

Abstract
Rheumatoid arthritis is a chronic inflammatory disease that leads to bone and cartilage erosion. The inhibition of osteoblast differentiation by the inflammatory factor TNF-α is critical for the pathogenesis of rheumatoid arthritis. To modulate TNF-α mediated inhibition of osteoblast differentiation is required to improve therapeutic efficacy of rheumatoid arthritis. Here, we explored the potential role of rocaglamide-A, a component of Aglaia plant, in osteoblast differentiation. Rocaglamide-A prevented TNF-α mediated inhibition of osteoblast differentiation, and promoted osteoblast differentiation directly, in both C2C12 and primary mesenchymal stromal cells. Mechanistically, Rocaglamide-A inhibited the phosphorylation of NF-κB component p65 protein and the accumulation of p65 in nucleus, which resulted in the diminished NF-κB responsible transcriptional activity. Oppositely, overexpression of p65 reversed rocaglamide-A’s protective effects on osteoblast differentiation. Collectively, rocaglamide-A protected and stimulated osteoblast differentiation via blocking NF-κB pathway. It suggests that rocaglamide-A may be a good candidate to develop as therapeutic drug for rheumatoid arthritis associated bone loss diseases.
Keywords: NF-κB, osteoblast differentiation, rocaglamide-A, TNF-α
INTRODUCTION
Rheumatoid arthritis (RA) is a chronic disease characterized by the erosion of bone and cartilage in the joints, and untreated RA leads to deformities and disability (Nanjundaiah et al., 2013). Tumor necrosis factor-α (TNF-α) is considered to be an important inflammatory factor that induces bone damage in RA (McInnes and Schett, 2007; Nanjundaiah et al., 2013) by promoting osteoclast differentiation, which results in bone resorption (Azuma et al., 2000; Redlich et al., 2002), and inhibiting osteoblast differentiation and bone formation (Walsh et al., 2009). TNF-α disrupts the balance of bone remodeling, which ultimately leads to bone loss.
In the adult skeleton, osteoblasts are derived from pluripotent mesenchymal stem cells (MSCs), which are able to differentiate into several cell types including osteoblasts, chondrocytes, myoblasts and adipocytes (Zaidi, 2007). Osteoblast differentiation from MSCs is important for adult bone maintenance because the differentiated osteoblast can continuously synthesize bone matrix to generate new bone (Zaidi, 2007). The coordinated stimulation provided by extracellular signaling and the expression of osteoblast transcription factors is necessary for appropriate osteoblast differentiation (Huang et al., 2007). Stimulation by growth factors, such as bone morphogenetic proteins (BMPs) and wingless-ints (Wnts), provides signals for the organization of osteoblast differentiation (Huang et al., 2007). Transcription factors, including runt-related transcription factor 2 (Runx2), osterix (Osx), and activating transcription factor 4 (ATF4), are capable of determining osteoblast differentiation by promoting the synthesis of osteoblast matrix (Karsenty et al., 2009).
The in vitro inhibition of osteoblast differentiation by TNF-α was first discovered by Canalis (1987). Later, in vitro experiments indicated that TNF-α inhibited the differentiation of mature osteoblasts from fetal calvarial precursor cells (Canalis, 1987), and an in vivo study from TNF-α or its p55 receptor gene-knockout mice demonstrated that the overproduction of TNF-α can reduce maximum peak bone mass due to decreased osteoblastic bone formation. This effect was derived from the downstream nuclear factor-κB (NF-κB) signaling pathway (Li et al., 2007). The inhibitory effect of NF-κB signaling on osteoblastic bone formation was demonstrated in 2009, when the specific inactivation of NF-κB signaling in osteoblasts was observed to promote greater bone mass in mice and rescue the bone loss observed in an ovariectomized (OVX) mouse model (Chang et al., 2009). Recently, NF-κB was found to inhibit osteoblast differentiation by attenuating canonical β-catenin signaling (Chang et al., 2013). These studies reveal that TNF-α and NF-κB suppress osteoblast differentiation and ultimately cause adult bone loss.
In recent years, natural bioactive components have attracted considerable attention as a new source of medicinal compounds. The crude extract of Aglaia plants has been used in traditional medicine for the treatment of inflammatory diseases. The active chemical compounds isolated from these plants are rocaglamides, which are derivatives of tetrahydrobenzofuran (Dreyer et al., 2001). In 2002, rocaglamide derivatives were recognized as potent inhibitors of NF-κB activation in T cells (Baumann et al., 2002). Later, rocaglamides were found to have immunosuppressive effects by inhibiting the expression of various inflammatory cytokines by circulating T cells through the suppression of NF-AT activity (Proksch et al., 2005). These insights provided molecular mechanisms to explain how rocaglamides suppress inflammation. Rocaglamides have also been found to possess anticancer activities through in vitro cell-line studies and in vivo mouse model studies (Kim et al., 2006; Lee et al., 1998). The mechanism for this anticancer activity was found to be the induction of cancer cell apoptosis and the inhibition of cancer cell proliferation (Bohnenstengel et al., 1999; Zhu et al., 2009).
As a chronic inflammatory disease, rheumatoid arthritis is associated with TNF-α and other cytokines, as well as inflammatory NF-κB activation. Although the anti-inflammatory rocaglamides exert a potent NF-κB-inhibitory effect on T cells, it remains unclear whether rocaglamides can regulate the inflammatory effect of rheumatoid arthritis. In the current study, we investigated the role of rocaglamide-A (ROC-A) on the regulation of osteoblast differentiation. We found that ROC-A potentiates osteoblast differentiation in two ways: it both prevents the TNF-α-induced inhibition of osteoblast differentiation and promotes osteoblast differentiation directly. Furthermore, we demonstrated that these functions of ROC-A are derived from the suppression of NF-κB signaling in osteoblasts.
MATERIALS AND METHODS
C2C12 cell cultures and the induction of osteoblast differentiation
The C2C12 mesenchymal cell line was obtained from the American Type Culture Collection (ATCC, USA). A monolayer culture was maintained in a growth medium containing Dulbecco’s Modified Eagle’s Medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS), 50 U/ml penicillin, and 50 mg/ml streptomycin (all from Hyclone, USA). The cultures were incubated in a humidified atmosphere at 37°C and 5% CO2. For experimental examination, the C2C12 cells were treated with growth medium supplemented with 200 ng/ml recombinant human BMP-2 (R&D Systems, USA), 10 ng/ml TNF-α (Peprotech) or indicated concentrations of rocaglamide-A (Santa Cruz Biotech, USA).
