

Abstract
Retinols are metabolized into retinoic acids by alcohol dehydrogenase (ADH) and retinaldehyde dehydrogenase (Raldh). However, their roles have yet to be clarified in hepatitis despite enriched retinols in hepatic stellate cells (HSCs). Therefore, we investigated the effects of retinols on Concanavalin A (Con A)-mediated hepatitis.
Con A was injected into wild type (WT), Raldh1 knock-out (Raldh1−/−), CCL2−/− and CCR2−/− mice. For migration study of regulatory T cells (Tregs), we used in vivo and ex vivo adoptive transfer systems. Blockade of retinol metabolism in mice given 4-methylpyrazole, an inhibitor of ADH, and ablated Raldh1 gene manifested increased migration of Tregs, eventually protected against Con A-mediated hepatitis by decreasing interferon-γ in T cells. Moreover, interferon-γ treatment increased the expression of ADH3 and Raldh1, but it suppressed that of CCL2 and IL-6 in HSCs. However, the expression of CCL2 and IL-6 was inversely increased upon the pharmacologic or genetic ablation of ADH3 and Raldh1 in HSCs. Indeed, IL-6 treatment increased CCR2 expression of Tregs. In migration assay, ablated CCR2 in Tregs showed reduced migration to HSCs. In adoptive transfer of Tregs in vivo and ex vivo, Raldh1-deficient mice showed more increased migration of Tregs than WT mice. Furthermore, inhibited retinol metabolism increased survival rate (75%) compared with that of the controls (25%) in Con A-induced hepatitis.
These results suggest that blockade of retinol metabolism protects against acute liver injury by increased Treg migration, and it may represent a novel therapeutic strategy to control T cell-mediated acute hepatitis.
Keywords: 4-methylpyrazole, concanavalin A, Hepatic stellate cells, interferon-gamma
INTRODUCTION
T cell-mediated liver injuries are prominent in a variety of liver diseases, including autoimmune hepatitis, viral hepatitis and alcoholic liver injury (Ichiki et al., 2005; Jaruga et al., 2004; Kita et al., 2001). Concanavalin A (Con A) treatment induces massive liver injury by enhancing the production of interferon-γ (IFN-γ) in T cells; therefore, it has been widely used for studying T cell-mediated hepatitis (Tiegs et al., 1992). However, CD4+CD25+Foxp3+ regulatory T cells (Tregs) can inhibit T cell-mediated liver injury by producing interleukin-10 (IL-10) or transforming growth factor-β (TGF-β). Moreover, decreased hepatic Tregs and suppressed IL-10 expression have been highlighted as important factors for the progress of inflammatory diseases, such as viral hepatitis (Choi et al., 2014; Leifeld et al., 2002; Li et al., 2012). Actually, the decreased frequency of circulating Tregs was observed in patients with acute hepatitis A, subsequently resulting in severe liver injury (Choi et al., 2014). Thus, Tregs and IL-10 are definitely negative regulators for T cell-mediated inflammation.
Although CC chemokine receptors (CCRs), such as CCR2, CCR4, CCR5, CCR6, and CCR7, might be essential for Treg migration into the inflamed area (Ding et al., 2012; Erhardt et al., 2007; Lafdil et al., 2009; Wei et al., 2008), the underlying mechanism of Treg migration in acute inflamed liver has not been clearly understood. In patients and murine models of acute fulminant hepatic failure, highly expressed CC chemokine ligand 2 (CCL2) recruits monocytes and macrophages via CC chemokine receptor 2 (CCR2)(Saiman and Friedman, 2012). However, CCR2 is also expressed on Tregs, and it is important in Treg migration into inflamed arthritic joints and draining lymph nodes (Bruhl et al., 2004; Zhang et al., 2009), suggesting possible involvement of CCR2 in Treg migration into an inflamed liver. The liver is a major storage organ of body retinoids (retinol and its metabolites), and hepatic stellate cells (HSCs) play an important role in retinol homeostasis by storing 70% of body retinols in their cytoplasm (Radaeva et al., 2007). Our recent studies have demonstrated that these retinols can be metabolized into retinaldehydes and then further oxidized into retinoic acids (RAs) by alcohol dehydrogenases (ADHs), especially ADH3 in HSCs, and retinaldehyde dehydrogenases (Raldhs), respectively (Radaeva et al., 2007; Yi et al., 2014). Additionally, retinol metabolism can be suppressed by 4-methylpyrazole (4-MP), a broad ADH inhibitor, during in vitro culture of HSCs (Jeong et al., 2011; Yi et al., 2014). Interestingly, other previous studies have reported that decreased hepatic retinol contents were observed in chemical-induced liver injuries, such as carbon tetrachloride (CCl4)-, thioacetamide- and alcohol-induced rodent models and in alcoholic patients, implicating increased retinoid metabolism in liver disease (Leo and Lieber, 1982; Natarajan et al., 2005; Yi et al., 2014). Moreover, as regulatory bystanders, HSCs are involved in Treg expansion and differentiation via IFN-γ-mediated stimulation and Raldh1-derived production of RAs, respectively (Dunham et al., 2013; Ichikawa et al., 2011; Yang et al., 2009). Nevertheless, it remains unclear whether retinoids can influence the interplay between HSCs and Tregs during acute liver injury. Thus, in the present study, we investigated the effects of retinol metabolism on Con A-induced acute hepatitis and how HSCs modulate the hepatic population and functionality of Tregs during acute liver inflammation.
MATERIALS AND METHODS
Animals
All animals were housed in accordance with the guidelines of the Care and Use of Laboratory Animals published by NIH, and all experimental procedures were approved by the Institutional Animal Care and Use Committee of KAIST. Male C57BL/6 wild type (WT), enhanced GFP (eGFP) transgenic, CCL2 knock-out (CCL2−/−), CCR2−/− and signal transducer and activator of transcription 1 (STAT1)−/− mice were purchased from the Jackson Laboratory. FoxP3-GFP transgenic mice were generated previously and were purchased from the Jackson Laboratory, which express both of GFP and FoxP3 that is specific marker of Tregs with the control of the endogenous promoter (Haribhai et al., 2007). B6/SJL (CD45.1) mice were purchased from Taconic. Raldh1−/− mice were generated previously (Ziouzenkova et al., 2007).
T cell-mediated hepatitis and inhibition of retinol metabolism
Acute hepatitis was induced by Con A (Sigma-Aldrich) injection into the tail vain (12 mg·kg−1). To block retinol metabolism, the mice were pre-treated with 4-MP (Sigma-Aldrich) (10 mg·kg−1), a broad ADH inhibitor, for 3 h before Con A treatment (Lopez-Valencia et al., 2007; Yi et al., 2014). To assess the survival rate, 30 mg·kg−1 of Con A was injected after 4-MP or vehicle treatment.
