

Abstract
Background & Aims
Gastrointestinal motility is regulated by enteric neural circuitry that includes enteric neurons and glia. Enteric glia monitor synaptic activity and exhibit responses to neurotransmitters that are encoded by intracellular calcium (Ca2+) signaling. What role evoked glial responses play in the neural regulation of gut motility is unknown. We tested how evoking Ca2+ signaling in enteric glia affects the neural control of intestinal motility.
Methods
We used a novel chemogenetic mouse model that expresses the designer receptor hM3Dq under the transcriptional control of the glial fibrillary acidic protein (GFAP) promoter (GFAP::hM3Dq mice) to selectively trigger glial Ca2+ signaling. We used in situ Ca2+ imaging and immunohistochemistry to validate this model, and we assessed gut motility by measuring pellet output and composition, colonic bead expulsion time, small intestinal transit time, total gut transit time, colonic migrating motor complex (CMMC) recordings, and muscle tension recordings.
Results
Expression of the hM3Dq receptor is confined to GFAP-positive enteric glia in the intestines of GFAP::hM3Dq mice. In these mice, application of the hM3Dq agonist clozapine-N-oxide (CNO) selectively triggers intracellular Ca2+ responses in enteric glia. Glial activation drove neurogenic contractions in the ileum and colon but had no effect on neurogenic relaxations. CNO enhanced the amplitude and frequency of CMMCs in ex vivo preparations of the colon, and CNO increased colonic motility in vivo. CNO had no effect on the composition of fecal matter, small intestinal transit, or whole gut transit.
Conclusions
Glial excitability encoded by intracellular Ca2+ signaling functions to modulate excitatory enteric circuits. Selectively triggering glial Ca2+ signaling might be a novel strategy to improve gut function in motility disorders.
Keywords: Autonomic, Chemogenetic, Enteric Nervous System, Intestine, Gut
Abbreviations used in this paper: ADP, adenosine diphosphate; BCH, bethanechol; CMMC, colonic migrating motor complex; DMEM, Dulbecco’s modified Eagle medium; DREADD, designer receptors exclusively activated by designer drugs; CNO, clozapine-N-oxide; EFS, electrical field stimulation; ENS, enteric nervous system; GFAP, glial fibrillary acidic protein; GPCR, G protein-coupled receptor; HA, hemagglutinin; LMMP, longitudinal muscle myenteric plexus; PGF2α, prostaglandin F2α; tTA, tetracycline-controlled transactivator protein; WT, wild type
Summary.
Enteric glia monitor neurotransmission in the gut but the significance of glial activity has remained unclear. We found that selectively triggering glial activity has the potential to drive excitatory neural programs that control gut motility.
Intestinal functions such as peristalsis require the coordinated activity of multiple cell types throughout the gut wall, including enteroendocrine cells, nerves, interstitial cells, and smooth muscle.1 Yet deciphering the exact contribution of individual cell types is extremely challenging because of the complex nature of tissue and the overlapping expression of many signaling molecules. Each cell type in the chain of command from mucosa to smooth muscle is clearly essential for the transduction of luminal cues into motor responses, and the ultimate integration and execution of motor patterns depends on the neural circuitry within the enteric nervous system (ENS).2 However, the exact roles of many classes of cells in the gut wall is still highly debated.3, 4, 5
One class of cells that have gained particular interest recently are the enteric glia.5 These astrocyte-like peripheral glial cells surround enteric neurons and play important roles in the maintenance of enteric neurocircuits. Indeed, enteric glia are tuned to detect neuronal activity6, 7, 8, 9, 10, 11 and express receptors for all major classes of enteric neurotransmitters.12 New data showing that enteric glia are activated by specific neural pathways13 and during physiologic patterns of ENS activity 9 raise the possibility that glial activity may be involved in the modulation of ENS circuits in addition to supporting neuronal health.14, 15, 16 In support, impairing glial functions with a metabolic toxin17 or the selective genetic ablation of glial channels involved in intercellular communication impairs gut motility.18 These studies suggest that glial functions are necessary for the maintenance of gut motility, but whether the observed changes reflect poor glial metabolic support of neurons or a change in active glial signaling is not known.
Most enteric glial receptors for neuroactive compounds are G protein-coupled receptors (GPCRs) and many of these couple to Gq and downstream intracellular signaling cascades that lead to elevations in intracellular Ca2+.8 These intracellular Ca2+ responses are largely considered central to many glial functions and are currently used as the main readout of glial activity10 but the significance of glial Ca2+ responses remains a matter of great debate.19 Thus, despite intense research investigating glial responsiveness to various mediators, major questions remain unresolved concerning the physiological significance glial Ca2+ responses and what information enteric glial Ca2+ responses encode.
New technologies have emerged in recent years that permit the activational control of individual populations of cells in complex tissue.20, 21 Importantly, these noninvasive techniques can be employed in vivo or in intact organs to assess the function of specific signaling pathways in defined cell populations such as enteric glia. Here, we used a chemogenetic approach to selectively trigger a glial Gq–GPCR signal cascade leading to intracellular Ca2+ responses22 to determine the effect of glial Ca2+-dependent signaling on the neural control of gut motility in vivo and ex vivo. Our results show that Ca2+-dependent activity within glial cells has a major effect on excitatory neuromuscular transmission in the colon and that the selective activation of Ca2+ signaling in glial cells drives neurogenic contractions. Importantly, our data provide the first conclusive evidence of active glial regulation of enteric neurotransmission. In addition, our findings raise the possibility that the selective modulation of glial cells could be a novel therapeutic approach to improve gut motility in functional gastrointestinal disorders such as slow transit constipation.
