DNMTs, promoting passive demethylation. They also facilitate active DNA demethylation because thymine DNA glycosylase (TDG) directly excise 5fC and 5caC, followed by base excision repair (BER) of the abasic site and replacement with an unmodified cytosine.
Enzymes of TET family (TET1, TET2 and TET3) belong to the superfamily of Fe2+- and 2-oxoglutarate (2OG)-dependent dioxygenases. All three TET proteins possess a highly conserved carboxy-terminal catalytic region that is composed of a cysteine-rich (Cys-rich) and a double-stranded β-helix (DSBH) domain (Fig. 1B) (Iyer et al., 2009; 2011; Tahiliani et al., 2009). TET proteins modify the methylation status of DNA by catalyzing consecutive oxidation of the methyl group of 5mC to form 5-hydroxymethylcytosine (5hmC), which in turn undergoes further oxidation by TET proteins into 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) (Fig. 1C) (He et al., 2011; Ito et al., 2011; Ko et al., 2010; Tahiliani et al., 2009). These oxidized methylcytosines are collectively termed ‘oxi-mCs’. When expressed in cells, the catalytic region alone is capable of oxidizing 5mCs in genome. TET proteins require ferrous iron Fe2+ (as an essential cofactor) and 2OG (as an obligatory co-substrate) which bind to the highly conserved ‘His-Xaa-Asp-(Xaa)n-His’ motif (Xaa refers to any amino acid) and Arg residues within the DSBH domain, respectively (Fig. 1C) (Hu et al., 2013; Tahiliani et al., 2009). Once Fe2+ and 2OG are incorporated into their cognate binding motifs in the active site, dioxygen (O2) binds to Fe2+ and oxidizes it to ferric intermediates (Fe3+), stimulating the oxidative decarboxylation of 2OG (to succinate and CO2) and the oxidation of substrate molecules (Fig. 2) (Shen et al., 2014).
Fig. 2.
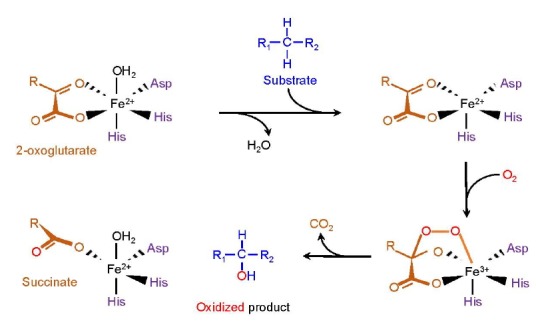
The catalytic reaction mediated by TET proteins. The catalytic residues within the DSBH domain bind 2OG and Fe2+. Incorporation of O2 yields ferric intermediate Fe3+, stimulating the substrate oxidation and oxidative decarboxylation of 2OG. The final products of this process are the oxidized product (oxi-mCs), succinate and CO2.
TET1 (also known as CXXC6) and TET3 additionally have the amino-terminal CXXC domain that belongs to the zinc finger type DNA-binding domain. This domain preferably recognizes unmethylated relative to methylated CpG dinucleotides in vitro and in cells (Xu et al., 2011b; 2012; Zhang et al., 2010). The TET2 gene does not contain exons that encode the CXXC domain but studies on the ontogeny of TET genes during vertebrate evolution proposed that after an ancestral TET gene underwent triplication to give rise to three different TET paralogs, a chromosomal inversion occurred in the TET2 gene, leading to the separation of the segments that encode the CXXC domain and catalytic domain (Iyer et al., 2009; 2011; Ko et al., 2013). Thus, the exon encoding ancestral CXXC domain of TET2 formed a distinct gene named IDAX (also known as CXXC4) (Fig. 1B). Similar to the linked CXXC domains of TET1 and TET3, the IDAX CXXC domain has a high affinity for unmethylated CpG sequences and thus is preferentially targeted to CpG islands (Ko et al., 2013), suggesting that TET proteins contribute to the maintenance of the hypomethylated status of CpG islands.
The early observation of the chromosomal translocation t(10;11)(q22;q23) that occurs rarely in patients with acute myeloid leukemia (AML) and acute lymphocytic leukemia (ALL) revealed that TET proteins may play a role in the pathogenesis of hematopoietic cancers (Lorsbach et al., 2003; Ono et al., 2002). As a result of this translocation, the segment on chromosome 10 that encodes the amino-terminal region of mixed-lineage leukemia (MLL) H3K4 methyltransferase is fused to the segment on chromosome 11 that encodes the DSBH domain of TET1 (Lorsbach et al., 2003; Ono et al., 2002). Loss-of-function mutations or deletions in TET2 gene are recurrent in a wide range of myeloid and lymphoid malignancies (Huang and Rao, 2014; Ko et al., 2015), and hypermethylation of the TET1 promoter leads to a decrease in its expression in B cell lymphoma (Cimmino et al., 2015). Moreover, the expression and function of TET proteins and/or their modulators are frequently dysregulated in a wide variety of cancers (Huang and Rao, 2014; Ko et al., 2015), significantly impairing 5mC oxidation in the genome. Despite incomplete comprehension, various Tet-deficient murine models have provided valuable insights into the physiological function of TET proteins in normal hematopoiesis together with their pathogenic functions in malignant hematopoiesis, establishing TET proteins as novel tumor suppressors that directly drive hematopoietic transformation upon dysregulation. In this review, we focus on various loss-of-function TET mutations and mechanisms leading to the dysregulation of TET expression and function in hematopoietic cancers. We also discuss the functional redundancy among different TET proteins, their association with other mutations, and the potential mechanisms by which TET loss-of-function promotes malignant transformation.
TET PROTEINS FACILITATE DNA DEMETHYLATION IN MAMMALS
DNA demethylation refers to the erasure of the methyl group from 5mC in DNA. This process occurs through at least two pathways, the passive or active demethylation pathways (Pastor et al., 2013; Shen et al., 2014; Wu and Zhang, 2014). As mentioned earlier, 5mC can be removed passively if newly synthesized DNA strands fail to be methylated again following replication when DNMT1 is absent or prevented from being recruited to these hemimethylated sites. TET proteins potentiate passive demethylation because all oxi-mCs are capable of interfering with maintenance methylation by antagonizing DNA binding of the DNMT1/UHRF1 complex, which is termed ‘oxi-mC-facilitated passive demethylation’ (Fig. 1C) (Hashimoto et al., 2012; Valinluck and Sowers, 2007).
It has long been known that the methyl group of 5mC can also be removed regardless of replication, particularly in the zygotic paternal genome or primordial germ cells, a process denoted active demethylation (Pastor et al., 2013; Shen et al., 2014). However, the mechanism that underlies the active demethylation of the mammalian genome has remained enigmatic until the TET enzyme function was identified recently (Tahiliani et al., 2009). TET proteins facilitate this replication-independent active demethylation in conjunction with DNA repair machinery (Fig. 1C); TET proteins iteratively oxidize 5mC to generate oxi-mCs, of which 5fC and 5caC are subject to removal by DNA repair enzyme thymine-DNA glycosylase (TDG), which triggers base excision repair that ultimately substitutes the resulting abasic sites with unmodified cytosines (Hashimoto et al., 2013; He et al., 2011; Maiti and Drohat, 2011; Zhang et al., 2012). TDG typically removes the mispaired thymine (generated as a result of 5mC deamination) from T:G mismatch but recent studies demonstrated that it also has affinity for 5fC and 5caC that are normally base-paired with G (He et al., 2011; Maiti and Drohat, 2011; Zhang et al., 2012). Additional mechanisms that are potentially implicated in the DNA demethylation but are relatively less characterized include decarboxylation of 5caC by an unidentified enzyme (Schiesser et al., 2012), 5mC removal by DNMT (Chen et al., 2012), and 5hmC deamination by AID (activation-induced deaminase)/APOBEC (apolipoprotein B mRNA-editing enzyme, catalytic polypeptide) family of cytidine deaminases followed by base excision repair (Guo et al., 2011).