Isolation and co-culturing of nTregs with HSCs
Natural Tregs (nTregs) were isolated using isolation kits (Miltenyi Biotec). For co-culturing nTregs with HSCs, using 96 well plates, 2 × 104 cells of isolated HSCs were co-cultured with 1 × 105 of nTregs cells with or without treatments of IFN-γ (5 ng·ml−1) in RPMI medium for several time points. HSCs were treated with 4-MP (0.5 mM) for 3 h before co-culturing. After co-culturing, nTregs were separated from HSCs by pipetting and then purified with positive selection using magnetic beads. The remaining HSCs were detached from the plate using trypsin-EDTA (Invitrogen). The collected cells were subjected to flow cytometry and quantitative real-time PCR analysis. For proliferation assay, isolated Tregs were labeled with carboxyfluorescein succinimidyl ester (CFSE) (Invitrogen) just before co-culturing with HSCs following the manufacture’s guideline.
Flow cytometry analyses
Isolated Tregs and liver mononuclear cells (MNCs) were analyzed using flow cytometry analyses. The collected cells were re-suspended in DPBS containing 0.5% BSA and 0.05% sodium azide. After washing, cells were stained for 30 min with various surface antibodies, such as anti-CD45, anti-CD3, anti-CD4, anti-CD8a, anti-CD11b, anti-CD25, anti-CD62L, anti-CD69, anti-NK1.1, anti-Gr1 and anti-F4/80 (eBioscience and BD Pharmingen). Each of these antibodies was conjugated with pacific blue, FITC, PE, PerCP-Cy 5.5, APC or PE-Cy7. To identify Treg cells, cells were stained with anti-FoxP3 (eBioscience) using a FoxP3 staining buffer kit (eBioscience). LSR II Flow Cytometer (BD Bioscience) and FlowJo software (Tree Star) were used to analyze the stained cells.
Adoptive transfer of eGFP nTreg
Tregs were isolated from eGFP-mice and transferred to WT mice which were treated Con A. At 3, 6 and 9 h after Con A treatment, 1 × 106 nTregs were injected to Con A-treated mice though tail vein. Mice were sacrificed 12 h later after Con A treatment and liver was extracted. Transferred Tregs were analyzed by immunofluorescence staining and flow cytometry. For immunofluorescence staining, frozen liver tissue were sliced to a thickness of 5 μm and stained with anti-desmin antibody and mounted with DAPI medium as described above. In flow cytometry analyses, proportion of GFP+ Tregs among total Tregs and total number of transferred Tregs were calculated at each time points.
Ex vivo migration assay of Tregs using closed circulation
After anesthesia, the liver, inferior vena cava (IVC) and portal vein were exposed, and the inferior portion of the IVC below the liver was ligated carefully. On the other hand, the portal vein and superior part of the IVC above liver were catheterized. Each catheter line was connected with an infusion pump, and the opposite ends of the lines were placed together in a tube filled with 2 × 106 of eGFP+ Tregs and IFN-γ (20 ng·ml−1). To maintain the same intramural pressure in the sinusoid with the live state, the flow rate was regulated as 2 ml/min (Xie et al., 2014). After closed circulation for 2 h, the liver was extracted and liver MNCs were isolated for flow cytometry. The mice and all equipment were kept in 37°C incubator during closed circulation. Details on the methodology are provided as online supplementary methods.
Chimeric mouse generation
Chimeric mice were generated as previously described (Yi et al., 2014). Briefly, after radiation, WT and Raldh1-deficient mice were infused with whole bone marrow cells (3 × 106 cells) of CD45.1+ WT mice.
Statistical analysis
Data are presented as the mean ± SEM. To compare values obtained from 2 or more groups, Student’s t-test or one-way analysis of variance was performed. Values of P < 0.05 were considered statistically significant. For more details, please see the Supporting information.
RESULTS
Inhibited retinol metabolism by 4-MP attenuates Con A-induced acute hepatitis and increases hepatic Tregs
As previously reported (Dunham et al., 2013; Mucida et al., 2007), we tested whether suppressed retinol metabolism of HSCs by 4-MP influences the differentiation of naïve T cells into Tregs. Treatments of CD3/CD28 antibodies to naïve T cells did not increase Treg differentiation (0.33%), whereas co-culturing with HSCs increased the differentiation of naïve T cells into Tregs (4.04%) because HSCs are a source of TGF-β1 and all-trans RAs. However, 4-MP treatment decreased Treg differentiation (2.27%) (Fig. 1A). In co-cultured HSCs, the gene expression of TGF-β1 and Raldh1, which is a major metabolizing enzyme for RA production (Ziouzenkova et al., 2007), was markedly suppressed by 4-MP (Fig. 1A). These findings also show that suppressed retinol metabolism in HSCs reduced the differentiation of naïve T cells into Tregs.
Fig. 1.

The 4-MP treatment attenuates Con A-induced hepatitis and increases hepatic Tregs. (A) Isolated naïve T cells were co-cultured with freshly isolated HSCs for 3 days with or without 4-MP (0.5 mM). The culture medium contained antibodies of CD3 (1 μg·ml−1), CD28 (1 μg·ml−1), IFN-γ (10 μg·ml−1) and IL-4 (10 μg·ml−1). Cultured naïve T cells and HSCs were subjected to flowcytometry and real-time PCR analyses, respectively. Con A (12 mg·kg−1) was injected into mice via the tail vein with or without pretreatment with 4-MP (10 mg·kg−1) 3 h earlier. (B) Gross findings and liver sections were stained with H&E 24 h after Con A injection. Yellow dotted lines indicate damaged areas. (C) Serum levels of ALT, IFN-γ and IL-10 were measured. (D) Population and total numbers of Tregs were analyzed by flow cytometry. (E) Real-time PCR analyses were performed with isolated liver MNCs. (F) Levels of phosphorylated STAT1 were determined by Western blot. Data represent the mean ± SEM (n = 8–12/group). *P < 0.05; **P < 0.01 compared with the corresponding control. Scale bars, 200 μm.