Primary bone marrow osteoblast cultures
To generate bone marrow-derived osteoblasts from mice, bone marrow cells were flushed from femoral and tibial bones and cultured in α-medium plus 20% fetal bovine serum for 7 days to generate mesenchymal stromal cells. The cells were then cultured in osteoblast inducing medium (ODM) (α-minimal essential medium containing 10% fetal bovine serum with 50 μg/ml ascorbic acid, 10 mM glycerophosphate) and treated with TNF-α (10 ng/ml) and/or indicated concentrations of rocaglamide-A (Santa Cruz Biotech, USA) for 21 days.
RNA harvest and real-time PCR
TRIzol reagent (Invitrogen) was used to isolate the total RNA according to the manufacturer’s instructions. Real-time PCR was performed with an ABI7900HT system using SYBR1 Premix Ex TaqTM (TaKaRa, China) after the reverse transcription reaction according to the manufacturer’s instructions. We used glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the internal control. Each sample was analyzed in triplicate. The primer sequences for C2C12 cells used in this study were as follows: GAPDH: forward 5′-GACTTCAACAGCAACTCCCAC-3′ and reverse 5′-TCCACCACCCTGTTGCTGTA-3′; ColI: forward 5′-GAGCTGGTGTAATGGGTCCT-3′, and reverse 5′-GAG ACCCAGGAAGACCTCTG-3′; Bsp: forward 5′-CAGGGAGG CAGTGACTCTTC-3′, reverse 5′-AGTGTGGAAAGTGTGGCG TT-3′; Ocn: forward 5′-AAGCAGGAGGGCAATAAGGT-3′ and reverse 5′-TTTGTAGGCGGTCTTCAAGC-3′; Runx2: forward 5′-GACTGTGGTTACCGTCATGGC-3′, reverse 5′-ACTTGGTT TTTCATAACAGCGGA-3′; ATF4: forward 5′-CCTGAACAGCGA AGTGTTGG-3′, reverse 5′-TGGAGAACCC- ATGAGGTTTCAA -3′; Osx: forward 5′-GGAAAGGAGGCACAAAGAAGC-3′, reverse 5′-CCCCTTAGGCACTAGGAGC-3′.
Western blotting
Cells were lysed on ice for 30 min in lysis buffer containing 50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1% Nonidet P-40, and 0.1% SDS supplemented with protease inhibitors (10 mg/ml leupeptin, 10 mg/ml pepstatin A, and 10 mg/ml aprotinin). Protein content was measured with the Pierce BCA reagent (PIERCE, USA) following the manufacturer’s protocol. Cytosolic and nuclear fractions were prepared with a Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, China) following the manufacturer’s protocol. For western blotting analysis, 20–40 mg of sample was resolved on a 12% SDS-PAGE gel and electro-transferred onto nitrocellulose membranes (Whatman, USA). Anti-GAPDH, anti-p65, anti-phosphorylated p65 and anti-Lamin B antibodies (all from Santa Cruz Biotech, USA) were used at 1:1000 dilutions. The antigen-antibody complexes were visualized using the enhanced chemiluminescence detection system (Millipore, USA), as recommended by the manufacturer.
Gene cloning of pcDNA3.1(−)-p65
For the overexpression of the gene p65 in C2C12 cells, the coding sequences were inserted into the expression vector pcDNA3.1(−) (Invitrogen). The DNA fragments were produced by a PCR method using the cDNA of C2C12 cells as DNA templates. The following primers were used for the PCR amplification: Forward: 5′-TAGCTCTAGAGCGGGGCCGGGAACGGG ACC-3′ (XbaI digestion); Reverse: 5′-GATCAAGCTTAAGACA TTTATTAGTTCAGA-3′ (HindIII digestion). This plasmid was transfected into cells using Lipo2000 (Invitrogen).
Alkaline phosphatase (ALP) activity
C2C12 cells were rinsed 3 times with ice-cold PBS, scraped from the dishes and suspended in ddH2O. Three cycles of freezing and thawing were then performed. ALP activity was determined at 405 nm using p-nitrophenyl phosphate (pNPP) (Sigma-Aldrich) as the substrate. A 50-ml aliquot of the sample was mixed with 50 ml of pNPP (1 mg/ml) in 1 M diethanolamine buffer containing 0.5 mM MgCl2 (pH 9.8) and incubated at 37°C for 15 min on a bench shaker. The reaction was stopped by the addition of 200 ml of 2 M NaOH per 200 ml of reaction mixture. Total protein content was determined by the BCA method with a protein assay kit (PIERCE, USA). ALP activity was presented as the fold changes over the non-loading group at the respective time points. All experiments were conducted in triplicate.
Alizarin Red S staining and calcium mineral content quantification
Alizarin Red S staining and calcium mineral content quantification were performed as described by Stanford et al. (1995). The cells were rinsed with calcium and phosphate-free saline solution, and fixed with ice-cold 70% ethanol for 1 h. After a brief wash with water, the cells were stained for 10 min with 40 mM Alizarin Red S solution (pH 4.2) at room temperature. The cells were rinsed four times with water followed by a 15-min wash with PBS (with rotation) to reduce nonspecific Alizarin Red S stain. Stained cultures were scanned by a quantitative distaining procedure using 10% (w/v) cetylpyridinium chloride in 10 mM sodium phosphate (pH 7.0), for 15 min at room temperature, and the Alizarin Red S concentrations were determined by absorbance measurements at 570 nm on Victor3TMV (PerkinElmer) using an Alizarin Red S standard curve in the same solution. The values were normalized to the control group.
Luciferase activity assays
pGL4.32[luc2P/NF-κB-RE/Hygro] plasmids were transfected into C2C12 cells for the determination of NF-κB activity. As an internal control, pRL Renilla luciferase (Rluc) control reporter vector plasmids were co-transfected into C2C12 cells with pGL4.32[luc2P/NF-κB-RE/Hygro] vector plasmids. The two plasmids were both purchased from Promega (USA) and were transfected into cells using Lipo2000 (Invitrogen). C2C12 cells were seeded at 5 × 104 per well in a 24-well plate in complete medium and were stimulated with 200 ng/ml BMP-2, 10 ng/ml TNF-α or rocaglamide-A at 24 h after plasmid transfection. Cells were harvested 48 h later and luciferase activities were measured using a dual luciferase system (Promega, USA).