DNA DEMETHYLATION-INDEPENDENT FUNCTIONS OF TET PROTEINS
Increasing evidence suggests that TET-generated oxi-mCs are not simple intermediates in DNA demethylation pathways. Instead, they have direct regulatory roles in gene expression by behaving as independent epigenetic modifications (Fig. 3). The extensive genome-wide mapping of oxi-mCs in various cell types revealed that these modified bases are strongly enriched at enhancers, promoters and gene bodies, with distinct effects on gene expression (Pastor et al., 2013; Wu and Zhang, 2014). In addition, 5hmC and 5fC are fairly stable bases that are diluted mainly through cell proliferation (Bachman et al., 2014; 2015). Compared with unmodified cytosine, the presence of 5hmC increased the thermodynamic stability of double-stranded DNA (Raiber et al., 2015). In contrast, 5fC did not affect thermodynamic stability but induced the conformational change of a DNA double helix, leading to an unusual structure (designated as ‘F-DNA’ by the authors) with characteristic helical underwinding (Raiber et al., 2015), raising the possibility that 5fC may affect local DNA supercoiling and packaging and hence modify the DNA’s capacity to accommodate proteins. Also, RNA polymerase II specifically recognized the 5-carboxyl or 5-carbonyl moieties of 5caC or 5fC, respectively, by forming a hydrogen bond (Wang et al., 2015a). This interaction induced transient Pol II pausing or stalling and reduced the rate of Pol II transcription elongation on gene bodies (Kellinger et al., 2012; Wang et al., 2015a). Moreover, all oxi-mCs were shown to be recognized by their unique protein readers including transcription factors, splicing factors, DNA repair enzymes and chromatin remodelers (Hashimoto et al., 2014; Iurlaro et al., 2013; Spruijt et al., 2013) and TET proteins were also shown to interact with a number of cellular proteins (Ko et al., 2015; Shen et al., 2014). These results collectively indicate that oxi-mCs potentially function as independent epigenetic marks regardless of DNA demethylation, and that the loss of these marks per se can directly affect expression of genes that are implicated in various aspects of cellular development and functions.
Fig. 3.
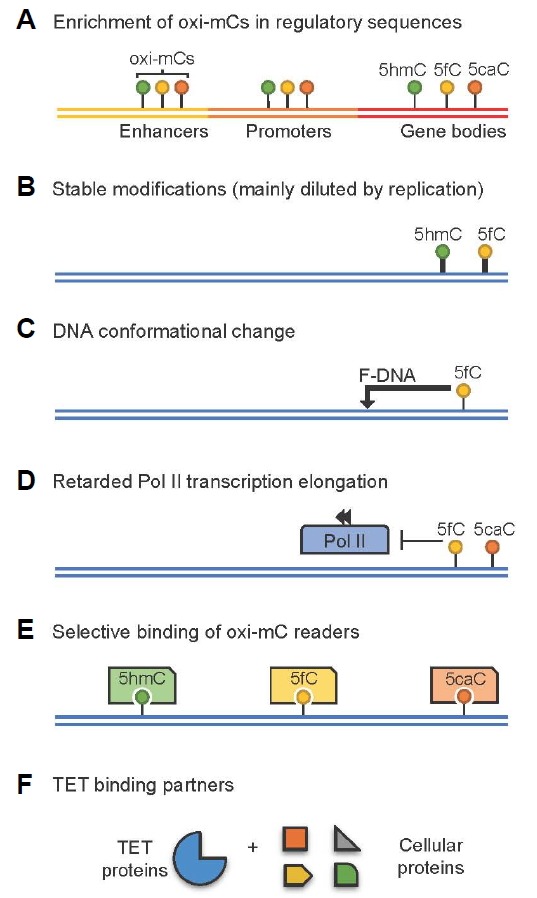
Oxidized methylcytosines(oxi-mCs) can function as independent epigenetic modifications. (A) Oxi-mCs are enriched at DNA sequences associated with gene expression control. (B) 5hmC and 5fC are suggested as stable modifications that are mainly diluted through cell proliferation. (C) 5fC induces conformational change of DNA to ‘F-DNA’. (D) 5fC and 5caC interact with RNA polymerase II and induce a pausing or retardation during transcription elongation. (E–F) Physical association of oxi-mCs (E) and TET proteins (F) with unique cellular proteins.
TET LOSS-OF-FUNCTION IN HEMATOPOIETIC TRANSFORMATION
TET1: friend or foe?
The potential function of TET1 in oncogenesis was first suggested in early studies in which TET1 was discovered as a partner gene that undergoes fusion with the MLL gene in rare cases of AML/ALL (Lorsbach et al., 2003; Ono et al., 2002). However, because cancer genome sequencing analyses rarely identified mutations in the TET1 (as well as TET3) coding sequences in hematologic cancers (Huang and Rao, 2014; Ko et al., 2015), its exact function in normal and malignant hematopoiesis remained unexplored until recent studies reported key roles of TET1 in hematopoietic transformation (Cimmino et al., 2015; Huang et al., 2013). Interestingly, TET1 seems to have distinct roles depending on the types of cancers. In MLL-rearranged leukemia, TET1 acts as an oncogene that facilitates leukemogenesis by being directly activated by MLL fusion proteins and upregulating the expression of key oncogenic target genes such as Hoxa9, Meis1 and Pbx3 (Huang et al., 2013).
By contrast, another study using mice with germline deletion of Tet1 showed that Tet1 may function as a tumor suppressor in the pathogenesis of B cell lymphoma (Cimmino et al., 2015). Ablation of Tet1 in mice led to lymphocytosis, enlargement of lymph nodes, splenomegaly, hepatomegaly, disrupted splenic architecture and infiltration of atypical lymphocytes into various organs such as the liver, lung and kidney, ultimately resulting in transplantable B cell lymphoma albeit at advanced age (Cimmino et al., 2015). Tet1 deficiency conferred a significant advantage on the ability of hematopoietic stem cells (HSCs) to reconstitute hematopoietic lineages in a cell-intrinsic manner, with a slight bias toward the B cell lineage. TET1 promoters tended to be hypermethylated and transcriptionally silenced in human non-Hodgkin B cell lymphoma (B-NHL). Tet1 deficiency significantly decreased the frequency of long-term HSCs in the bone marrow and induced skewing toward the B cell lineage at early stage as shown by an increased frequency of lymphoid-primed multipotent progenitors (LMPPs) (Cimmino et al., 2015). Loss of Tet1 in hematopoietic stem/progenitor cells (HSPCs) altered transcriptional programs particularly for B lineage specification, chromosome maintenance, transcription and DNA repair. Tet1-deficient pro-B cells were able to self-renew more efficiently in semi-solid medium, compared with wild-type cells. They also displayed accumulation of DNA damage (γH2AX staining) presumably due to reduced expression of genes implicated in DNA damage repair (Cimmino et al., 2015). Whole-exome sequencing of the tumors that developed in Tet1-deficient mice identified numerous co-existing mutations that are also recurrently found in patients with B cell lymphoma (Cimmino et al., 2015). However, how Tet1 plays distinct roles in the development of myeloid or lymphoid malignancies; how Tet1 loss induces alterations in the differentiation and function of HSCs, gene expression and DNA repair; and how these phenotypes are linked to oncogenic transformation remain to be determined.
High prevalence of TET2 mutations in hematopoietic malignancies
In contrast to TET1 and TET3, TET2 undergoes somatic mutations very frequently in hematopoietic malignancies (Huang and Rao, 2014; Ko et al., 2015). Early studies showed that the chromosome 4q24 recurrently undergoes microscopic deletions and copy number-neutral loss-of-heterozygosity (also referred to as uniparental disomy) in patients with various myeloid malignancies such as myelodysplastic syndrome (MDS), myeloproliferative neoplasm (MPN), acute myeloid leukemia (AML) and chronic myelomonocytic leukemia (CMML) (Delhommeau et al., 2009; Langemeijer et al., 2009; Viguie et al., 2005). TET2 was initially described as a tumor suppressor because the deleted regions on 4q24 in these patients contained only TET2 gene. Somatic mutations in the TET2 coding regions were very common in patients with either a heterozygous deletion or a copy number-neutral loss of heterozygosity (Delhommeau et al., 2009; Langemeijer et al., 2009). Following these reports, extensive whole-exome sequencing was performed in a wide range of myeloid malignancies and as a result, TET2 was established as one of the most frequently mutated genes in myeloid malignancies (Huang and Rao, 2014; Ko et al., 2015). However, the prognostic value of TET2 mutations in myeloid malignancies is still not clearly defined (Huang and Rao, 2014; Ko et al., 2015). Most of the TET2 missense mutations tend to be clustered in two highly conserved regions within the catalytic domain, some of which are directly targeting key catalytic residues that interact with Fe2+ and 2OG and impair the normal enzymatic activity of TET2 (Ko et al., 2010). TET2 mutations in clinical samples also correlate well with a profound reduction in the global level of 5hmC (Ko et al., 2010; Konstandin et al., 2011; Pronier et al., 2011). These results collectively indicate that TET2 undergoes inactivating loss-of-function mutations during malignant myeloid transformation. Depending on the mutational status of TET2, myeloid cancer patients can be classified into two groups (i.e. 5hmChi vs. 5hmClow groups) at the molecular level (Ko et al., 2010). Future studies will resolve whether these groups display any differences in disease progression or therapeutic responses, and whether 5hmC levels can be used as a novel diagnostic or prognostic marker.