Next, we examined the roles of retinoids in Con A-induced acute hepatitis. Retinol metabolizing enzyme ADH was inhibited by pretreatment with 4-MP 3 h before Con A injection. Unexpectedly in the gross and histological observation, 4-MP-treated mice showed decreased liver injury compared to that of vehicle-treated mice at 24 h (Fig. 1B). Consistent with those findings, serum levels of alanine aminotransferase (ALT) and IFN-γ were substantially reduced, whereas anti-inflammatory cytokine IL-10 increased in 4-MP-treated mice compared to the levels in vehicle-treated mice (Fig. 1C). Then, we investigated the effects of 4-MP on the population of hepatic immune cells. Pretreatment with 4-MP did not affect most of the population of hepatic immune cells (0 h), whereas 4-MP treatment increased the Treg population at 12 and 24 h after Con A treatment compared to those in vehicle-treated mice (Fig. 1D and Supplementary Fig. 1A). In addition, the counted numbers of Tregs per liver weight were increased in 4-MP-treated mice compared to those in vehicle-treated mice concomitant with decreased populations of Gr1+CD11b+ cells and F4/80+CD11b+ cells (Fig. 1D, Supplementary Fig. 1B). However, the populations and total numbers of CD4+ and CD8+ T cells showed no difference between the vehicle- and 4-MP-treated groups (Supplementary Figs. 1B and 1C). Furthermore, Con A-mediated increased expression of CD69, an activation marker of T cells, was not affected by treatment with 4-MP excluding 4-MP-mediated direct suppression of CD4+ T cells (Supplementary Fig. 1D).
In isolated liver MNCs, the gene expression of IFN-γ was decreased but that of Foxp3 and IL-10 was increased in 4-MP-treated mice compared to that in vehicle-treated mice (Fig. 1E). Concomitant with decreased IFN-γ levels, phosphorylation of STAT1 (pSTAT1) was reduced in the livers of 4-MP-treated mice compared to that of vehicle-treated mice (Fig. 1F). All of these findings show that hepatic Tregs are remarkably increased by 4-MP, subsequently leading to the attenuation of Con A-mediated liver injury.
HSCs increase the gene expression of CCR2 and IL-10 in Tregs in the presence of 4-MP and IFN-γ
To identify the location of Tregs in Con A-mediated hepatitis, FoxP3-GFP transgenic mice were used. Interestingly, we observed that most of the Tregs showed closer contact with the HSCs of 4-MP-treated mice than those of vehicle-treated mice (Fig. 2A). Given that inhibited retinol metabolism by 4-MP increased the interaction between Tregs and HSCs in liver, we examined whether HSCs might have some effects on the function of Tregs. Thus, we co-cultured freshly isolated nTregs with retinol-enriched HSCs for 24 h. To mimic a Con A-mediated inflammatory condition, IFN-γ and 4-MP were added during co-culturing. Comparing with nTreg only, co-culturing with HSCs remarkably increased the Foxp3 expression of nTregs, but neither treatments nor co-treatments with IFN-γ and 4-MP had any effect on the Foxp3 expression and proliferation of nTregs during co-culture with HSCs (Fig. 2B). Similar findings were observed in the co-culture system for up to 72 h (Supplementary Fig. 2A). In contrast to those findings, in the presence of IFN-γ during co-culturing, IFN-γ suppressed CCL2 expression in HSCs, but 4-MP treatment significantly increased the gene expression of CCL2 and IL-6 in HSCs and that of CCR2 and IL-10 in nTregs, respectively (Fig. 2C). However, in the absence of IFN-γ, these expressions in Tregs and HSCs were not changed by 4-MP. These findings suggest that IFN-γ negatively regulates the expression of CCL2 and IL-6. In addition, increased super-natant levels of IL-10 in the co-culture were decreased by IFN-γ, whereas 4-MP treatment increased IL-10 levels even in the presence of IFN-γ (Supplementary Fig. 2B). However, in the presence of IFN-α, the gene expression of CCR2 and IL-10 in nTregs was not changed by 4-MP (Supplementary Fig. 2C). To determine the mechanism of increased expression of CCR2 and IL-10 in nTregs, we tested the effects of IL-6 and IL-10 on Tregs as previously reported (Kubota et al., 2010; Murai et al., 2009). In quantitative PCR analyses, treatment with IL-6 and IL-10 increased the expression of IL-10, but CCR2 expression in Tregs was only enhanced by IL-6 (Fig. 2D). All of these data show that 4-MP-mediated inhibition of retinol metabolism in HSCs positively regulates the function of nTregs in the presence of the IFN-γ signaling pathway.
Fig. 2.

Expression of CCR2 and IL-10 increases significantly in Tregs co-cultured with HSCs in the presence of IFN-γ and 4-MP. (A) Twelve hours after Con A treatment to Foxp3-GFP reporter mice (n = 4/group), liver sections were stained with desmin (red) for HSCs and DAPI (blue) for nuclei, revealing close contact (white arrow) between Tregs and HSCs. (B, C) Isolated nTregs were co-cultured with HSCs for 24 h with or without 4-MP (0.5 mM) ± IFN-γ (5 ng·ml−1). Cultured nTregs were stained with antibodies to Foxp3 and CD25 or labeled with CFSE for a proliferation assay. The expression levels of CCR2, IL-10, CCL2 and IL-6 in co-cultured nTregs and HSCs were determined by quantitative real-time PCR. (D) Expression levels of IL-10 and CCR2 were analyzed by quantitative real-time PCR in WT nTregs treated with IL-6 (20 ng·ml−1) or IL-10 (20 ng·ml−1). Data represent the mean ± SEM of three independent experiments. *P < 0.05; **P < 0.01 compared with the corresponding control. Scale bars, 25 μm.
Retinoid deficiency in HSCs increases migration of Tregs in a CCL2/CCR2-dependent manner
Because ADH3 and Raldh1 are known as main retinol-metabolizing enzymes in HSCs (Ichikawa et al., 2011; Yi et al., 2014), we checked the altered expression of ADH3 and Raldh1 in vivo and in vitro. At 12 h of sacrifice, HSCs treated with Con A showed increased expression of ADH3 but decreased retinol droplet contents compared to those of the control HSCs, while 4-MP treatment suppressed ADH3 expression but increased retinol droplet contents in HSCs compared with those of Con A-treated HSCs (Fig. 3A and Supplementary Fig. 3A). In quantitative PCR analyses, the gene expression of ADH3 and Raldh1 in HSCs isolated from Con A-treated mice increased approximately 20-fold and 80-fold, respectively, compared to those of the control HSCs, while the expression of these enzymes was remarkably suppressed in the HSCs of 4-MP-treated mice (Fig. 3B). With direct treatment of HSCs in vitro with IFN-γ and/or 4-MP, the content of all-trans RA (atRA) and the expression of ADH3 and Raldh1 in HSCs were markedly up-regulated by IFN-γ (Figs. 3C and 3D); however, the expression of CCL2 and IL-6 was decreased or unchanged in IFN-γ-treated HSCs (Fig. 3D). In contrast, interestingly, 4-MP treatment suppressed the increased expression of ADH3 and Raldh1; conversely, it up-regulated the expression of CCL2 and IL-6 in both freshly isolated and IFN-γ-treated HSCs (Figs. 3B and 3D). These findings were quite similar to those for co-cultured HSCs in vitro as shown in Fig. 2C.