Electrophoretic mobility shift assay
C2C12 cells were treated with 200 ng/ml BMP-2, 10 ng/ml TNF-α, or the indicated concentrations of ROC-A or were left untreated. Nuclear extracts were prepared from 1 × 107 cells, and electrophoretic mobility shift assay (EMSA) was carried out as previously described (Li et al., 1991; Steer et al., 2000). Nuclear proteins (4 μg) were pre-incubated for 10 minutes at room temperature with 0.5 μg of poly(dI-dC) (Amersham Pharmacia Biotech) in a binding buffer (4% Ficoll, 20 mM HEPES [pH 7.9], 1 mM EDTA, 1 mM dithiothreitol, 50 mM KCl, 0.05% IGEPAL CA-630) to give a final reaction volume of 10 μl. A double-stranded NF-κB consensus oligonucleotide probe 5′-GGGCATGGGAATTTCCAACTC-3′ (0.25 pmol) with overhanging 5′-G, which had been fill-in labeled with [α-32P]dCTP (Amersham Pharmacia Biotech) using the Klenow fragment of E. coli DNA polymerase I (Promega), was then added. After 10 min of incubation, samples were loaded onto a 4% polyacrylamide gel containing 0.25 × Tris-Borate-EDTA buffer, which had been pre-run for 2 h in the same buffer. Gels were then exposed to Kodak X-ray film using a single intensifying screen.
MTT and caspase-3 activity assays
Cell viability was determined by MTT [3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide] Assay. MTT was purchased from Sigma-Aldrich (USA). Culturing medium was removed and cells were washed 3 times with PBS. Cells were treated with 5 mg/ml MTT salts and incubated at 37°C for 2 h. The absorbance at 570 nm was measured. Apoptosis was characterized by a caspase-3 activity assay using a caspase fluorescent assay kit (BD Biosciences-Clontech, USA) according to the manufacturer’s instructions.
Statistical analysis
Each assay was performed no fewer than 3 times. Each testing group contained more than 3 samples (n > 3). The data were expressed as the mean ± SD. Student’s t-test was used to test the statistical significance between two groups. For more than two groups, an ANOVA was used to determine which groups were significantly different. A p value < 0.05 was considered to be statistically significant.
RESULTS
Rocaglamide-A blocks the inhibition of osteoblast differentiation from TNF-α
Because the high dosage of ROC-A may induce cell apoptosis, we treated C2C12 cells with different doses of ROC-A to define the appropriate concentrations. According to the previous reports, the mammalian cell survival or proliferation could be determined by the MTT method (Mosmann, 1983); thus, we used the MTT assay to test the cell viability after seventy-two hours of ROC-A treatment. Compared to the control (C2C12 cells treated with 200 ng/ml BMP-2 and 10 ng/ml TNF-α), the doses ranging from 20 to 100 nM of ROC-A did not affect cell viability significantly. However, 140 or 180 nM of ROC-A led to reduced viable cell numbers (*p < 0.05, n > 3), as shown in Fig. 1B. Caspase-3 activity assay indicated a significant promotion of apoptosis in the C2C12 cells treated with 140 or 180 nM of ROC-A. However, there were no notable changes in the groups treated with 20 to 100 nM ROC-A, as shown in Fig. 1C. We then examined whether ROC-A affected the inhibition of osteoblast differentiation by TNF-α. Consistent with previous reports, ALP activity was suppressed by 10 ng/ml TNF-α. However, ALP activity could be rescued by 30 or 50 nM ROC-A treatment. (*p < 0.05, **p < 0.01, n > 3), as shown in Fig. 1D. Next, we found that the expression of other osteoblast-specific marker genes, including type I collagen (ColI), osteocalcin (Ocn) and bone sialoprotein (Bsp), as shown in Fig. 1E, and the osteoblast transcription factors, including Runx2, Osx and ATF4, as shown in Fig. 1F, could also be rescued by 50 nM ROC-A treatment, as revealed by the real-time PCR method (*p < 0.05, **p < 0.01, n > 3).
Fig. 1.

Rocaglamide-A prevented the TNF-α induced inhibition of osteoblast differentiation in C2C12 cells. (A) The chemical structure of rocaglamide-A (ROC-A). (B, C) The MTT [3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide] assay was used to test the cell viability. The results revealed that concentrations of 20 to 100 nM ROC-A caused no significant decrease in cell viability; however, concentrations of 140 or 180 nM ROC-A led to a notable decrease of cell viability (n > 3, *p < 0.05). Concentrations of 20 to 100 nM ROC-A caused no significant elevation of caspase-3 activity; however, 140 or 180 nM ROC-A treatment led to a notable increase of caspase-3 activity (n > 3, *p < 0.05, **p < 0.01) (D) Concentrations of 30 or 50 nM ROC-A rescued the suppression of alkaline phosphatase (ALP) activity from 10 ng/ml tumor necrosis factor-α (TNF-α): the cells treated with 50 nM ROC-A exhibited a 7.3 ± 1.9-fold promotion of ALP activity compared to the control group (n > 3, **p < 0.01). (E) At a concentration of 50 nM, ROC-A significantly protected the gene expression of type I collagen (ColI), osteocalcin (Ocn) and bone sialoprotein (Bsp) from the suppression of 10 ng/ml TNF-α (n > 3, *p < 0.05, **p < 0.01). (F) At a concentration of 50 nM, ROC-A significantly protected the gene expression of runt-related transcription factor 2 (Runx2), osterix (Osx), and activating transcription factor 4 (ATF4) from the suppression of 10 ng/mL TNF-α (n > 3, *p < 0.05, **p < 0.01).
Rocaglamide-A promotes BMP-2-induced osteoblast differentiation from C2C12 cells
As NF-κB signaling was proven to have endogenous inhibitory effects on osteoblast differentiation, we proposed that the potent NF-κB inhibitor ROC-A may directly promote osteoblast differentiation and bone formation. Our results indicated that 50 nM ROC-A can promote ALP activity, as shown in Fig. 2A. In Figs. 2B and 2C, the expressions of the extracellular matrix genes Col I, Ocn and Bsp and the transcription factor genes Runx2, Osx and ATF4 (*p < 0.05, **p < 0.01, n > 3) were all promoted.
Fig. 2.