One of the prominent features of TET2 loss-of-function mutations is their ability to cause developmental skewing during differentiation of HSPCs. CD34+ HSPCs derived from MPN patients bearing TET2 mutations efficiently reconstituted the hematopoietic system in nonobese diabetic mice with severe combined immunodeficiency (NOD-SCID), with a bias toward myeloid cells at the expense of lymphoid cells (Delhommeau et al., 2009). Similarly, murine and human HSPCs depleted of TET2 also preferentially differentiated toward monocyte/macrophage lineages over the other lineages in vitro (Chang et al., 2014; Ko et al., 2010; 2011; Pronier et al., 2011). However, the molecular basis behind this biased differentiation is still ambiguous.
In addition to myeloid malignancies, TET2 mutations are also common in lymphoid malignancies, particularly among patients with peripheral T cell lymphoma (PTCL) such as angioimmunoblastic T cell lymphoma (AITL: 33∼76%) and peripheral T cell lymphoma, not otherwise specified (PTCL-NOS: 20∼38%) (Lemonnier et al., 2012; Odejide et al., 2014; Quivoron et al., 2011). These patients frequently harbor TET2 mutations together with mutations in RHOA, DNMT3A and IDH2 genes (Couronne et al., 2012; Lemonnier et al., 2012; Odejide et al., 2014; Palomero et al., 2014; Sakata-Yanagimoto et al., 2014). TET2 mutations correlate with derivation of T follicular helper cells and are associated with advanced-stage diseases and poorer clinical outcomes (Lemonnier et al., 2012). However, little is known about whether TET2 mutations have any prognostic value in lymphoma. Given the rare occurrence of lymphomas in Tet-deficient murine models, acquisition of additional mutations may be important to drive lymphomagenesis. The pathophysiologic function of TET2 mutations in lymphoma development and the outcome of their genetic interactions with other mutations warrant further investigation.
Lessons from murine models with individual deficiency of TET genes
In order to model a human disease containing TET2 loss-of-function mutation, various groups have generated a variety of murine models by either conditionally deleting critical exons of Tet2 or adopting insertional mutagenesis [reviewed in (Ko et al., 2015)]. Despite distinct gene targeting strategies, all the Tet2-disrupted mice exhibited very similar hematopoietic phenotypes in vivo. Deficiency of Tet2 in adult mice as well as fetal livers resulted in amplification of the cell fraction (known as LSK, Lin− c-Kit+ Sca-1+) that is enriched in HSCs (Ko et al., 2011; Kunimoto et al., 2012; 2014; Li et al., 2011; Moran-Crusio et al., 2011; Quivoron et al., 2011; Shide et al., 2012). Under competitive conditions, cells isolated from the bone marrow or fetal liver in Tet2-deficient mice were more efficient in reconstituting hematopoietic lineages in a cell-autonomous manner, compared with wild-type cells (Ko et al., 2011; Kunimoto et al., 2012; Li et al., 2011; Moran-Crusio et al., 2011; Quivoron et al., 2011). Similarly, serial transplantation of Tet2-deficient LSK CD150+ fetal liver cells showed augmented hematopoietic repopulation capacity (Kunimoto et al., 2014). Tet2 ablation seemed to increase serial replating capacity of HSPCs in colony formation assays in vitro (Kunimoto et al., 2014; Moran-Crusio et al., 2011; Quivoron et al., 2011). Taken together, these results imply that Tet2 normally restricts the expansion, differentiation and function of HSCs. Several studies have argued that Tet2 deficiency increases HSC self-renewal. However, the term ‘self-renewal’ is often misinterpreted in a manner that sometimes can be confused with ‘serial repopulating capacity’. Strictly speaking, HSC ‘self-renewal’ refers to the process by which HSCs undergo divisions to give rise to identical HSCs to perpetuate themselves throughout life. Therefore, the impact of Tet2 deficiency on HSC self-renewal has yet to be directly tested. More rigorous test that directly quantifies amplified HSCs, instead of measuring peripheral blood chimerism, after serial transplantation of purified HSCs is required, as exemplified by a recent study that confirmed an augmented HSC self-renewal upon loss of Dnmt3a (Challen et al., 2012).
Notably, both homozygous and heterozygous deletion of Tet2 gene can predispose mice to the development of CMML-like myeloid disease in several models, characterized by myeloid hyperplasia, leukocytosis associated monocytosis and neutrophilia, extramedullary hematopoiesis and enlargement of the spleen and liver (Li et al., 2011; Moran-Crusio et al., 2011; Quivoron et al., 2011). However, the mechanistic links between Tet2 loss and myeloid transformation are still lacking. Considering the increase of HSC number and repopulating capacity upon Tet2 deficiency, additional studies including limiting dilution assays might be of significant help to determine whether Tet2 deficiency promotes leukemogenesis by conferring a stem cell advantage on leukemia initiating cells.
It is now generally accepted that Tet2 loss-of-function facilitates myeloid transformation. However, for reasons that are not entirely clear, only part of the Tet2-deficient mice strains sporadically developed full-blown malignancy with a very long latency and incomplete penetrance (Huang and Rao, 2014; Ko et al., 2015). Some strains of Tet2-deficient mice did not develop disease, implying that the deletion of Tet2 alone is not sufficient to drive hematopoietic malignancies (Quivoron et al., 2011). Furthermore, TET2 mutations observed in elderly people with clonal hematopoiesis were not always associated with an overt hematological malignancy (Busque et al., 2012). Considering that TET2 mutations frequently occur at an early phase of oncogenesis in MDS, CMML and MPN and seem to contribute to clonal expansion of leukemic cells (Delhommeau et al., 2009; Haferlach et al., 2014; Itzykson et al., 2013; Papaemmanuil et al., 2013), TET2 mutations are likely to represent an early oncogenic event that facilitates the initiation of transformation, and then the resulting TET2-mutated pre-malignant cells acquire additional mutations in order to progress to a more malignant phenotype (Huang and Rao, 2014; Ko et al., 2015), in line with the ‘second hit’ hypothesis in cancer development. In support of this hypothesis, analyses of the mutational landscape in human cancers showed that TET2 mutations are indeed frequently found in conjunction with other mutations such as mutations in splicing factors SRSF2, SF3B1 and ZRSR2, and the polycomb proteins EZH2 and ASXL1 in MDS and CMML (Gelsi-Boyer et al., 2010; Haferlach et al., 2014; Hanssens et al., 2014; Meggendorfer et al., 2012; Muto et al., 2013). They also co-exist with KITD816V or FLT3ITD (ITD refers to internal tandem duplication) mutations in systemic mastocytosis (Tefferi et al., 2009; Traina et al., 2012) or AML (Patel et al., 2012), respectively. In murine models, concurrent depletion of Tet2 together with either Asxl1 or Ezh2 hastened the development of myeloid disease, recapitulating characteristics of human MDS (Abdel-Wahab et al., 2013; Muto et al., 2013), and the co-existence of KITD816V or Flt3ITD led to a more aggressive form of systemic mastocytosis (De Vita et al., 2014) or AML (Shih et al., 2015), respectively. Thus, accumulation of secondary mutations in a TET2-deficient background may affect the disease type, progression and latency of tumorigenesis.
Other than the expansion of HSPCs and sporadic occurrences of full-blown malignancy, Tet2 deficiency in mice results in relatively mild hematopoietic phenotypes and all hematopoietic lineages differentiate normally. In murine hematopoietic cells, Tet2 and Tet3 mRNAs are both highly expressed and their expression levels are dynamically regulated during differentiation (Ko et al., 2010; Li et al., 2011; Moran-Crusio et al., 2011). Loss of either Tet2 or Tet3 results in merely a modest decrease in levels of genomic 5hmC (Ko et al., 2011; 2015). Like Tet2 deficiency, conditional ablation of Tet3 with a hematopoietic-specific Vav-iCre transgene leads to an alteration in the frequency of HSCs and augments their repopulating capacity under competitive condition (Ko et al., 2015). However, the overall hematopoiesis takes place normally even in the absence of Tet3 (Ko et al., 2015). These results suggest that Tet2 and Tet3 may have overlapping functions and can compensate for one another. Further studies involving simultaneous loss of Tet2 and Tet3 in the hematopoietic system will resolve this issue.
POTENTIAL MECHANISMS UNDERLYING ONCOGENESIS DRIVEN BY TET DYSREGULATION
So far, numerous studies strongly suggest that TET2 is a bonafide tumor suppressor in the pathogenesis of myeloid cancers. However, most of these studies are descriptive with no detailed explanation about the underlying mechanisms. It is clear that Tet2 deficiency not only alters homeostasis, differentiation and function of the HSCs, but also elevates 5mC levels at specific genomic loci by impairing 5mC consumption. However, whether these aberrations contribute to oncogenic transformation still remains unclear. Also, the impact of TET loss on changes of DNA methylation status might be more complex than we expect because the oxi-mCs can function independent of DNA demethylation as described earlier.