Fig. 3.
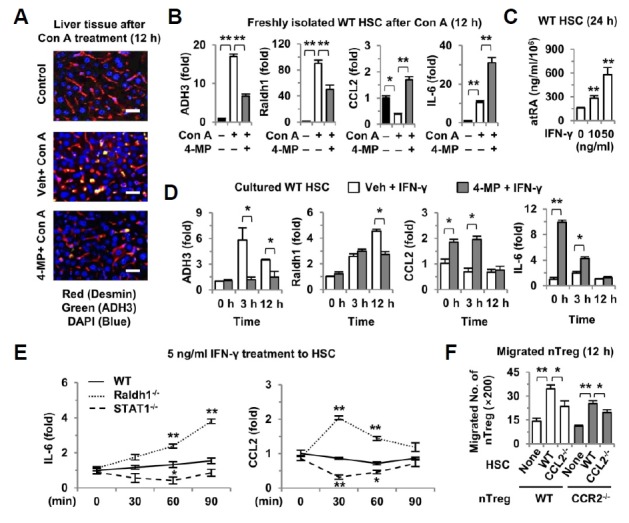
Inhibition of IFN-γ-mediated retinol metabolic pathway in HSCs increases Tregs migration. (A) Twelve hours after Con A and 4-MP treatment (n = 4/group), liver sections were stained with antibodies to ADH3 (green) and desmin (red) as well as staining with DAPI (blue). (B) Expression levels of ADH3, Raldh1, CCL2 and IL-6 were determined by quantitative real-time PCR in freshly isolated HSCs from Con A- and 4-MP-treated mice (n = 3/group). (C) Cytosolic levels of all-trans RA (atRA) were measured in WT HSCs following IFN-γ treatment. (D) WT HSCs were treated with IFN-γ (5 ng·ml−1) ± 4-MP (0.5 mM) until 12 h, and then the expression levels of ADH3, Raldh1, CCL2 and IL-6 were analyzed by quantitative real-time PCR. (E) Freshly isolated HSCs from WT, Radh1- and STAT1-deficient mice were treated with IFN-γ (5 ng·ml−1) for several time periods, and the expression levels of IL-6 and CCL2 were analyzed by quantitative real-time PCR. (F) Migration assay was performed using a 12-transwell insert with 3 μm pores. In the lower chamber, 1 × 105 HSCs were plated. Then, 2 × 105 Tregs were added to the upper chamber and were incubated for 12 h. Migrated nTregs were counted in 5 different areas under ×200 magnification. Data represent the mean ± SEM of three independent experiments. *P < 0.05; **P < 0.01 compared with the corresponding control. Scale bars, 25 μm.
Based on our findings and those of previous studies (Bruhl et al., 2004; Fujimoto et al., 2011; Kubota et al., 2010; Murai et al., 2009), we further investigated the IFN-γ-mediated retinoid signaling pathways in HSCs and the migration mechanism of CCR2-expressing Tregs to CCL2-expressing HSCs. In HSCs isolated from WT, STAT1−/− and Raldh1−/− mice, IFN-γ treatment slightly increased the expression of IL-6 but not that of CCL2 in WT HSCs (Fig. 3E). In contrast, the gene expression of IL-6 and CCL2 decreased in STAT1−/− HSCs, whereas it increased remarkably in Raldh1−/− HSCs compared with that in WT HSCs (Fig. 3E). These findings strongly suggest that there are at least two IFN-γ signaling pathways (STAT1-dependent and Raldh1-dependent) within HSCs, and that IFN-γ-mediated retinol metabolism negatively regulates the expression of CCL2 and IL-6 in HSCs. Next, we examined whether the expression of CCL2 in HSCs is important for the migration of CCR2-expressing Tregs (Supplementary Fig. 3B). As shown in Supplementary Figs. S3C and S3D, small numbers of Tregs treated with 4-MP or IFN-γ migrated to HSCs, while co-treatment with 4-MP and IFN-γ significantly enhanced Treg migration compared to those of other types of treatments, which might be due in part to the increased expression of CCL2 in HSCs. This hypothesis was confirmed by the results shown in Fig. 3F. CCL2−/− HSCs induced decreased migration of WT Tregs compared to that of WT HSCs, and conversely fewer CCR2−/− Tregs migrated to WT HSCs than did WT Tregs, where migration was more suppressed in CCL2−/− HSCs (Fig. 3F). All of these data show that inhibition of the IFN-γ-mediated activation of retinol metabolism in HSCs increases Treg migration partly in a CCR2/CCL2-dependent manner.
Enhanced migration of Tregs in Raldh1-deficient mice ameliorates Con A-mediated acute liver injury
To confirm the relationship between the Treg migration and retinol metabolism of HSCs in acute hepatitis, as shown in Fig. 3, WT, CCL2−/−, CCR2−/− and Raldh1−/− mice were sacrificed at 12 h after Con A injection. In the gross and histological findings, CCL2−/− and CCR2−/− mice showed severe liver injuries and more apoptotic bodies than did WT mice, whereas liver injuries in Raldh1−/− mice were decreased compared to those in WT mice (Figs. 4A and 4B). Similarly, serum levels of ALT and IFN-γ were higher, and IL-10 level was lower in CCL2−/− and CCR2−/− mice than in WT mice (Fig. 4C). In contrast, Raldh1−/− mice showed lower levels of ALT and IFN-γ but enhanced levels of IL-10 compared to WT mice (Fig. 4C). In flow cytometry analyses, the population and number of Tregs were significantly decreased in CCL2−/− and CCR2−/− mice compared to those in WT and Raldh1−/− mice, but the total number of Tregs in Raldh1−/− mice was higher than in WT mice (Fig. 4D).
Fig. 4.