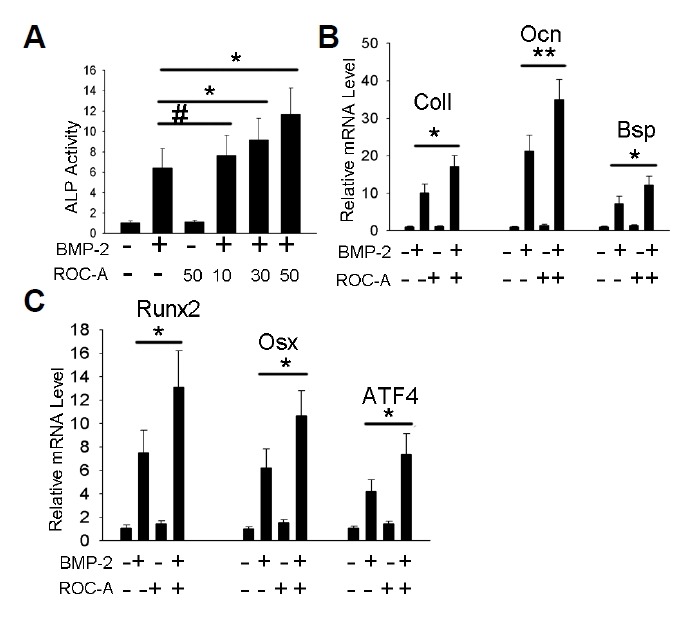
Rocaglamide-A directly promoted osteoblast differentiation in C2C12 cells. (A) Concentrations of 30 or 50 nM ROC-A significantly promoted ALP activity compared to the bone morphogenetic protein-2 (BMP-2) group (n > 3, *p < 0.05). (B) At a concentration of 50 nM, ROC-A notably promoted the gene expression of ColI, Ocn and Bsp compared to the BMP-2 group (n > 3, *p < 0.05, **p < 0.01). (C) At a concentration of 50 nM, ROC-A promoted the gene expression of Runx2, Osx and ATF4 compared to the BMP-2 group (n > 3, *p < 0.05).
Rocaglamide-A potentiates osteoblast differentiation from primary mesenchymal stromal cells
The results above indicated that ROC-A potentiates BMP-2-induced C2C12 osteoblast differentiation; however, whether ROC-A exerts the same effects for the primary cell is yet to be elucidated. To examine this possibility, we generated an osteoblast differentiation model with the primary mesenchymal stromal cells. Bone marrow mesenchymal stromal cells from 4-8-month-old mice were cultured and osteoblast differentiation was induced with osteoblast induction medium (ODM). In the group treated with the combination of ODM and 10 ng/ml TNF-α, the activity of ALP and the expression of Col I, Ocn and Bsp were notably inhibited, as shown in Figs. 3A and 3B; the expression of transcription factor genes, including Runx2, Osx and ATF4, was also found to be inhibited significantly, as shown in Fig. 3C. However, in the group treated with ROC-A, the inhibitory effects of TNF-α were blocked. The ALP activity and the expression of Col I, Ocn and Bsp were significantly rescued (*p < 0.05, **p < 0.01, ***p < 0.001, n > 3), as shown in Figs. 3A and 3B; in Fig. 3C, the expression of transcription factor genes, including Runx2, Osx and ATF4, were notably rescued by ROC-A (**p < 0.01, n > 3). Additionally, in the presence of ROC-A, the ODM induced a significantly higher level of ALP activity (*p < 0.05, n > 3), as shown in Fig. 3A. In Figs. 3B and 3C, the gene expression of Col I, Ocn and Bsp (*p < 0.05, n > 3), as well as the transcription factor genes Runx2, Osx and ATF4 (*p < 0.05, n > 3), were found to be stimulated. Surprisingly, we also observed that ROC-A promoted all the osteoblast markers directly, without the presence of the ODM. Compared to the control group (cells were maintained with only the growth medium), the presence of ROC-A promoted significantly higher ALP activity (*p < 0.05, n > 3), as shown in Fig. 3A. In Figs. 3B and 3C, the gene expression of Col I, Ocn and Bsp (*p < 0.05, n > 3), as well as the transcription factor genes Runx2, Osx and ATF4 (*p < 0.05, n > 3), were all directly promoted. These results indicated that ROC-A can block TNF-α-induced osteoblast inhibition and directly stimulate osteoblast differentiation in the primary mesenchymal stromal cells.
Fig. 3.
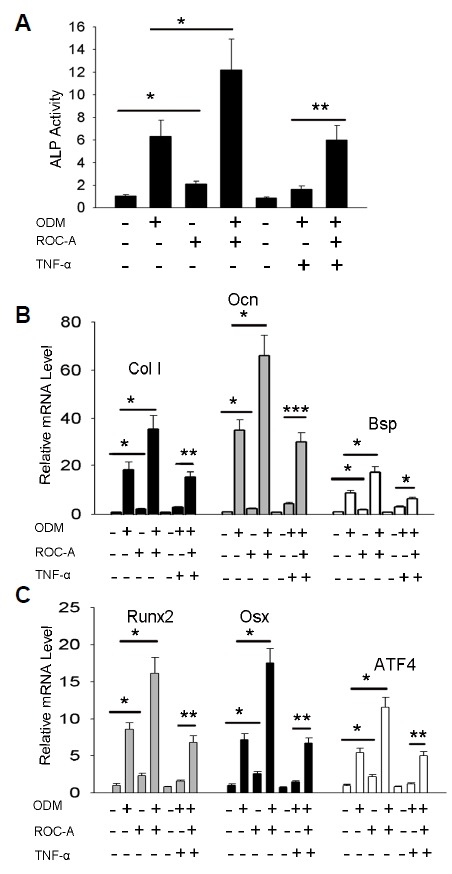
Rocaglamide-A potentiates osteoblast differentiation from primary mesenchymal stromal cells. (A) The 30 nM ROC-A blocked the suppression of ALP activity from 10 ng/ml TNF-α, and 30 nM ROC-A stimulated the osteoblast inducing medium (ODM)-induced mesenchymal stromal cell ALP activity; furthermore, 30 nM ROC-A treatment alone stimulated ALP activity (n > 3, *p < 0.05, **p < 0.01). (B) At a concentration of 30 nM, ROC-A blocked the suppression of the gene expression of ColI, Ocn and Bsp from 10 ng/ml TNF-α, and 30 nM ROC-A stimulated the ODM-induced mesenchymal stromal cell gene expression of ColI, Ocn and Bsp; furthermore, 30 nM ROC-A treatment alone stimulated the gene expression of ColI, Ocn and Bsp (n > 3, *p < 0.05, **p < 0.01, ***p < 0.001). (C) The 30 nM ROC-A blocked the suppression of gene expression of Runx2, Osx and ATF4 from 10 ng/ml TNF-α, and 30 nM ROC-A stimulated the ODM-induced mesenchymal stromal cell gene expression of Runx2, Osx and ATF4; furthermore, the 30 nM ROC-A treatment alone stimulated the gene expression of Runx2, Osx and ATF4 (n > 3, *p < 0.05, **p < 0.01).