Aberrant gene expression and function are hallmarks of cancers and it was proposed that altered DNA methylation in TET-deficient cells would disrupt transcriptional programs that have pivotal regulatory roles in normal hematopoiesis. However, recent global approaches that involve genome-wide mapping of 5mC-marked regions combined with global gene expression profiles of wild-type and Tet-deficient embryonic stem cells or hematopoietic cells indicate that the changes in DNA methylation upon Tet loss do not directly account for altered patterns in gene expression (Challen et al., 2012; Hon et al., 2014; Huang et al., 2014; Lu et al., 2014). The conventional view regarding the abnormal DNA methylation patterns in cancers (global hypomethylation and localized CpG island hypermethylation) was proposed mainly based on the bisulfite sequencing, which does not discriminate between 5mC and 5hmC (Huang et al., 2010; Pastor et al., 2013). Considering a substantial loss of 5hmC in cancers, the apparent global hypomethylation in cancers may reflect reduced levels of 5hmC rather than 5mC. Thus, the cancer-associated DNA methylation patterns and their correlation with gene transcription need to be reevaluated using more advanced techniques that allows for genome-wide mapping of individual cytosine derivatives. Furthermore, the question regarding whether loss of TET catalytic activity, TET protein scaffolding function or both are responsible for hematopoietic transformation is also not clearly resolved, so there remains a possibility that the TET catalytic activity-independent mechanisms could also contribute to oncogenic transformation by altering chromatin architecture and gene expression through interactions with various cellular proteins, which will be discussed in more detail in a later section. Since the identification of factors leading to TET dysfunction and oncogenesis remains critical for the development of more safe and effective cancer therapy, the molecular basis of oncogenesis upon loss of TET function deserves further exploration.
NON-MUTATIONAL MECHANISMS THAT INACTIVATE TET PROTEINS
The mechanisms of TET protein regulation are of intense interest as TET protein expression and the global level of their primary catalytic product, 5hmC, is significantly diminished in many cancers (Huang and Rao, 2014; Ko et al., 2015). Given that TET proteins interact with a variety of proteins and that they are subject to regulation by various modulators, it is not surprising that the expression and activity of TET proteins can be targeted by non-mutational mechanisms during the onset and/or progression of cancers (Fig. 4). Indeed, a significant proportion of myeloid cancer patients who do not have somatic mutations in any TET genes had substantially reduced levels of 5hmC (Ko et al., 2010; Konstandin et al., 2011; Pronier et al., 2011), illustrating that wild-type TET proteins can become non-functional in these cancers. Even though mutations in TET1 and TET3 coding sequences are infrequent in hematopoietic malignancies, this does not necessarily mean that they are functional. Understanding of the various mechanisms that lead to the silencing of TET proteins in cancers will increase the options for manipulating TET activity or expression, and provide a more solid basis to restore normal levels of TET proteins and oxi-mCs in cancers, which may be beneficial for cancer therapy.
Fig. 4.
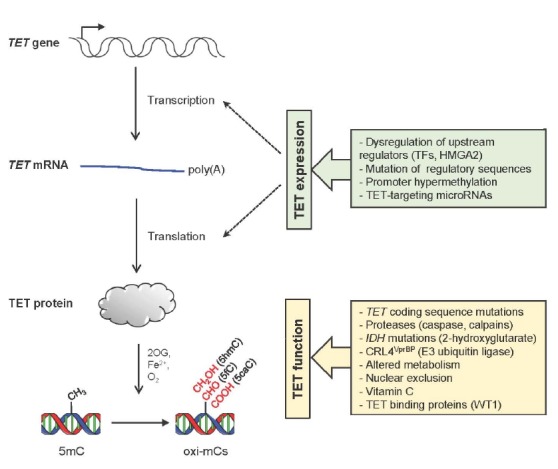
Non-mutational modulation of TET expression and function. The expression and function of TET proteins are regulated on many levels. For details, please refer to the text.
Regulation of TET mRNA or protein expression
The expression of TET proteins is regulated at multiple levels (Fig. 4). Mutations that lead to changes in the key regulatory sequences of TET genes such as promoters or enhancers can alter transcription by differentially regulating the recruitment of transcription regulators. Similarly, dysregulation of genes that encode essential upstream regulators can also affect TET expression. For instance, depletion of the high mobility group AT-hook 2 (HMGA2), a chromatin remodeling factor that is highly expressed in some cancers but not in most normal cells, upregulated TET1 expression in breast cancer cells (Sun et al., 2013). Thereafter, TET1 stimulated the expression of its downstream target homeobox A (HOXA) genes by being directly targeted to their promoters to induce demethylation, preventing breast tumor growth, invasion and metastasis. Promoter CpG hypermethylation can also affect TET expression as reported in the TET1 promoter in B cell lymphomas (Cimmino et al., 2015) or the TET2 promoter in some gliomas and MPN (Chim et al., 2010; Kim et al., 2011).
Furthermore, a number of microRNAs repressed TET expression at the post-transcriptional level. One representative example is the oncogenic microRNA miR-22 that is highly expressed in myeloid malignancies (Song et al., 2013a; 2013b). Mimicking Tet2 deficiency, ectopic expression of miR-22 in mice led to HSC expansion and enhanced their repopulating activity and serial replating capacity, ultimately resulting in MDS-like hematopoietic malignancies that progress to myeloid leukemia (Song et al., 2013a). The disease phenotype was transplantable when miR-22-expressing LSK cells were transferred into wild-type recipient mice. The aberrant hematopoietic phenotypes and transformation induced by miR-22 expression were reversed by enforced expression of Tet2, demonstrating that Tet2 is a central downstream target of miR-22. MiR-22 also reduced genomic 5hmC levels in breast cancer cells by directly binding to the 3′ untranslated region (3′UTR) of all TET mRNAs (Song et al., 2013b). Interestingly, miR-22 promoted the mesenchymal-epithelial transition (MET), mammary tumor development, and metastasis by downregulating TET expression, which in turn led to promoter methylation and suppression of the downstream miR-200 family members that are known to control various genes implicated in MET and metastasis (Song et al., 2013b). In another study utilizing high-throughput screening, more than 30 miRNAs including miR-125b, miR-29a, miR-29b, miR-26a, miR-26b, miR-101 and miR-7 were shown to diminish TET levels (Cheng et al., 2013). Enforced expression of Tet2-targeting miRNAs mimicked Tet2 loss as it induced the expansion of myeloid lineage cells and CMML-like phenotypes, which was rescued by expressing Tet2 that lacks 3′UTR. Repression of TET expression by miR-26a was also shown to promote pancreatic differentiation (Fu et al., 2013).
TET expression is also controlled at the post-translational level. For example, a recent study showed that TET2 undergoes monoubiquitylation by the CRL4VprBP complex, which increases the DNA binding activity of TET2 (Nakagawa et al., 2015). Several leukemia-associated TET2 mutations were shown to impair CRL4VprBP-mediated monoubiquitylation. Additionally, calpains, a family of Ca2+-dependent cysteine proteinases, were shown to control the steady state level of TET proteins in embryonic stem cells (Wang and Zhang, 2014). Calpain 1 (encoded by Capn1) catalyzed the proteolysis of Tet1 and Tet2 proteins in undifferentiated embryonic stem cells, and calpain 2 (encoded by Capn2) degraded Tet3 in differentiated cells (Wang and Zhang, 2014). Interestingly, the expression of calpains is often aberrantly regulated in cancers. Similarly, caspases, another cysteine protease similar to calpains, were also reported to cleave Tet proteins (Ko et al., 2013).
Regulation of TET protein function
IDH1 and IDH2 are essential components of cellular metabolism that catalyze oxidative decarboxylation of isocitrate in the tricarboxylic acid (TCA) cycle to produce 2OG, a key intermediate in cellular energy metabolism. Genes encoding these proteins frequently undergo somatic heterozygous mutations in a majority of glioblastomas (∼75%) and a subset of myeloid leukemias (∼20%) or solid cancers (Abbas et al., 2010; Cairns et al., 2012; Gross et al., 2010; Parsons et al., 2008; Thol et al., 2010; Yan et al., 2009). Interestingly, specific arginine residues in the active site of IDH enzymes (R132 in IDH1 and R140 or R172 in IDH2 proteins) are frequently mutated, and the resulting mutant IDH enzymes gain a novel enzymatic function to reduce 2OG to oncometabolite 2-hydroxyglutarate (2HG) (Dang et al., 2009; Figueroa et al., 2010; Ward et al., 2010). 2HG functions as a potent inhibitor of TET proteins that competes with 2OG in vitro and in vivo (Xu et al., 2011a). Mice expressing mutant Idh1 or Idh2 showed aberrant hematopoietic phenotypes such as the expansion of HSPCs, splenomegaly, anemia and extramedullary hematopoiesis, although IDH mutations alone were not enough to drive full-blown leukemias unless they were combined with other mutations such as HoxA9, Meis1 or Flt3ITD mutations (Kats et al., 2014; Sasaki et al., 2012). Intriguingly, continuous production of 2HG by mutant IDH proteins seemed to be critical for the maintenance of leukemic cells as suppression of mutant IDH expression or pharmacological inhibition of 2HG production eliminated the pathogenic features by promoting differentiation of leukemic cells. Additionally, because TET proteins are critically dependent on the presence of co-substrates 2OG and O2 as well as a cofactor Fe2+, the activity of TET proteins is sensitive to alterations in the levels of energy metabolites (succinate, fumarate and 2HG), redox status, oxygen availability and iron homeostasis. Ascorbate (vitamin C) is also another cofactor that is presumed to help maintain Fe2+ in its reduced form for full TET catalytic activity (Blaschke et al., 2013; Minor et al., 2013; Yin et al., 2013).