Raldh1 deficiency attenuates Con A-mediated liver injury via increased migration of Tregs. WT, CCL2-, CCR2-, and Raldh1-deficient mice (n = 6/group) were sacrificed 12 h after Con A (12 mg/kg) treatment. (A) Representative gross findings of whole livers and liver sections stained with H&E and TUNEL. (B) Numbers of apoptotic bodies in liver sections were counted in 5 different areas under ×400 magnification. (C) Serum levels of ALT, IFN-γ and IL-10 were measured. (D) Frequency and total numbers of hepatic Tregs from liver MNCs of mice were analyzed by flow cytometry. (E) For adoptive transfer of nTregs in vivo, eGFP+ nTregs (1 × 106) were transferred through the tail vein at 3, 6 and 9 h after Con A treatment, and then mice (n = 4/time point) were sacrificed 12 h after Con A treatment. For ex vivo, isolated eGFP+ nTregs (2 × 106) and IFN-γ (20 ng·ml−1) were co-circulated for 2 h through a closed circulation system from the portal vein to the inferior vena cava. (n = 4/group) (F) Migrated eGFP+ nTregs were counted from isolated liver MNCs. Nine hours after Con A treatment, liver sections were stained with desmin (red) for HSCs and DAPI (blue) for nuclei. (G) Representative frequency and numbers of migrated eGFP+ nTregs. (H) WT nTregs were co-cultured with Raldh1-deficient HSCs for 24 h in the presence of IFN-γ (5 ng·ml−1). Expression levels of CCR2 and IL-10 in Tregs, and CCL2 and IL-6 in HSCs were determined by quantitative real-time PCR. Data represent the mean ± SEM of three independent experiments. *P < 0.05; **P < 0.01 compared with the corresponding control. Scale bar, 200 μm (A) and 25 μm (F)
To further confirm Treg migration, we performed in vivo and ex vivo adoptive transfer experiments by using eGFP-expressing nTregs (Fig. 4E). In Con A-induced acute liver injury, more adoptively transferred eGFP+ Tregs migrated and were in close contact with the HSCs of Raldh1−/− mice than those in WT mice (Fig. 4F). Similarly, in IFN-γ-mediated liver injury, more eGFP+ Tregs migrated into livers of Raldh1−/− mice and were in close contact with HSCs than those of WT mice in the closed circulation system (Fig. 4G and Supplementary Fig. 4). In the in vitro co-culture of Raldh1−/− HSCs with WT nTregs, IFN-γ treatment enhanced the expression of CCL2 and IL-6 in Raldh1−/− HSCs compared to that in Raldh1−/− HSCs without IFN-γ treatment (Fig. 4H). In addition, in the presence of IFN-γ, WT nTregs co-cultured with Raldh1−/− HSCs showed enhanced expression of CCR2 and IL-10 compared to that of Tregs in the absence of IFN-γ (Fig. 4H). Collectively, these data suggest that Raldh1 deficiency might be partly involved in the enhanced migration and interaction of Tregs with HSCs in a CCL2/CCR2-dependent manner, which attenuates Con A-mediated liver injury by increased IL-10 production.
Raldh1-deficient chimeric mice are resistant to Con A-mediated liver injury via increased migration of Tregs
To further investigate the effects of Raldh1-deficient HSCs on the migration of Tregs and Con A-mediated liver injury, we produced chimeric mice by transplantation of WT bone marrow to WT mice (WTWT) and Raldh1−/− mice (Raldh1WT) (Fig. 5A). At 8 weeks after the transplantation of WT bone marrow (CD45.1), over 80% of liver MNCs and T cells had been replaced by donor cells (CD45.2) (Fig. 5A and Supplementary Fig. 5A). In addition, freshly isolated HSCs from Raldh1WT mice did not show expression of the Raldh1 gene compared with WTWT mice, whereas the liver MNCs of Raldh1WT mice strongly expressed the Raldh1 gene (Fig. 5A and Supplementary Fig. 5B). In the gross and histological findings, liver injuries and apoptotic bodies were significantly increased in WTWT mice compared to those in Raldh1WT mice (Fig. 5B and Supplementary Fig. 5C). In WTWT mice, serum levels of ALT and IFN-γ were significantly increased, and the IL-10 level was decreased compared to Raldh1WT mice (Fig. 5C). Coincidently, the expression of IFN-γ was decreased, and the expression of IL-10 was increased in the liver MNCs of Raldh1WT mice compared with those in WTWT mice (Fig. 5D). In flow cytometry analyses, the number of CD4+CD25+Foxp3+ Tregs was remarkably increased in Raldh1WT mice compared to that in WTWT mice (Fig. 5E). These findings suggest that Raldh1 deficiency in HSCs could attenuate Con A-induced liver injury via increased hepatic Tregs.
Fig. 5.
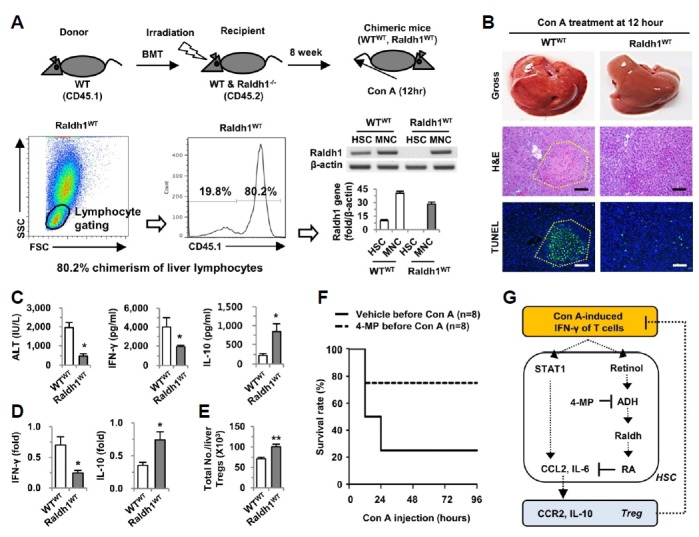
Blocking of the retinol metabolic pathway ameliorates Con A-mediated liver injury, subsequently augmenting the survival rate. (A) Chimeric mice were generated by transplantation of WT bone marrow to WT mice (WTWT) and Raldh1-deficient (Raldh1−/−) mice (Raldh1WT). Eight weeks after bone marrow transplantation, isolated liver MNCs and HSCs were analyzed for chimerism with flow cytometry or RT- and real-time PCR. (B) Eight weeks after bone marrow transplantation, mice (n = 5/group) were injected with Con A (12 mg·kg−1) for 12 h. Gross findings of whole livers and liver sections stained with H&E and TUNEL. Yellow dotted lines indicate damaged areas. (C) Serum levels of ALT, IFN-γ and IL-10 were measured. (D) Real-time PCR analyses were performed with isolated liver MNCs. (E) Total numbers of hepatic Tregs from liver MNCs of mice were analyzed by flow cytometry. (F) Ninety-six hour survival rates were evaluated in mice (n = 8/group) treated with a lethal dose of Con A (30 mg·kg−1). (G) Schematic diagram of the beneficial effects of 4-MP in Con A-mediated acute hepatitis in mice. Data are representative of three independent experiments. *P < 0.05; **P < 0.01 compared with the corresponding control. Scale bar, 100 μm.