Rocaglamide-A stimulates osteoblast mineralization
To confirm that whether ROC-A affects the osteoblast mineralization, we performed Alizarin Red S staining experiments. Primary mesenchymal stromal cells were treated for 21 days and stained with Alizarin Red S to visually detect the presence of mineralization. Again, the results showed a significantly lower degree of mineralization in the TNF-α inhibition group compared to the ODM group; however, the inhibition from TNF-α was blocked by ROC-A (**p < 0.01, n > 3), as shown in Figs. 4A and 4B. Furthermore, as shown in Figs. 4C and 4D, ROC-A stimulated the mineralization in the primary mesenchymal stromal cells compared to the ODM group (*p < 0.05, n > 3).
Fig. 4.
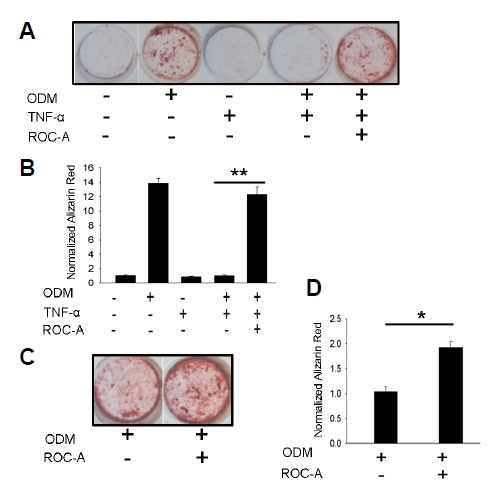
Rocaglamide-A affects the formation of mineralized osteoblast nodules. (A) The 30 nM ROC-A blocked the suppression of nodule formation from 10 ng/ml TNF-α; (B) The quantification of Fig. 4A (n > 3, **p < 0.01). (C) 30 nM ROC-A stimulated the ODM induced mesenchymal stromal cell nodule formation; (D) The quantification of Fig. 4C (n > 3, *p < 0.05).
Rocaglamide-A inhibits the activation of NF-κB signaling
Upon finding that ROC-A can protect against the TNF-α-induced inhibition of osteoblast differentiation and can directly promote osteoblast differentiation, we explored the molecular mechanism for how ROC-A regulates osteoblasts. Because ROC-A was identified as a potent inhibitor of NF-κB activation, we hypothesized that ROC-A exerts positive effects on osteoblasts through the suppression of NF-κB signaling. First, we examined the NF-κB protein p65 via western blotting to detect whether ROC-A could prevent its translocation to the nucleus in C2C12 cells. We found that stimulation with 10 ng/ml of TNF-α resulted in p65 accumulation in the nucleus. However, the p65 accumulation in the nucleus was found to be inhibited in the group treated with 10, 30, or 50 nM ROC-A, as shown in Fig. 5A. We then used the luciferase method to assess whether ROC-A could attenuate TNF-α-induced NF-κB activity. The pGL4.32[luc2P/NF-κB-RE/Hygro] plasmid system, which contains a consensus NF-κB response element, was used in C2C12 cells to determine the NF-κB signal-induced transcriptional activity. As shown in Fig. 5. (B), the results demonstrated that 10 ng/ml TNF-α treatment caused a notable elevation in NF-κB luciferase activity in C2C12 cells and that this activation of luciferase activity was reduced significantly by ROC-A treatment in a dose-dependent manner (*p < 0.05, **p < 0.01, n > 3), suggesting that the activation of NF-κB was inhibited. Because the activation of NF-κB signaling required the phosphorylation of the NF-κB protein p65, the level of phospho-p65 (p-p65) was examined as well. As shown in Fig. 5C, the level of p-p65 was found to be stimulated by 10 ng/ml of TNF-α in either the cytoplasm or the nucleus; however, this stimulation was blocked by 10, 30, or 50nM ROC-A treatment. Furthermore, an electrophoretic mobility shift assay was performed to determine the NF-κB activation. With TNF-α treatment (10 ng/ml), the binding complex of NF-κB protein and the double-stranded NF-κB consensus oligonucleotide probe was promoted; however, this NF-κB protein-oligonucleotide probe complex was reduced by treatment with ROC-A in a dose-dependent manner, as shown in Fig. 5D.
Fig. 5.
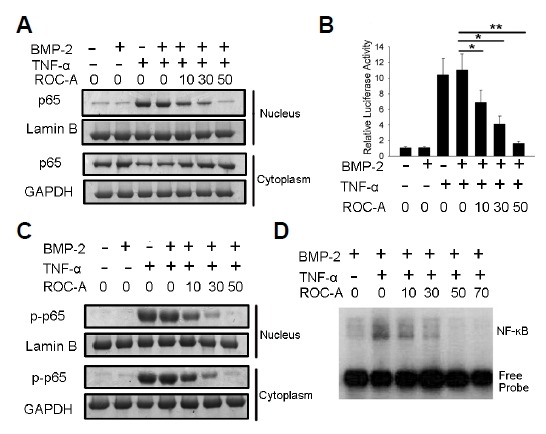
(A) The nuclear p65 protein was stimulated by the treatment of 10 ng/ml TNF-α, however, was blocked by 10, 30 or 50 nM ROC-A treatment. (B) NF-κB luciferase activity was inhibited significantly by treatment with 10, 30 or 50 nM ROC-A (n > 3, *p < 0.05, **p < 0.01). (C) The level of p-p65 was found to be stimulated by 10 ng/ml of TNF-α in either the cytoplasm or the nucleus, however, this stimulation was blocked by 10, 30 or 50 nM ROC-A treatment. The overexpression of p65 was confirmed by the western blotting experiment. (D) In the electrophoretic mobility shift assay (EMSA) experiment, with the treatment of TNF-α (10 ng/ml), the binding complex of the NF-κB protein and the double-stranded NF-κB consensus oligonucleotide probe was promoted; however, this NF-κB protein-oligonucleotide probe complex was reduced by treatment with ROC-A in a dose-dependent manner.