TET protein function is also controlled by physical associations with other cellular proteins. A variety of proteins were shown to bind to TET proteins including WT1, IDAX, PGC7 and IkBζ (Ko et al., 2015; Shen et al., 2014). Dysregulation of these binding proteins can potentially lead to inactivation of TET proteins by impairing their chromatin targeting. For instance, WT1 was identified as one of key interacting partners for TET proteins (Rampal et al., 2014; Wang et al., 2015b). WT1 recruited TET2 to specific chromatin regions, particularly promoters, to activate TET2 target genes and thus WT1 mutations led to functional inactivation of TET2, impairing TET2 target gene expression (Rampal et al., 2014). WT1 was also necessary for TET2 to suppress the growth and the colony formation of leukemic cells (Wang et al., 2015b). In line with these observations, WT1 mutations were mutually exclusive with mutations in TET2 and IDH genes in leukemias.
TET2 protein levels can also be regulated through association with another interaction partner, IDAX (Ko et al., 2013). IDAX was originally described as an inhibitor of the canonical Wnt/β-catenin signaling (Hino et al., 2001). Through a direct interaction, IDAX directly bound to the catalytic domain of TET2 via its CXXC domain and seemed to recruit TET2 to chromatin regions with high to medium density of unmethylated CpG dinucleotides, such as promoters and CpG islands. Intriguingly, high level of IDAX led to the activation of caspases that degraded TET2 proteins (Ko et al., 2013). IDAX is reported to be dysregulated in cancers; IDAX overexpression was reported in villous adenomas of the colon where it would contribute to the depletion of 5hmCs by promoting TET2 degradation (Paez et al., 2014). By contrast, homozygous deletion of IDAX was reported in an aggressive renal cell carcinoma (Kojima et al., 2009). In this case, the loss of IDAX would disrupt normal genomic targeting of TET2, resulting in aberrant expression of TET2 target genes.
In innate immune cells such as dendritic cells and macrophages, Tet2 was shown to repress Il6 transcription in the late phase of the lipopolysaccharide induced inflammatory response (Zhang et al., 2015). Interestingly, Tet2 was recruited to the Il6 promoter by a transcription factor IkBζ, which in turn additionally recruited Hdac2 to induce Il6 transrepression through histone deacetylation. In addition to these regulatory mechanisms, TET3 can be localized in the cytoplasm as well as in the nucleus (Arioka et al., 2012; Zhang et al., 2014). Therefore, it is likely to be subjected to regulation by signals that impair nuclear translocation. However, it is unclear whether subcellular localization of TET3 is altered in cancers, and therefore future studies are required to resolve this point.
CONCLUSION
Epigenetic modifications are generated and erased in a highly dynamic and reversible manner. Therefore, genetic aberrations leading to epimutations that are directly implicated in the initiation and progression of cancers represent an attractive target for cancer therapy. It is well documented that there is a direct causal link between TET dysregulation/mutations, impaired hematopoiesis and malignant hematopoietic transformation. Thus, it would be of great importance to assess whether manipulation of 5mC oxidation status by targeting TET enzymes displays clinical efficacy in cancer patients. In order for these trials to succeed, we essentially need comprehensive understanding of the mechanisms that lead to the dysregulation of TET protein expression and activity during oncogenic transformation. Moreover, further identification of proteins or small molecules that physically or functionally interact with TET enzymes, together with detailed dissection of the molecular basis of their actions are necessary. The knowledge gained through these research will allow us to devise more sophisticated strategies to specifically manipulate TET activities, thereby facilitating the development of more efficient, clinically promising therapeutic avenues to treat human cancers. Since the inactivation of TET function is presumed to be a general molecular basis implicated in the development of diverse solid cancers including cancers of lung, breast, colon, intestine, prostate, liver, brain and skin, an anti-cancer therapy targeting TET activity will be applicable over a broad spectrum of human cancers.
Acknowledgments
We apologize for the many studies that were not discussed here because of limited space. This work was supported by the research fund (1.150084) of Ulsan National Institute of Science and Technology (to M.K.).
REFERENCES
- Abbas S., Lugthart S., Kavelaars F.G., Schelen A., Koenders J.E., Zeilemaker A., van Putten W.J., Rijneveld A.W., Lowenberg B., Valk P.J. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood. 2010;116:2122–2126. doi: 10.1182/blood-2009-11-250878. [DOI] [PubMed] [Google Scholar]
- Abdel-Wahab O., Gao J., Adli M., Dey A., Trimarchi T., Chung Y.R., Kuscu C., Hricik T., Ndiaye-Lobry D., Lafave L.M., et al. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J. Exp. Med. 2013;210:2641–2659. doi: 10.1084/jem.20131141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arioka Y., Watanabe A., Saito K., Yamada Y. Activation-induced cytidine deaminase alters the subcellular localization of Tet family proteins. PLos One. 2012;7:e45031. doi: 10.1371/journal.pone.0045031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arita K., Ariyoshi M., Tochio H., Nakamura Y., Shirakawa M. Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature. 2008;455:818–821. doi: 10.1038/nature07249. [DOI] [PubMed] [Google Scholar]
- Avvakumov G.V., Walker J.R., Xue S., Li Y.J., Duan S.L., Bronner C., Arrowsmith C.H., Dhe-Paganon S. Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature. 2008;455:822–825. doi: 10.1038/nature07273. [DOI] [PubMed] [Google Scholar]
- Bachman M., Uribe-Lewis S., Yang X.P., Williams M., Murrell A., Balasubramanian S. 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat. Chem. 2014;6:1049–1055. doi: 10.1038/nchem.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachman M., Uribe-Lewis S., Yang X.P., Burgess H.E., Iurlaro M., Reik W., Murrell A., Balasubramanian S. 5-Formylcytosine can be a stable DNA modification in mammals. Nat. Chem. Biol. 2015;11:555–557. doi: 10.1038/nchembio.1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaschke K., Ebata K.T., Karimi M.M., Zepeda-Martinez J.A., Goyal P., Mahapatra S., Tam A., Laird D.J., Hirst M., Rao A., et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature. 2013;500:222–226. doi: 10.1038/nature12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostick M., Kim J.K., Esteve P.O., Clark A., Pradhan S., Jacobsen S.E. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007;317:1760–1764. doi: 10.1126/science.1147939. [DOI] [PubMed] [Google Scholar]
- Busque L., Patel J.P., Figueroa M.E., Vasanthakumar A., Provost S., Hamilou Z., Mollica L., Li J., Viale A., Heguy A., et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet. 2012;44:1179–1181. doi: 10.1038/ng.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns R.A., Iqbal J., Lemonnier F., Kucuk C., de Leval L., Jais J.P., Parrens M., Martin A., Xerri L., Brousset P., et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood. 2012;119:1901–1903. doi: 10.1182/blood-2011-11-391748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challen G.A., Sun D.Q., Jeong M., Luo M., Jelinek J., Berg J.S., Bock C., Vasanthakumar A., Gu H.C., Xi Y.X., et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2012;44:23–31. doi: 10.1038/ng.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y.I., Damnernsawad A., Allen L.K., Yang D., Ranheim E.A., Young K.H., Zhang J.F., Kong G.Y., Wang J.Y., Liu Y.G., et al. Evaluation of allelic strength of human TET2 mutations and cooperation between Tet2 knockdown and oncogenic Nras mutation. Br. J. Haematol. 2014;166:461–465. doi: 10.1111/bjh.12871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.C., Wang K.Y., Shen C.K. The mammalian de novo DNA methyltransferases DNMT3A and DNMT3B are also DNA 5-hydroxymethylcytosine dehydroxymethylases. J. Biol. Chem. 2012;287:33116–33121. doi: 10.1074/jbc.C112.406975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J.J., Guo S.Q., Chen S.N., Mastriano S.J., Liu C.C., D'Alessio A.C., Hysolli E., Guo Y.W., Yao H., Megyola C.M., et al. An extensive network of TET2-targeting microRNAs regulates malignant hematopoiesis. Cell Rep. 2013;5:471–481. doi: 10.1016/j.celrep.2013.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chim C.S., Wan T.S., Fung T.K., Wong K.F. Methylation of TET2, CBL and CEBPA in Ph-negative myeloproliferative neoplasms. J. Clin. Pathol. 2010;63:942–946. doi: 10.1136/jcp.2010.080218. [DOI] [PubMed] [Google Scholar]