Finally, we investigated whether 4-MP treatment can augment the survival rate in Con A-induced fulminant hepatitis in mice. In accord with our data above, Con A-induced liver injury was less obvious in mice pre-treated with 4-MP than in the control mice, leading to an approximately 75% survival rate in the 4-MP-treated mice compared to a 25% survival rate in the control mice (Fig. 5F). Taken together, all of these results suggest that 4-MP could be applied as a preventative treatment, and retinol metabolizing enzymes such as ADH3 and Raldh1 in HSCs might represent novel therapeutic targets to control T cell (IFN-γ)-mediated acute hepatitis.
DISCUSSION
Recent emerging evidence suggests that retinoids, including retinol and its metabolites, play various functions in obesity-related metabolic disorders and in the regulation of immune cells (Hall et al., 2011; Ziouzenkova et al., 2007). Moreover, decreased levels of hepatic retinol with elevated retinol metabolites have been reported in animal models and in patients with liver damage (Leo and Lieber, 1982; Natarajan et al., 2005). Furthermore, the mechanism of liver damage and fibrosis induced by hypervitaminosis A remains unknown, although hepatic injury from vitamin A toxicity has been known for more than a century (Geubel et al., 1991; Nollevaux et al., 2006). Therefore, we investigated the involvement of retinol metabolism in the pathogenesis of Con-A mediated acute hepatitis. In the present study, we observed that the inhibition of IFN-γ-mediated retinol metabolism enhanced the gene expression of CCL2 and IL-6 in HSCs, leading to alleviation of liver inflammation via the recruitment and activation of CCR2-expressing Tregs. We have summarized these findings in Fig. 5G.
Although the anti-viral, anti-fibrotic and anti-regenerative effects of IFN-γ on the liver through the activation of STAT1 have been well documented (Hong et al., 2002; Ichiki et al., 2005; Jeong et al., 2011; Schuppan and Kim, 2013), The contribution of IFN-γ-mediated signals to the immunomodulatory effects on HSCs remains elusive. Several studies have suggested that HSCs are involved in the induction and expansion of immunosuppressive cells, such as myeloid-derived suppressor cells and Tregs in an IFN-γ or RA-dependent manner (Chou et al., 2011; Ichikawa et al., 2011; Yang et al., 2009; Yi et al., 2014). Moreover, a recent study reported that IFN-γ-induced MHC-class II molecule expression in endothelial cells enhanced the recruitment of Tregs into inflamed lymph node (Fu et al., 2014). However, the underlying mechanism of IFN-γ effects on the interplay between HSCs and Tregs is still unclear. Interestingly, in the present study, we observed that IFN-γ stimulated the retinol metabolism of HSCs by increasing expression of ADH3 and Raldh1 in vivo and in vitro (Fig. 3). We also observed that 4-MP-mediated inhibition of retinol metabolism or Raldh1-deficiency increased the expression of CCL2 and IL-6 in HSCs (Figs. 3D and 4H), while STAT1 deficiency decreased this expression in the presence of IFN-γ (Fig. 3E). These findings suggest that there are at least two IFN-γ signaling pathways within HSCs (Fig. 5G); IFN-γ-mediated activation of STAT1 signaling might be a positive regulator in the expression of CCL2 and IL-6 in HSCs, whereas the IFN-γ-mediated retinol metabolic pathway is the opposite in this regard. However, HSCs did not stimulate Treg proliferation except for the increased expression of Foxp3 during co-culturing (Fig. 2B).
Tregs act as a negative regulator in inflammation, and in particular, RAs are an important factor for the induction of Tregs from naïve T cells (Mucida et al., 2007). However, we observed that retinol metabolism suppressed by 4-MP or by Raldh1 deficiency increased the Treg population during acute liver inflammation, suggesting that the increase in hepatic Tregs might be due to the enhanced migration of Tregs but not the differentiation of naïve T cells in acute liver injury. More interestingly, infiltrated or adoptively transferred Tregs were in close contact with HSCs, which was more prominent with 4-MP treatment or in Raldh1 deficiency in vivo. Among the C-C chemokine receptors, the expression of CCR2, CCR4, CCR5 and CCR7 is critically involved in the migration of Tregs to inflamed tissues (Ding et al., 2012). Interestingly, CCR2 expression by T cells and Tregs played an important role in down-regulating the collagen-induced arthritis and T cell-mediated hepatitis of mice, in which increased IFN-γ levels were significantly reduced (Ajuebor et al., 2003; Bruhl et al., 2004). In the present study, in 4-MP-treated and Raldh1-deficient HSCs, the in vivo and in vitro expression of CCL2, a specific ligand for CCR2 (Schuppan and Kim, 2013), was remarkably increased in the presence of IFN-γ. These findings are similar to those of previous reports demonstrating that IFN-γ treatment increased CCL2 expression in cultured human HSCs, whereas RAs repressed the lipopolysaccharide-mediated transcription of CCL2 and IL-6 in several cell lines, including fibroblast (Austenaa et al., 2009; Marra et al., 1993). In terms of the effects of IL-6 on Tregs, previous studies have reported that IL-6 inhibits the differentiation of naïve T cells into Tregs but controls the expansion of nTregs indirectly (Fujimoto et al., 2011; Kubota et al., 2010). In the present study, we identified a novel function of IL-6 for CCR2 induction in nTregs in vitro (Fig. 2D). In addition, Tregs co-cultured with 4-MP-treated or Raldh1-deficient HSCs showed increased expression of CCR2, most likely due to increased IL-6 production by HSCs (Figs. 2C and 4H). Furthermore, we demonstrated that the expression of CCL2 and CCR2 was a critical factor for the migration of Tregs to HSCs in vitro and in vivo (Figs. 3F and 4D), consequently increasing the survival rate of mice by preventing Con A-induced severe liver injury (Fig. 5F).
In conclusion, our study suggests that IFN-γ-mediated retinol metabolism is involved in the pathogenesis of Con A-mediated liver injury via down-regulation of the interplay between HSCs and Tregs, subsequently accelerating liver injury. However, Con A-mediated liver injury could be prevented by the inhibition of retinol metabolism. Therefore, our findings crucially suggest that retinol metabolism could be a therapeutic target for improving T cell-mediated liver injury, such as viral hepatitis and autoimmune disease.