Rocaglamide-A potentiates osteoblast differentiation by inhibiting NF-κB signaling activation
In a further study, we tried to confirm whether the inhibition of NF-κB activation contributed to the ROC-A derived protection and promotion of osteoblast differentiation. According to the previous report, the NF-κB activity could be promoted by the overexpression of the NF-κB component p65 (Collett and Campbell, 2006). Therefore, we hypothesized that the overexpression of p65 may rescue the ROC-A-induced NF-κB activity inhibition. If the rescue of NF-κB activity by p65 overexpression can reverse the potentiation from ROC-A in osteoblast differentiation, it can be deduced that ROC-A potentiates osteoblast differentiation by inhibiting NF-κB activity. The NF-κB component p65 protein was overexpressed by the transfection of pcDNA3.1(−)-p65 in C2C12 cells. p65 protein expression was confirmed by western blotting, as shown in Fig. 6A. The cells treated with 10 ng/ml of TNF-α caused the accumulation of p65 protein in the nucleus (lane 3), and the cells with p65 overexpression presented p65 accumulation in the nucleus as well (lane 4). Therefore, NF-κB activity can be stimulated by p65 overexpression. In the group of p65-overexpressing cells treated with TNF-α, the accumulation of p65 in the nucleus was notably promoted (lane 5), compared to the single TNF-α stimulation group (lane 3). In the group treated with 50 nM ROC-A and TNF-α, the accumulation of p65 in the nucleus was inhibited (lane 6); however, in the p65-overexpressing cells treated with the same amount of ROC-A, the nuclear accumulation of p65 remained, even though it was reduced (lane 7). This result indicated that p65 overexpression can stimulate the accumulation of p65 in the nucleus and partially rescue the ROC-A-induced inhibition of the nucleus translocation of p65 protein. To obtain more evidence, we analyzed the activity of NF-κB with the luciferase method to confirm this hypothesis. As shown in Fig. 6B, the luciferase assay results demonstrated that p65 overexpression stimulated NF-κB transcriptional activity and partially rescued the NF-κB activity from ROC-A-induced inhibition. To confirm whether the rescue of NF-κB activity can impact osteoblast differentiation, we performed an ALP activity assay and examined the expression of osteoblast marker genes. The ALP activity results indicated that, consistent with the previous results, 50 nM ROC-A significantly protected the ALP activity. However, in the group that overexpressed p65, ROC-A had a relatively smaller effect on the rescue of ALP activity, as shown in Fig. 6C (*p < 0.05, **p < 0.01, n > 3). Additionally, an examination of the expression of the extracellular matrix genes Col I, Ocn and Bsp (*p < 0.05, **p < 0.01, n > 3) revealed similar effects, as shown in Figs. 6D–6F. These results demonstrated that the inhibition of NF-κB activation by ROC-A is the mechanism behind its potentiation of osteoblast differentiation.
Fig. 6.
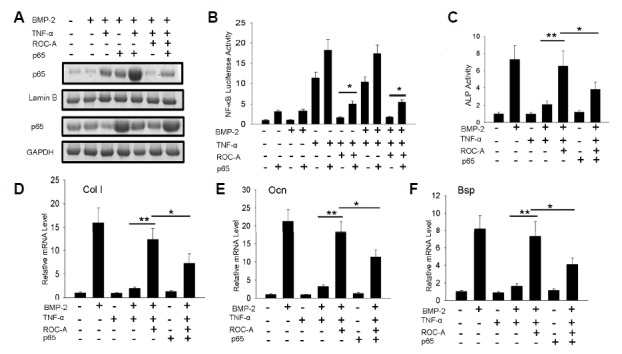
(A) The Western blotting experiment indicated that p65 overexpression stimulated the p65 nuclear accumulation, and partially rescue the translocation of p65 from the 50 nM ROC-A inhibition; (B) The luciferase assay demonstrated that p65 overexpression stimulated NF-κB transcriptional activity, and partially rescued the NF-κB activity from 50 nM ROC-A inhibition. (C) At a concentration of 50 nM, ROC-A prevented the suppression of ALP activity from 10 ng/ml TNF-α; however, this effect was reduced by the overexpression of p65 (n > 3, *p < 0.05, **p < 0.01). (D–F) At a concentration of 50 nM, ROC-A prevented the suppression of ColI, Ocn and Bsp gene expression by 10 ng/ml TNF-α, however, this effect was reduced by the overexpression of p65 (n > 3, *p < 0.05, **p < 0.01).
DISCUSSION
The inflammatory factor TNF-α has been shown to play a critical role in the pathogenesis of bone loss diseases. In the case of rheumatoid arthritis, TNF-α causes bone loss in the axial and appendicular skeleton (Goldring and Gravallese, 2000). RA is a chronic inflammatory disease characterized by progressive joint destruction. With joint deterioration, patients suffer from pain and loss of function, and they often have decreased quality of life and high mortality (Nanjundaiah et al., 2013). TNF-α has been found in the synovial fluid and the synovium of RA patients. TNF-α was found to play a central role in RA-associated bone degradation by disrupting normal bone remodeling (Nanjundaiah et al., 2013). TNF-α has biphasic effects that disrupt the balance of bone remodeling, including the promotion of osteoclastic bone resorption (Azuma et al., 2000) and the impairment of osteoblastic bone formation (Walsh et al., 2009). The inhibition of osteoblast differentiation by TNF-α contributes to the impairment of bone formation, which ultimately results in the failure of new bone generation. It has been shown that TNF-α inhibits osteoblast differentiation and bone formation by activating the NF-κB signaling pathway (Chang et al., 2009; Li et al., 2007).
Rocaglamide-A is one of the active chemical forms of rocaglamide, which is extracted from the Aglaia plant. Aglaia has been used in traditional medicine to treat inflammatory diseases and was recently found to have anticancer activity. The molecular mechanism for the anti-inflammatory activity of rocaglamides was identified in the last 20 years: it can target T cells and reduce the production of various cytokines, as well as inhibit NF-κB activation in T cells. However, it remains unclear whether rocaglamides have an anti-inflammatory effect in RA. In the current study, we examined the role of ROC-A in regulating osteoblast differentiation by blocking its inhibition by TNF-α and NF-κB. In the previous reports, BMP-2-induced C2C12 osteoblast differentiation was used extensively to model the in vivo osteoblast differentiation from mesenchymal stem cells (Mukai et al., 2007). Additionally, TNF-α-induced inhibition of C2C12 was used to model the in vivo inhibition of osteoblast differentiation in inflammatory diseases, including RA (Lee et al., 2010). In the current research, C2C12 is commonly used to model osteoblast differentiation in RA. Based on the C2C12 osteoblast differentiation inhibition system, our results proved that ROC-A can protect ALP activity and osteoblast extracellular marker gene expression against impairment by TNF-α. Furthermore, evidence from the gene expression of the three transcription factors Runx2, Osx and ATF4 demonstrated that the expression of all of these osteoblast marker genes was protected from TNF-α-induced inhibition, suggesting that ROC-A protected osteoblast differentiation by blocking the inhibition from TNF-α. Due to the importance of TNF-α-induced osteoblast inhibition in RA, the blockade of this process by ROC-A may benefit RA patients by maintaining normal osteoblast differentiation and new bone formation. Our experiments also demonstrated that ROC-A can promote BMP-2-induced osteoblast differentiation, suggesting that ROC-A likely inhibited endogenous C2C12 NF-κB activity to facilitate osteoblast differentiation. ROC-A may therefore stimulate osteoblast differentiation towards the generation of new bone. We additionally examined whether ROC-A could block NF-κB activity in our C2C12 cell system. We observed that ROC-A prevented the accumulation of the NF-κB protein p65 in the nucleus. Additionally, the TNF-α-induced promotion of phospho-p65 was inhibited by ROC-A. Through the luciferase activity assay and EMSA experiment, we confirmed that ROC-A inhibited the activation of NF-κB transcriptional activity, suggesting that ROC-A inhibited the activation of the NF-κB signal in C2C12 cells. Furthermore, a p65 overexpression experiment demonstrated that the inhibition of NF-κB by ROC-A contributed to the rescue of osteoblast differentiation.