- Cimmino L., Dawlaty M.M., Ndiaye-Lobry D., Yap Y.S., Bakogianni S., Yu Y., Bhattacharyya S., Shaknovich R., Geng H., Lobry C., et al. TET1 is a tumor suppressor of hematopoietic malignancy. Nat. Immunol. 2015;16:653–662. doi: 10.1038/ni.3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couronne L., Bastard C., Bernard O.A. TET2 and DNMT3A mutations in human T-cell lymphoma. N Engl. J. Med. 2012;366:95–96. doi: 10.1056/NEJMc1111708. [DOI] [PubMed] [Google Scholar]
- Dang L., White D.W., Gross S., Bennett B.D., Bittinger M.A., Driggers E.M., Fantin V.R., Jang H.G., Jin S., Keenan M.C., et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vita S., Schneider R.K., Garcia M., Wood J., Gavillet M., Ebert B.L., Gerbaulet A., Roers A., Levine R.L., Mullally A., et al. Loss of function of TET2 cooperates with constitutively active KIT in murine and human models of mastocytosis. PLoS One. 2014;9:e96209. doi: 10.1371/journal.pone.0096209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delhommeau F., Dupont S., Della Valle V., James C., Trannoy S., Masse A., Kosmider O., Le Couedic J.P., Robert F., Alberdi A., et al. Mutation in TET2 in myeloid cancers. N Engl. J. Med. 2009;360:2289–2301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- Figueroa M.E., Abdel-Wahab O., Lu C., Ward P.S., Patel J., Shih A., Li Y., Bhagwat N., Vasanthakumar A., Fernandez H.F., et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X.H., Jin L., Wang X.C., Luo A., Hu J.K., Zheng X.W., Tsark W.M., Riggs A.D., Ku H.T., Huang W.D. MicroRNA-26a targets ten eleven translocation enzymes and is regulated during pancreatic cell differentiation. Proc. Natl. Acad. Sci. USA. 2013;110:17892–17897. doi: 10.1073/pnas.1317397110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelsi-Boyer V., Trouplin V., Roquain J., Adelaide J., Carbuccia N., Esterni B., Finetti P., Murati A., Arnoulet C., Zerazhi H., et al. ASXL1 mutation is associated with poor prognosis and acute transformation in chronic myelomonocytic leukaemia. Br. J. Haematol. 2010;151:365–375. doi: 10.1111/j.1365-2141.2010.08381.x. [DOI] [PubMed] [Google Scholar]
- Gross S., Cairns R.A., Minden M.D., Driggers E.M., Bittinger M.A., Jang H.G., Sasaki M., Jin S.F., Schenkein D.P., Su S.S.M., et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 2010;207:339–344. doi: 10.1084/jem.20092506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J.U., Su Y.J., Zhong C., Ming G.L., Song H.J. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haferlach T., Nagata Y., Grossmann V., Okuno Y., Bacher U., Nagae G., Schnittger S., Sanada M., Kon A., Alpermann T., et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28:241–247. doi: 10.1038/leu.2013.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanssens K., Brenet F., Agopian J., Georgin-Lavialle S., Damaj G., Cabaret L., Chandesris M.O., de Sepulveda P., Hermine O., Dubreuil P., et al. SRSF2-p95 hotspot mutation is highly associated with advanced forms of mastocytosis and mutations in epigenetic regulator genes. Haematologica. 2014;99:830–835. doi: 10.3324/haematol.2013.095133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H., Horton J.R., Zhang X., Bostick M., Jacobsen S.E., Cheng X.D. The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature. 2008;455:826–829. doi: 10.1038/nature07280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H., Liu Y., Upadhyay A.K., Chang Y., Howerton S.B., Vertino P.M., Zhang X., Cheng X. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res. 2012;40:4841–4849. doi: 10.1093/nar/gks155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H., Zhang X., Cheng X.D. Selective excision of 5-carboxylcytosine by a thymine DNA glycosylase mutant. J. Mol. Biol. 2013;425:971–976. doi: 10.1016/j.jmb.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H., Olanrewaju Y.O., Zheng Y., Wilson G.G., Zhang X., Cheng X.D. Wilms tumor protein recognizes 5-carboxylcytosine within a specific DNA sequence. Genes Dev. 2014;28:2304–2313. doi: 10.1101/gad.250746.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y.F., Li B.Z., Li Z., Liu P., Wang Y., Tang Q., Ding J., Jia Y., Chen Z., Li L., et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hino S., Kishida S., Michiue T., Fukui A., Sakamoto I., Takada S., Asashima M., Kikuchi A. Inhibition of the Wnt signaling pathway by Idax, a novel Dvl-binding protein. Mol. Cell Biol. 2001;21:330–342. doi: 10.1128/MCB.21.1.330-342.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon G.C., Song C.X., Du T.T., Jin F.L., Selvaraj S., Lee A.Y., Yen C.A., Ye Z., Mao S.Q., Wang B.A., et al. 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation. Mol. Cell. 2014;56:286–297. doi: 10.1016/j.molcel.2014.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L.L., Li Z., Cheng J.D., Rao Q.H., Gong W., Liu M.J., Shi Y.J.G., Zhu J.Y., Wang P., Xu Y.H. Crystal structure of TET2-DNA complex: insight into TET-mediated 5mC oxidation. Cell. 2013;155:1545–1555. doi: 10.1016/j.cell.2013.11.020. [DOI] [PubMed] [Google Scholar]
- Huang Y., Rao A. Connections between TET proteins and aberrant DNA modification in cancer. Trends Genet. 2014;30:464–474. doi: 10.1016/j.tig.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Pastor W.A., Shen Y.H., Tahiliani M., Liu D.R., Rao A. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLos One. 2010;5:e8888. doi: 10.1371/journal.pone.0008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H., Jiang X., Li Z., Li Y., Song C.X., He C., Sun M., Chen P., Gurbuxani S., Wang J., et al. TET1 plays an essential oncogenic role in MLL-rearranged leukemia. Proc. Natl. Acad. Sci. USA. 2013;110:11994–11999. doi: 10.1073/pnas.1310656110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Chavez L., Chang X., Wang X., Pastor W.A., Kang J., Zepeda-Martinez J.A., Pape U.J., Jacobsen S.E., Peters B., et al. Distinct roles of the methylcytosine oxidases Tet1 and Tet2 in mouse embryonic stem cells. Proc. Natl. Acad. Sci. USA. 2014;111:1361–1366. doi: 10.1073/pnas.1322921111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S., Shen L., Dai Q., Wu S.C., Collins L.B., Swenberg J.A., He C., Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzykson R., Kosmider O., Renneville A., Morabito M., Preudhomme C., Berthon C., Ades L., Fenaux P., Platzbecker U., Gagey O., et al. Clonal architecture of chronic myelomonocytic leukemias. Blood. 2013;121:2186–2198. doi: 10.1182/blood-2012-06-440347. [DOI] [PubMed] [Google Scholar]
- Iurlaro M., Ficz G., Oxley D., Raiber E.A., Bachman M., Booth M.J., Andrews S., Balasubramanian S., Reik W. A screen for hydroxymethylcytosine and formylcytosine binding proteins suggests functions in transcription and chromatin regulation. Genome Biol. 2013;14:R119. doi: 10.1186/gb-2013-14-10-r119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer L.M., Tahiliani M., Rao A., Aravind L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle. 2009;8:1698–1710. doi: 10.4161/cc.8.11.8580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer L.M., Abhiman S., Aravind L. Natural history of eukaryotic DNA methylation systems. Prog. Mol. Biol. Transl. Sci. 2011;101:25–104. doi: 10.1016/B978-0-12-387685-0.00002-0. [DOI] [PubMed] [Google Scholar]
- Kats L.M., Reschke M., Taulli R., Pozdnyakova O., Burgess K., Bhargava P., Straley K., Karnik R., Meissner A., Small D., et al. Proto-oncogenic role of mutant IDH2 in leukemia initiation and maintenance. Cell Stem Cell. 2014;14:329–341. doi: 10.1016/j.stem.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellinger M.W., Song C.X., Chong J., Lu X.Y., He C., Wang D. 5-formylcytosine and 5-carboxylcytosine reduce the rate and substrate specificity of RNA polymerase II transcription. Nat. Struct. Mol. Biol. 2012;19:831–833. doi: 10.1038/nsmb.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.H., Pierscianek D., Mittelbronn M., Vital A., Mariani L., Hasselblatt M., Ohgaki H. TET2 promoter methylation in low-grade diffuse gliomas lacking IDH1/2 mutations. J. Clin. Pathol. 2011;64:850–852. doi: 10.1136/jclinpath-2011-200133. [DOI] [PubMed] [Google Scholar]