Supplementary Information
Acknowledgments
The authors thank Drs. Gregg Duester for his kindness gifts of Raldh1-deficient mice, and Dr. Hyojin Ko for excellent technical assistance. This work was supported by Korea Mouse Phenotyping Project (NRF-2014M3A9D5A01073556) of the Ministry of Science, ICT and Future Planning through the National Research Foundation, grants of the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (A111345 and A111498), a grant from the Next-Generation BioGreen 21 Program (No. PJ009957), Rural Development Administration, Republic of Korea.
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Ajuebor M.N., Hogaboam C.M., Le T., Swain M.G. C-C chemokine ligand 2/monocyte chemoattractant protein-1 directly inhibits NKT cell IL-4 production and is hepatoprotective in T cell-mediated hepatitis in the mouse. J. Immunol. 2003;170:5252–5259. doi: 10.4049/jimmunol.170.10.5252. [DOI] [PubMed] [Google Scholar]
- Austenaa L.M., Carlsen H., Hollung K., Blomhoff H.K., Blomhoff R. Retinoic acid dampens LPS-induced NF-kappaB activity: results from human monoblasts and in vivo imaging of NF-kappaB reporter mice. J. Nutr. Biochem. 2009;20:726–734. doi: 10.1016/j.jnutbio.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Bruhl H., Cihak J., Schneider M.A., Plachy J., Rupp T., Wenzel I., Shakarami M., Milz S., Ellwart J.W., Stangassinger M., et al. Dual role of CCR2 during initiation and progression of collagen-induced arthritis: evidence for regulatory activity of CCR2+ T cells. J. Immunol. 2004;172:890–898. doi: 10.4049/jimmunol.172.2.890. [DOI] [PubMed] [Google Scholar]
- Choi Y.S., Lee J., Lee H.W., Chang D.Y., Sung P.S., Jung M.K., Park J.Y., Kim J.K., Lee J.I., Park H., et al. Liver injury in acute hepatitis A is associated with decreased frequency of regulatory T cells caused by Fas-mediated apoptosis. Gut. 2014;64:1303–1313. doi: 10.1136/gutjnl-2013-306213. [DOI] [PubMed] [Google Scholar]
- Chou H.S., Hsieh C.C., Yang H.R., Wang L., Arakawa Y., Brown K., Wu Q., Lin F., Peters M., Fung J.J., et al. Hepatic stellate cells regulate immune response by way of induction of myeloid suppressor cells in mice. Hepatology. 2011;53:1007–1019. doi: 10.1002/hep.24162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y., Xu J., Bromberg J.S. Regulatory T cell migration during an immune response. Trends Immunol. 2012;33:174–180. doi: 10.1016/j.it.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham R.M., Thapa M., Velazquez V.M., Elrod E.J., Denning T.L., Pulendran B., Grakoui A. Hepatic stellate cells preferentially induce Foxp3+ regulatory T cells by production of retinoic acid. J. Immunol. 2013;190:2009–2016. doi: 10.4049/jimmunol.1201937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhardt A., Biburger M., Papadopoulos T., Tiegs G. IL-10, regulatory T cells, and Kupffer cells mediate tolerance in concanavalin A-induced liver injury in mice. Hepatology. 2007;45:475–485. doi: 10.1002/hep.21498. [DOI] [PubMed] [Google Scholar]
- Fu H., Kishore M., Gittens B., Wang G., Coe D., Komarowska I., Infante E., Ridley A.J., Cooper D., Perretti M., et al. Self-recognition of the endothelium enables regulatory T-cell trafficking and defines the kinetics of immune regulation. Nat. Commun. 2014;5:3436. doi: 10.1038/ncomms4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M., Nakano M., Terabe F., Kawahata H., Ohkawara T., Han Y., Ripley B., Serada S., Nishikawa T., Kimura A., et al. The influence of excessive IL-6 production in vivo on the development and function of Foxp3+ regulatory T cells. J. Immunol. 2011;186:32–40. doi: 10.4049/jimmunol.0903314. [DOI] [PubMed] [Google Scholar]
- Geubel A.P., De Galocsy C., Alves N., Rahier J., Dive C. Liver damage caused by therapeutic vitamin A administration: estimate of dose-related toxicity in 41 cases. Gastroenterology. 1991;100:1701–1709. doi: 10.1016/0016-5085(91)90672-8. [DOI] [PubMed] [Google Scholar]
- Hall J.A., Grainger J.R., Spencer S.P., Belkaid Y. The role of retinoic acid in tolerance and immunity. Immunity. 2011;35:13–22. doi: 10.1016/j.immuni.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haribhai D., Lin W., Relland L.M., Truong N., Williams C.B., Chatila T.A. Regulatory T cells dynamically control the primary immune response to foreign antigen. J. Immunol. 2007;178:2961–2972. doi: 10.4049/jimmunol.178.5.2961. [DOI] [PubMed] [Google Scholar]
- Hong F., Jaruga B., Kim W.H., Radaeva S., El-Assal O.N., Tian Z., Nguyen V.A., Gao B. Opposing roles of STAT1 and STAT3 in T cell-mediated hepatitis: regulation by SOCS. J. Clin. Invest. 2002;110:1503–1513. doi: 10.1172/JCI15841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa S., Mucida D., Tyznik A.J., Kronenberg M., Cheroutre H. Hepatic stellate cells function as regulatory bystanders. J. Immunol. 2011;186:5549–5555. doi: 10.4049/jimmunol.1003917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichiki Y., He X.S., Shimoda S., Ishibashi H., Keeffe E.B., Rossaro L., Gershwin M.E. T cell immunity in hepatitis B and hepatitis C virus infection: implications for autoimmunity. Autoimmun. Rev. 2005;4:82–95. doi: 10.1016/j.autrev.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Jaruga B., Hong F., Kim W.H., Sun R., Fan S., Gao B. Chronic alcohol consumption accelerates liver injury in T cell-mediated hepatitis: alcohol disregulation of NF-kappaB and STAT3 signaling pathways. Am. J. Physiol. Gastrointest Liver Physiol. 2004;287:G471–479. doi: 10.1152/ajpgi.00018.2004. [DOI] [PubMed] [Google Scholar]
- Jeong W.I., Park O., Suh Y.G., Byun J.S., Park S.Y., Choi E., Kim J.K., Ko H., Wang H., Miller A.M., et al. Suppression of innate immunity (natural killer cell/interferon-gamma) in the advanced stages of liver fibrosis in mice. Hepatology. 2011;53:1342–1351. doi: 10.1002/hep.24190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita H., Mackay I.R., Van De Water J., Gershwin M.E. The lymphoid liver: considerations on pathways to autoimmune injury. Gastroenterology. 2001;120:1485–1501. doi: 10.1053/gast.2001.22441. [DOI] [PubMed] [Google Scholar]