Bone morphogenetic proteins (BMPs) are proved by the Food and Drug Association (FDA) in wide clinical use due to its capability of robust bone formation promotion; however, BMPs are found to induce various clinical side effects, including the increased incidence of clinically relevant postoperative prevertebral swelling problems (Smucker et al., 2006); ectopic bone formation in the spinal canal with potential neurologic compromise (Wong et al., 2008);.direct stimulation of bone resorption (Kaneko et al., 2000); and the induction of structurally abnormal bone and inflammation (Zara et al., 2011), etc. Thus, the application of BMPs to induce bone formation in RA patients should be cautious. In the current study, our results indicated that ROC-A may exert positive effect for bone formation in RA patients without the combination of BMPs treatment. Firstly, although rhBMP-2 was used in our C2C12 cell system, in the mouse primary mesenchymal stromal cell system, no BMPs were used. Our results demonstrated that ROC-A is capable of potentiating osteoblast differentiation from primary stromal cells (Figs. 3 and 4). Furthermore, in Fig. 3, our results indicated that ROC-A stimulated the ALP activity and gene expression of osteoblast markers, even without the treatment with osteoblast induction medium, suggesting that the single ROC-A treatment can promote primary stromal cell differentiation. Secondly, in the in vivo bone remodeling, the osteoblast differentiation of mesenchymal stromal cells are stimulated by a diversity of endogenous growth factors, including BMPs, TGF-β, and Wnts (Huang et al., 2007). The concentrations of such endogenous growth factors may be regulated appropriately and spontaneously in human body. In RA, the osteoblast inhibition from TNF-α may depend on the inhibition of BMPs/TGF-β-Smads (Yamazaki et al., 2009) and Wnt-β-cantenin (Chang et al., 2013) signaling by the activation of the NF-κB pathway. The inactivation of NF-κB by ROC-A may exert a protective effect via the rescue of in vivo endogenous Smads and the β-cantenin signal pathways. With the existence of the endogenous growth factors, the additional artificial treatment with BMPs should be non-essential.
In summary, ROC-A potentiates osteoblast differentiation by blocking TNF-α-mediated inhibition and directly promotes osteoblast differentiation. These effects are caused by the inhibition of NF-κB activity. For the first time, our study has revealed the role of ROC-A as a protective and stimulating compound for osteoblast differentiation. We prospect that ROC-A may have a positive effect on in vivo osteoblast differentiation and bone anabolism, after which ROC-A may be a lead compound in the development of therapeutic agents to promote anabolic bone metabolism and cure RA-derived bone loss in the future.
REFERENCES
- Azuma Y., Kaji K., Katogi R., Takeshita S., Kudo A. Tumor necrosis factor-alpha induces differentiation of and bone resorption by osteoclasts. J. Biol. Chem. 2000;275:4858–4864. doi: 10.1074/jbc.275.7.4858. [DOI] [PubMed] [Google Scholar]
- Baumann B., Bohnenstengel F., Siegmund D., Wajant H., Weber C., Herr I., Debatin K.M., Proksch P., Wirth T. Rocaglamide derivatives are potent inhibitors of NF-kappa B activation in T-cells. J. Biol. Chem. 2002;277:44791–44800. doi: 10.1074/jbc.M208003200. [DOI] [PubMed] [Google Scholar]
- Bohnenstengel F.I., Steube K.G., Meyer C., Nugroho B.W., Hung P.D., Kiet L.C., Proksch P. Structure activity relationships of antiproliferative rocaglamide derivatives from Aglaia species (Meliaceae) Z. Naturforsch. C. 1999;54:55–60. [PubMed] [Google Scholar]
- Canalis E. Effects of tumor necrosis factor on bone formation in vitro. Endocrinology. 1987;121:1596–1604. doi: 10.1210/endo-121-5-1596. [DOI] [PubMed] [Google Scholar]
- Chang J., Wang Z., Tang E., Fan Z., McCauley L., Franceschi R., Guan K., Krebsbach P.H., Wang C.Y. Inhibition of osteoblastic bone formation by nuclear factor-kappaB. Nat. Med. 2009;15:682–689. doi: 10.1038/nm.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J., Liu F., Lee M., Wu B., Ting K., Zara J.N., Soo C., Al Hezaimi K., Zou W., Chen X., et al. NF-kappaB inhibits osteogenic differentiation of mesenchymal stem cells by promoting beta-catenin degradation. Proc. Natl. Acad. Sci. USA. 2013;110:9469–9474. doi: 10.1073/pnas.1300532110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collett G.P., Campbell F.C. Overexpression of p65/RelA potentiates curcumin-induced apoptosis in HCT116 human colon cancer cells. Carcinogenesis. 2006;27:1285–1291. doi: 10.1093/carcin/bgi368. [DOI] [PubMed] [Google Scholar]
- Dreyer M., Nugroho B.W., Bohnenstengel F.I., Ebel R., Wray V., Witte L., Bringmann G., Muhlbacher J., Herold M., Hung P.D., et al. New insecticidal rocaglamide derivatives and related compounds from Aglaia oligophylla. J. Nat. Prod. 2001;64:415–420. doi: 10.1021/np000123x. [DOI] [PubMed] [Google Scholar]
- Goldring S.R., Gravallese E.M. Pathogenesis of bone erosions in rheumatoid arthritis. Curr. Opin. Rheumatol. 2000;12:195–199. doi: 10.1097/00002281-200005000-00006. [DOI] [PubMed] [Google Scholar]
- Huang W., Yang S., Shao J., Li Y.P. Signaling and transcriptional regulation in osteoblast commitment and differentiation. Front. Biosci. 2007;12:3068–3092. doi: 10.2741/2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko H., Arakawa T., Mano H., Kaneda T., Ogasawara A., Nakagawa M., Toyama Y., Yabe Y., Kumegawa M., Hakeda Y. Direct stimulation of osteoclastic bone resorption by bone morphogenetic protein (BMP)-2 and expression of BMP receptors in mature osteoclasts. Bone. 2000;27:479–486. doi: 10.1016/s8756-3282(00)00358-6. [DOI] [PubMed] [Google Scholar]