- Ko M., Huang Y., Jankowska A.M., Pape U.J., Tahiliani M., Bandukwala H.S., An J., Lamperti E.D., Koh K.P., Ganetzky R., et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–843. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M., Bandukwala H.S., An J., Lamperti E.D., Thompson E.C., Hastie R., Tsangaratou A., Rajewsky K., Koralov S.B., Rao A. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc. Natl. Acad. Sci. USA. 2011;108:14566–14571. doi: 10.1073/pnas.1112317108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M., An J., Bandukwala H.S., Chavez L., Aijo T., Pastor W.A., Segal M.F., Li H.M., Koh K.P., Lahdesmaki H., et al. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature. 2013;497:122–126. doi: 10.1038/nature12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M., An J., Pastor W.A., Koralov S.B., Rajewsky K., Rao A. TET proteins and 5-methylcytosine oxidation in hematological cancers. Immunol. Rev. 2015;263:6–21. doi: 10.1111/imr.12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima T., Shimazui T., Hinotsu S., Joraku A., Oikawa T., Kawai K., Horie R., Suzuki H., Nagashima R., Yoshikawa K., et al. Decreased expression of CXXC4 promotes a malignant phenotype in renal cell carcinoma by activating Wnt signaling. Oncogene. 2009;28:297–305. doi: 10.1038/onc.2008.391. [DOI] [PubMed] [Google Scholar]
- Konstandin N., Bultmann S., Szwagierczak A., Dufour A., Ksienzyk B., Schneider F., Herold T., Mulaw M., Kakadia P.M., Schneider S., et al. Genomic 5-hydroxymethylcytosine levels correlate with TET2 mutations and a distinct global gene expression pattern in secondary acute myeloid leukemia. Leukemia. 2011;25:1649–1652. doi: 10.1038/leu.2011.134. [DOI] [PubMed] [Google Scholar]
- Kunimoto H., Fukuchi Y., Sakurai M., Sadahira K., Ikeda Y., Okamoto S., Nakajima H. Tet2 disruption leads to enhanced self-renewal and altered differentiation of fetal liver hematopoietic stem cells. Sci. Rep. 2012;2:273. doi: 10.1038/srep00273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunimoto H., Fukuchi Y., Sakurai M., Takubo K., Okamoto S., Nakajima H. Tet2-mutated myeloid progenitors possess aberrant in vitro self-renewal capacity. Blood. 2014;123:2897–2899. doi: 10.1182/blood-2014-01-552471. [DOI] [PubMed] [Google Scholar]
- Langemeijer S.M.C., Kuiper R.P., Berends M., Knops R., Aslanyan M.G., Massop M., Stevens-Linders E., van Hoogen P., van Kessel A.G., Raymakers R.A.P., et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat. Genet. 2009;41:838–842. doi: 10.1038/ng.391. [DOI] [PubMed] [Google Scholar]
- Lemonnier F., Couronne L., Parrens M., Jais J.P., Travert M., Lamant L., Tournillac O., Rousset T., Fabiani B., Cairns R.A., et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with T-FH-like features and adverse clinical parameters. Blood. 2012;120:1466–1469. doi: 10.1182/blood-2012-02-408542. [DOI] [PubMed] [Google Scholar]
- Li Z., Cai X., Cai C.L., Wang J., Zhang W., Petersen B.E., Yang F.C., Xu M. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. 2011;118:4509–4518. doi: 10.1182/blood-2010-12-325241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorsbach R.B., Moore J., Mathew S., Raimondi S.C., Mukatira S.T., Downing J.R. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23) Leukemia. 2003;17:637–641. doi: 10.1038/sj.leu.2402834. [DOI] [PubMed] [Google Scholar]
- Lu F.L., Liu Y.T., Jiang L., Yamaguchi S., Zhang Y. Role of Tet proteins in enhancer activity and telomere elongation. Genes Dev. 2014;28:2103–2119. doi: 10.1101/gad.248005.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti A., Drohat A.C. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J. Biol. Chem. 2011;286:35334–35338. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meggendorfer M., Roller A., Haferlach T., Eder C., Dicker F., Grossmann V., Kohlmann A., Alpermann T., Yoshida K., Ogawa S., et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML) Blood. 2012;120:3080–3088. doi: 10.1182/blood-2012-01-404863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor E.A., Court B.L., Young J.I., Wang G.F. Ascorbate Induces Ten-Eleven Translocation (Tet) Methylcytosine Dioxygenase-mediated Generation of 5-Hydroxymethylcytosine. J. Biol. Chem. 2013;288:13669–13674. doi: 10.1074/jbc.C113.464800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran-Crusio K., Reavie L., Shih A., Abdel-Wahab O., Ndiaye-Lobry D., Lobry C., Figueroa M.E., Vasanthakumar A., Patel J., Zhao X., et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto T., Sashida G., Oshima M., Wendt G.R., Mochizuki-Kashio M., Nagata Y., Sanada M., Miyagi S., Saraya A., Kamio A., et al. Concurrent loss of Ezh2 and Tet2 cooperates in the pathogenesis of myelodysplastic disorders. J. Exp. Med. 2013;210:2627–2639. doi: 10.1084/jem.20131144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T., Lv L., Nakagawa M., Yu Y.B., Yu C., D'Alessio A.C., Nakayama K., Fan H.Y., Chen X., Xiong Y. CRL4(VprBP) E3 Ligase Promotes Monoubiquitylation and Chromatin Binding of TET Dioxygenases. Mol. Cell. 2015;57:247–260. doi: 10.1016/j.molcel.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odejide O., Weigert O., Lane A.A., Toscano D., Lunning M.A., Kopp N., Kim S., van Bodegom D., Bolla S., Schatz J.H., et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood. 2014;123:1293–1296. doi: 10.1182/blood-2013-10-531509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono R., Taki T., Taketani T., Taniwaki M., Kobayashi H., Hayashi Y. LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10;11)(q22;q23) Cancer Res. 2002;62:4075–4080. [PubMed] [Google Scholar]
- Ooi S.K.T., O'Donnell A.H., Bestor T.H. Mammalian cytosine methylation at a glance. J. Cell Sci. 2009;122:2787–2791. doi: 10.1242/jcs.015123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez D., Gerger A., Zhang W., Yang D., Labonte M.J., Benhanim L., Kahn M., Lenz F., Lenz C., Ning Y., et al. Association of common gene variants in the WNT/beta-catenin pathway with colon cancer recurrence. Pharmacogenomics J. 2014;14:142–150. doi: 10.1038/tpj.2013.20. [DOI] [PubMed] [Google Scholar]
- Palomero T., Couronne L., Khiabanian H., Kim M.Y., Ambesi-Impiombato A., Perez-Garcia A., Carpenter Z., Abate F., Allegretta M., Haydu J.E., et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat. Genet. 2014;46:166–170. doi: 10.1038/ng.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E., Gerstung M., Malcovati L., Tauro S., Gundem G., Van Loo P., Yoon C.J., Ellis P., Wedge D.C., Pellagatti A., et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122:3616–3627. doi: 10.1182/blood-2013-08-518886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons D.W., Jones S., Zhang X.S., Lin J.C.H., Leary R.J., Angenendt P., Mankoo P., Carter H., Siu I.M., Gallia G.L., et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor W.A., Aravind L., Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat. Rev. Mol. Cell Biol. 2013;14:341–356. doi: 10.1038/nrm3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel J.P., Gonen M., Figueroa M.E., Fernandez H., Sun Z.X., Racevskis J., Van Vlierberghe P., Dolgalev I., Thomas S., Aminova O., et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl. J. Med. 2012;366:1079–1089. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronier E., Almire C., Mokrani H., Vasanthakumar A., Simon A., Mor B.D.R.M., Masse A., Le Couedic J.P., Pendino F., Carbonne B., et al. Inhibition of TET2-mediated conversion of 5-methylcytosine to 5-hydroxymethylcytosine disturbs erythroid and granulomonocytic differentiation of human hematopoietic progenitors. Blood. 2011;118:2551–2555. doi: 10.1182/blood-2010-12-324707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quivoron C., Couronne L., Della Valle V., Lopez C.K., Plo I., Wagner-Ballon O., Do Cruzeiro M., Delhommeau F., Arnulf B., Stern M.H., et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20:25–38. doi: 10.1016/j.ccr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Raiber E.A., Murat P., Chirgadze D.Y., Beraldi D., Luisi B.F., Balasubramanian S. 5-Formylcytosine alters the structure of the DNA double helix. Nat. Struct. Mol. Biol. 2015;22:44–49. doi: 10.1038/nsmb.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampal R., Alkalin A., Madzo J., Vasanthakumar A., Pronier E., Patel J., Li Y.S., Ahn J.H., Abdel-Wahab O., Shih A., et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 2014;9:1841–1855. doi: 10.1016/j.celrep.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata-Yanagimoto M., Enami T., Yoshida K., Shiraishi Y., Ishii R., Miyake Y., Muto H., Tsuyama N., Sato-Otsubo A., Okuno Y., et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014;46:171–175. doi: 10.1038/ng.2872. [DOI] [PubMed] [Google Scholar]