- Kubota N., Ebihara T., Matsumoto M., Gando S., Seya T. IL-6 and IFN-alpha from dsRNA-stimulated dendritic cells control expansion of regulatory T cells. Biochem. Biophys. Res. Commun. 2010;391:1421–1426. doi: 10.1016/j.bbrc.2009.12.081. [DOI] [PubMed] [Google Scholar]
- Lafdil F., Wang H., Park O., Zhang W., Moritoki Y., Yin S., Fu X.Y., Gershwin M.E., Lian Z.X., Gao B. Myeloid STAT3 inhibits T cell-mediated hepatitis by regulating T helper 1 cytokine and interleukin-17 production. Gastroenterology. 2009;137:2125–2135. e2121–2122. doi: 10.1053/j.gastro.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leifeld L., Cheng S., Ramakers J., Dumoulin F.L., Trautwein C., Sauerbruch T., Spengler U. Imbalanced intrahepatic expression of interleukin 12, interferon gamma, and interleukin 10 in fulminant hepatitis B. Hepatology. 2002;36:1001–1008. doi: 10.1053/jhep.2002.35532. [DOI] [PubMed] [Google Scholar]
- Leo M.A., Lieber C.S. Hepatic vitamin A depletion in alcoholic liver injury. N Engl. J. MedM. 1982;307:597–601. doi: 10.1056/NEJM198209023071006. [DOI] [PubMed] [Google Scholar]
- Li J., Qiu S.J., She W.M., Wang F.P., Gao H., Li L., Tu C.T., Wang J.Y., Shen X.Z., Jiang W. Significance of the balance between regulatory T (Treg) and T helper 17 (Th17) cells during hepatitis B virus related liver fibrosis. PLoS One. 2012;7:e39307. doi: 10.1371/journal.pone.0039307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Valencia V., Rangel P., Rodriguez S., Hernandez-Munoz R. Involvement of alcohol and aldehyde dehydrogenase activities on hepatic retinoid metabolism and its possible participation in the progression of rat liver regeneration. Biochem. Pharmacol. 2007;73:586–596. doi: 10.1016/j.bcp.2006.10.021. [DOI] [PubMed] [Google Scholar]
- Marra F., Valente A.J., Pinzani M., Abboud H.E. Cultured human liver fat-storing cells produce monocyte chemotactic protein-1. Regulation by proinflammatory cytokines. J. Clin. Invest. 1993;92:1674–1680. doi: 10.1172/JCI116753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucida D., Park Y., Kim G., Turovskaya O., Scott I., Kronenberg M., Cheroutre H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- Murai M., Turovskaya O., Kim G., Madan R., Karp C.L., Cheroutre H., Kronenberg M. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat. Immunol. 2009;10:1178–1184. doi: 10.1038/ni.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan S.K., Thomas S., Ramachandran A., Pulimood A.B., Balasubramanian K.A. Retinoid metabolism during development of liver cirrhosis. Arch. Biochem. Biophys. 2005;443:93–100. doi: 10.1016/j.abb.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Nollevaux M.C., Guiot Y., Horsmans Y., Leclercq I., Rahier J., Geubel A.P., Sempoux C. Hypervitaminosis A-induced liver fibrosis: stellate cell activation and daily dose consumption. Liver Int. 2006;26:182–186. doi: 10.1111/j.1478-3231.2005.01207.x. [DOI] [PubMed] [Google Scholar]
- Radaeva S., Wang L., Radaev S., Jeong W.I., Park O., Gao B. Retinoic acid signaling sensitizes hepatic stellate cells to NK cell killing via upregulation of NK cell activating ligand RAE1. Am. J. Physiol. Gastrointest Liver Physiol. 2007;293:G809–816. doi: 10.1152/ajpgi.00212.2007. [DOI] [PubMed] [Google Scholar]
- Saiman Y., Friedman S.L. The role of chemokines in acute liver injury. Front. Physiol. 2012;3:213. doi: 10.3389/fphys.2012.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuppan D., Kim Y.O. Evolving therapies for liver fibrosis. J. Clin. Invest. 2013;123:1887–1901. doi: 10.1172/JCI66028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiegs G., Hentschel J., Wendel A. A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J. Clin. Invest. 1992;90:196–203. doi: 10.1172/JCI115836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H.X., Chuang Y.H., Li B., Wei H., Sun R., Moritoki Y., Gershwin M.E., Lian Z.X., Tian Z. CD4+ CD25+ Foxp3+ regulatory T cells protect against T cell-mediated fulminant hepatitis in a TGF-beta-dependent manner in mice. J. Immunol. 2008;181:7221–7229. doi: 10.4049/jimmunol.181.10.7221. [DOI] [PubMed] [Google Scholar]
- Xie C., Wei W., Zhang T., Dirsch O., Dahmen U. Monitoring of systemic and hepatic hemodynamic parameters in mice. J. Vis. Exp. 2014;4:e51955. doi: 10.3791/51955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.R., Chou H.S., Gu X., Wang L., Brown K.E., Fung J.J., Lu L., Qian S. Mechanistic insights into immunomodulation by hepatic stellate cells in mice: a critical role of interferon-gamma signaling. Hepatology. 2009;50:1981–1991. doi: 10.1002/hep.23202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi H.S., Lee Y.S., Byun J.S., Seo W., Jeong J.M., Park O., Duester G., Haseba T., Kim S.C., Park K.G., et al. Alcohol dehydrogenase III exacerbates liver fibrosis by enhancing stellate cell activation and suppressing natural killer cells in mice. Hepatology. 2014;60:1044–1053. doi: 10.1002/hep.27137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N., Schroppel B., Lal G., Jakubzick C., Mao X., Chen D., Yin N., Jessberger R., Ochando J.C., Ding Y., et al. Regulatory T cells sequentially migrate from inflamed tissues to draining lymph nodes to suppress the alloimmune response. Immunity. 2009;30:458–469. doi: 10.1016/j.immuni.2008.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziouzenkova O., Orasanu G., Sharlach M., Akiyama T.E., Berger J.P., Viereck J., Hamilton J.A., Tang G., Dolnikowski G.G., Vogel S., et al. Retinaldehyde represses adipogenesis and diet-induced obesity. Nat. Med. 2007;13:695–702. doi: 10.1038/nm1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.