- Karsenty G., Kronenberg H.M., Settembre C. Genetic control of bone formation. Annu. Rev. Cell Dev. Biol. 2009;25:629–648. doi: 10.1146/annurev.cellbio.042308.113308. [DOI] [PubMed] [Google Scholar]
- Kim S., Salim A.A., Swanson S.M., Kinghorn A.D. Potential of cyclopenta[b]benzofurans from Aglaia species in cancer chemotherapy. Anticancer Agents Med. Chem. 2006;6:319–345. doi: 10.2174/187152006777698123. [DOI] [PubMed] [Google Scholar]
- Lee S.K., Cui B., Mehta R.R., Kinghorn A.D., Pezzuto J.M. Cytostatic mechanism and antitumor potential of novel 1H-cyclopenta[b]benzofuran lignans isolated from Aglaia elliptica. Chem. Biol. Interact. 1998;115:215–228. doi: 10.1016/s0009-2797(98)00073-8. [DOI] [PubMed] [Google Scholar]
- Lee H.L., Yi T., Woo K.M., Ryoo H.M., Kim G.S., Baek J.H. Msx2 mediates the inhibitory action of TNF-alpha on osteoblast differentiation. Exp. Mol. Med. 2010;42:437–445. doi: 10.3858/emm.2010.42.6.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.C., Ross J., Scheppler J.A., Franza B.R., Jr. An in vitro transcription analysis of early responses of the human immunodeficiency virus type 1 long terminal repeat to different transcriptional activators. Mol. Cell Biol. 1991;11:1883–1893. doi: 10.1128/mcb.11.4.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Li A., Strait K., Zhang H., Nanes M.S., Weitzmann M.N. Endogenous TNFalpha lowers maximum peak bone mass and inhibits osteoblastic Smad activation through NF-kappaB. J. Bone Miner Res. 2007;22:646–655. doi: 10.1359/jbmr.070121. [DOI] [PubMed] [Google Scholar]
- McInnes I.B., Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007;7:429–442. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Mukai T., Otsuka F., Otani H., Yamashita M., Takasugi K., Inagaki K., Yamamura M., Makino H. TNF-alpha inhibits BMP-induced osteoblast differentiation through activating SAPK/JNK signaling. Biochem. Biophys. Res. Commun. 2007;356:1004–1010. doi: 10.1016/j.bbrc.2007.03.099. [DOI] [PubMed] [Google Scholar]
- Nanjundaiah S.M., Astry B., Moudgil K.D. Mediators of inflammation-induced bone damage in arthritis and their control by herbal products. Evid. Based Complement Alternat. Med. 2013;2013:518094. doi: 10.1155/2013/518094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proksch P., Giaisi M., Treiber M.K., Palfi K., Merling A., Spring H., Krammer P.H., Li-Weber M. Rocaglamide derivatives are immunosuppressive phytochemicals that target NF-AT activity in T cells. J. Immunol. 2005;174:7075–7084. doi: 10.4049/jimmunol.174.11.7075. [DOI] [PubMed] [Google Scholar]
- Redlich K., Hayer S., Ricci R., David J.P., Tohidast-Akrad M., Kollias G., Steiner G., Smolen J.S., Wagner E.F., Schett G. Osteoclasts are essential for TNF-alpha-mediated joint destruction. J. Clin. Invest. 2002;110:1419–1427. doi: 10.1172/JCI15582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smucker J.D., Rhee J.M., Singh K., Yoon S.T., Heller J.G. Increased swelling complications associated with off-label usage of rhBMP-2 in the anterior cervical spine. Spine. 2006;31:2813–2819. doi: 10.1097/01.brs.0000245863.52371.c2. [DOI] [PubMed] [Google Scholar]
- Stanford C.M., Jacobson P.A., Eanes E.D., Lembke L.A., Midura R.J. Rapidly forming apatitic mineral in an osteoblastic cell line (UMR 106-01 BSP) J. Biol. Chem. 1995;270:9420–9428. doi: 10.1074/jbc.270.16.9420. [DOI] [PubMed] [Google Scholar]
- Steer J.H., Kroeger K.M., Abraham L.J., Joyce D.A. Glucocorticoids suppress tumor necrosis factor-alpha expression by human monocytic THP-1 cells by suppressing transactivation through adjacent NF-kappa B and c-Jun-activating transcription factor-2 binding sites in the promoter. J. Biol. Chem. 2000;275:18432–18440. doi: 10.1074/jbc.M906304199. [DOI] [PubMed] [Google Scholar]
- Walsh N.C., Reinwald S., Manning C.A., Condon K.W., Iwata K., Burr D.B., Gravallese E.M. Osteoblast function is compromised at sites of focal bone erosion in inflammatory arthritis. J. Bone Miner Res. 2009;24:1572–1585. doi: 10.1359/jbmr.090320. [DOI] [PubMed] [Google Scholar]
- Wong D.A., Kumar A., Jatana S., Ghiselli G., Wong K. Neurologic impairment from ectopic bone in the lumbar canal: a potential complication of off-label PLIF/TLIF use of bone morphogenetic protein-2 (BMP-2) Spine J. 2008;8:1011–1018. doi: 10.1016/j.spinee.2007.06.014. [DOI] [PubMed] [Google Scholar]
- Yamazaki M., Fukushima H., Shin M., Katagiri T., Doi T., Takahashi T., Jimi E. Tumor necrosis factor alpha represses bone morphogenetic protein (BMP) signaling by interfering with the DNA binding of Smads through the activation of NF-kappaB. J. Biol. Chem. 2009;284:35987–35995. doi: 10.1074/jbc.M109.070540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi M. Skeletal remodeling in health and disease. Nat. Med. 2007;13:791–801. doi: 10.1038/nm1593. [DOI] [PubMed] [Google Scholar]
- Zhu J.Y., Giaisi M., Kohler R., Muller W.W., Muhleisen A., Proksch P., Krammer P.H., Li-Weber M. Rocaglamide sensitizes leukemic T cells to activation-induced cell death by differential regulation of CD95L and c-FLIP expression. Cell Death Differ. 2009;16:1289–1299. doi: 10.1038/cdd.2009.42. [DOI] [PubMed] [Google Scholar]
- Zara J.N., Siu R.K., Zhang X., Shen J., Ngo R., Lee M., Li W., Chiang M., Chung J., Kwak J., et al. High doses of bone morphogenetic protein 2 induce structurally abnormal bone and inflammation in vivo. Tissue Eng. Part A. 2011;17:1389–1399. doi: 10.1089/ten.tea.2010.0555. [DOI] [PMC free article] [PubMed] [Google Scholar]