- Sasaki M., Knobbe C.B., Munger J.C., Lind E.F., Brenner D., Brustle A., Harris I.S., Holmes R., Wakeham A., Haight J., et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. 2012;488:656–659. doi: 10.1038/nature11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiesser S., Hackner B., Pfaffeneder T., Muller M., Hagemeier C., Truss M., Carell T. Mechanism and stem-cell activity of 5-carboxycytosine decarboxylation determined by isotope tracing. Angew. Chem. Int. Ed. Engl. 2012;51:6516–6520. doi: 10.1002/anie.201202583. [DOI] [PubMed] [Google Scholar]
- Sharif J., Muto M., Takebayashi S.I., Suetake I., Iwamatsu A., Endo T.A., Shinga J., Mizutani-Koseki Y., Toyoda T., Okamura K., et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007;450:908–912. doi: 10.1038/nature06397. [DOI] [PubMed] [Google Scholar]
- Shen L., Song C.X., He C., Zhang Y. Mechanism and function of oxidative reversal of DNA and RNA methylation. Annu. Rev. Biochem. 2014;83:585–614. doi: 10.1146/annurev-biochem-060713-035513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shide K., Kameda T., Shimoda H., Yamaji T., Abe H., Kamiunten A., Sekine M., Hidaka T., Katayose K., Kubuki Y., et al. TET2 is essential for survival and hematopoietic stem cell homeostasis. Leukemia. 2012;26:2216–2223. doi: 10.1038/leu.2012.94. [DOI] [PubMed] [Google Scholar]
- Shih A.H., Jiang Y.W., Meydan C., Shank K., Pandey S., Barreyro L., Antony-Debre I., Viale A., Socci N., Sun Y.M., et al. Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell. 2015;27:502–515. doi: 10.1016/j.ccell.2015.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S.J., Ito K., Ala U., Kats L., Webster K., Sun S.M., Jongen-Lavrencic M., Manova-Todorova K., Teruya-Feldstein J., Avigan D.E., et al. The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell Stem Cell. 2013a;13:87–101. doi: 10.1016/j.stem.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S.J., Poliseno L., Song M.S., Ala U., Webster K., Ng C., Beringer G., Brikbak N.J., Yuan X., Cantley L.C., et al. MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell. 2013b;154:311–324. doi: 10.1016/j.cell.2013.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruijt C.G., Gnerlich F., Smits A.H., Pfaffeneder T., Jansen P.W., Bauer C., Munzel M., Wagner M., Muller M., Khan F., et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146–1159. doi: 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- Sun M., Song C.X., Huang H., Frankenberger C.A., Sankarasharma D., Gomes S., Chen P., Chen J.J., Chada K.K., He C., et al. HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth and metastasis. Proc. Natl. Acad. Sci. USA. 2013;110:9920–9925. doi: 10.1073/pnas.1305172110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M.M., Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- Tahiliani M., Koh K.P., Shen Y., Pastor W.A., Bandukwala H., Brudno Y., Agarwal S., Iyer L.M., Liu D.R., Aravind L., et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tefferi A., Levine R.L., Lim K.H., Abdel-Wahab O., Lasho T.L., Patel J., Finke C.M., Mullally A., Li C.Y., Pardanani A., et al. Frequent TET2 mutations in systemic mastocytosis: clinical, KITD816V and FIP1L1-PDGFRA correlates. Leukemia. 2009;23:900–904. doi: 10.1038/leu.2009.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thol F., Weissinger E.M., Krauter J., Wagner K., Damm F., Wichmann M., Gohring G., Schumann C., Bug G., Ottmann O., et al. IDH1 mutations in patients with myelodysplastic syndromes are associated with an unfavorable prognosis. Haematologica. 2010;95:1668–1674. doi: 10.3324/haematol.2010.025494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traina F., Visconte V., Jankowska A.M., Makishima H., O'Keefe C.L., Elson P., Han Y.C., Hsieh F.H., Sekeres M.A., Mali R.S., et al. Single nucleotide polymorphism array lesions, TET2, DNMT3A, ASXL1 and CBL mutations are present in systemic mastocytosis. PLos One. 2012;7:e43090. doi: 10.1371/journal.pone.0043090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valinluck V., Sowers L.C. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007;67:946–950. doi: 10.1158/0008-5472.CAN-06-3123. [DOI] [PubMed] [Google Scholar]
- Viguie F., Aboura A., Bouscary D., Ramond S., Delmer A., Tachdjian G., Marie J.P., Casadevall N. Common 4q24 deletion in four cases of hematopoietic malignancy: early stem cell involvement? Leukemia. 2005;19:1411–1415. doi: 10.1038/sj.leu.2403818. [DOI] [PubMed] [Google Scholar]
- Wang Y., Zhang Y. Regulation of TET protein stability by Calpains. Cell Rep. 2014;6:278–284. doi: 10.1016/j.celrep.2013.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.F., Zhou Y., Xu L., Xiao R., Lu X.Y., Chen L., Chong J., Li H.R., He C., Fu X.D., et al. Molecular basis for 5-carboxycytosine recognition by RNA polymerase II elongation complex. Nature. 2015a;523:621–625. doi: 10.1038/nature14482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.P., Xiao M.T., Chen X.F., Chen L.L., Xu Y.P., Lv L., Wang P., Yang H., Ma S.H., Lin H.P., et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol. Cell. 2015b;57:662–673. doi: 10.1016/j.molcel.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward P.S., Patel J., Wise D.R., Abdel-Wahab O., Bennett B.D., Coller H.A., Cross J.R., Fantin V.R., Hedvat C.V., Perl A.E., et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Zhang Y. Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell. 2014;156:45–68. doi: 10.1016/j.cell.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W., Yang H., Liu Y., Yang Y., Wang P., Kim S.H., Ito S., Yang C., Wang P., Xiao M.T., et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011a;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.F., Wu F.Z., Tan L., Kong L.C., Xiong L.J., Deng J., Barbera A.J., Zheng L.J., Zhang H.K., Huang S., et al. Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol. Cell. 2011b;42:451–464. doi: 10.1016/j.molcel.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.F., Xu C., Kato A., Tempel W., Abreu J.G., Bian C.B., Hu Y.G., Hu D., Zhao B., Cerovina T., et al. Tet3 CXXC domain and dioxygenase activity cooperatively regulate key genes for Xenopus eye and neural development. Cell. 2012;151:1200–1213. doi: 10.1016/j.cell.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H., Parsons D.W., Jin G.L., McLendon R., Rasheed B.A., Yuan W.S., Kos I., Batinic-Haberle I., Jones S., Riggins G.J., et al. IDH1 and IDH2 Mutations in Gliomas. N Engl. J. Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin R., Mao S.Q., Zhao B., Chong Z., Yang Y., Zhao C., Zhang D., Huang H., Gao J., Li Z., et al. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J. Am. Chem. Soc. 2013;135:10396–10403. doi: 10.1021/ja4028346. [DOI] [PubMed] [Google Scholar]
- Zhang H.K., Zhang X., Clark E., Mulcahey M., Huang S., Shi Y.G. TET1 is a DNA-binding protein that modulates DNA methylation and gene transcription via hydroxylation of 5-methylcytosine. Cell Res. 2010;20:1390–1393. doi: 10.1038/cr.2010.156. [DOI] [PubMed] [Google Scholar]
- Zhang L., Lu X., Lu J., Liang H., Dai Q., Xu G.L., Luo C., Jiang H., He C. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat. Chem. Biol. 2012;8:328–330. doi: 10.1038/nchembio.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Liu X.G., Gao W.Q., Li P.S., Hou J.L., Li J.W., Wong J.M. Differential regulation of the ten-eleven translocation (TET) family of dioxygenases by O-linked β-N-acetylglucosamine transferase (OGT) J. Biol. Chem. 2014;289:5986–5996. doi: 10.1074/jbc.M113.524140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Zhao K., Shen Q., Han Y., Gu Y., Li X., Zhao D., Liu Y., Wang C., Zhang X., et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015;525:389–393. doi: 10.1038/nature15252. [DOI] [PMC free article] [PubMed] [Google Scholar]