Materials and Methods
Animals
All experimental protocols were approved by the Michigan State University Institutional Animal Care and Use Committee (IACUC). The GFAP::hM3Dq transgenic mice were a gift from Dr. Ken McCarthy (University of North Carolina at Chapel Hill) and were bred for experiments as heterozygotes at Michigan State University.23 The GFAP::tTA mice (lines 67, 78, and 110) were a gift from Dr. Brian Popko24 (University of Chicago) and were bred with tetO::hM3Dq mice [Tg(tetO-CHRM3*)1Blr/J; Jackson Laboratory, Bay Harbor, MA; RRID: IMSR_JAX:014093] at Michigan State University to obtain double transgenic mice. Mice of both sexes, aged 8–12 weeks, were used for experiments and wild-type (WT) littermates served as experimental controls. Genotyping was performed by the Research Technology Support Facility at Michigan State University. The mice were maintained in a temperature-controlled environment on a 12-hour light/dark cycle, with access to tap water and regular chow ad libitum.
Whole-Mount Immunohistochemistry
Whole-mount preparations of the ileal and colonic myenteric plexus were prepared from Zamboni’s-fixed tissue and processed for immunohistochemical analysis with the antibodies shown in Table 1 as previously described elsewhere.25 Briefly, fixed tissue was pinned flat in a Sylgard (Dow-Corning, Midland, MI)-coated Petri dish, and the mucosa, submucosa, and circular muscle were removed with forceps to expose the myenteric plexus. The resulting longitudinal muscle myenteric plexus (LMMP) tissue preparations underwent three 10-minute washes in 0.1% Triton X-100 in phosphate-buffered saline (PBS-Triton) followed by a 45-minute incubation in blocking solution (containing 4% normal goat or normal donkey serum, 0.4% Triton X-100 and 1% bovine serum albumin). Preparations were incubated in primary antibodies overnight at room temperature and secondary antibodies for 2 hours at room temperature (in blocking solution) before mounting. Dual-labeling with antibodies raised in the same host was performed using a horseradish peroxidase–goat anti-rabbit IgG and Alexa Fluor 568 tyramide signal amplification kit (Life Technologies, Grand Island, NY) following the manufacturer’s instructions. Images were acquired through the 20× [PlanFluor, 0.75 numerical aperture (n.a.)] objective of an upright epifluorescence microscope (Nikon Eclipse Ni; Nikon, Melville, NY) with a Retiga 2000R camera (QImaging, Surrey, BC, Canada) controlled by QCapture Pro 7.0 (QImaging) or through the 60× (Plan-Apochromat, 1.42 n.a.) oil-immersion objective of an inverted Fluoview FV1000 confocal microscope (Olympus, Center Valley, PA).
Table 1.
Details of Primary and Secondary Antibodies
| Antibody | Source | Dilution | RRID |
|---|---|---|---|
| Primary antibodies | |||
| Goat anti-c-Kit | R&D Systems, Minneapolis, MN | 1:200 | AB_354750 |
| Chicken anti-GFAP | Abcam, Cambridge, MA | 1:1000 | AB_304558 |
| Rabbit anti-HA | Cell Signaling Technology, Danvers, MA | 1:500 | AB_1549585 |
| Invitrogen, Carlsbad, CA | 1:250 | AB_87935 | |
| Anti-human Hu C/D | Molecular Probes, Eugene, OR | 1:200 | AB_1500232 |
| Rabbit anti-S100beta | Abcam, Cambridge, MA | 1:2000 | AB_882426 |
| Secondary antibodies | |||
| Goat anti-rabbit Alexa Fluor 488 | Invitrogen | 1:400 | AB_10562715 |
| Donkey anti-goat Alexa Fluor 568 | Invitrogen | 1:400 | AB_10564097 |
| Goat anti-chicken Alexa Fluor 568 | Invitrogen | 1:400 | AB_10584483 |
| Goat anti-rabbit Alexa Fluor 568 | Invitrogen | 1:400 | AB_10563566 |
| Streptavidin conjugated Alexa Fluor 594 | Jackson ImmunoResearch, West Grove, PA | 1:400 | AB_2337250 |
RRID, Research Resource Identifiers, Antibody Registry (http://antibodyregistry.org).
Calcium Imaging
Live whole-mounts of the ileal and colonic myenteric plexus were prepared for Ca2+ imaging as described by Fried and Gulbransen.26 Briefly, distal ileal and colonic segments were collected in ice-cold Dulbecco’s modified Eagle medium (DMEM) and transferred to Sylgard-coated, open diamond shaped bath recording chambers and then opened along the mesenteric border, pinned flat, and microdissected. LMMP preparations were incubated for 15 minutes at room temperature in an enzyme mixture consisting of 150 U/mL Collagenase type II and 1 U/mL Dispase (Life Technologies) dissolved in DMEM before gentle trituration. LMMPs were loaded in the dark for 45 minutes at 37°C (5% CO2, 95% air) with 4 μM Fluo-4 AM, 0.02% Pluronic F-127 and 200 μM water-soluble Probenecid (Life Technologies) in DMEM. LMMPs were washed three times with DMEM and incubated with 200 μM probenecid in DMEM 15 minutes to de-esterify before imaging. Images were acquired every 1–2 seconds (s) through the 40× water-immersion objective (LUMPlan N, 0.8 n.a.) of an upright Olympus BX51WI fixed-stage microscope (Olympus, Tokyo, Japan) using IQ2 software and a Neo sCMOS camera (Andor, South Windsor, CT). Whole mounts were superfused with Krebs buffer (37°C) at 2–3 mL min−1.
Contractility Studies
Isometric muscle tension recordings were performed in longitudinally–oriented segments of distal colon and ileum under 1 g passive tension. Muscle strips were affixed to a force transducer (Grass Instruments, Quincy, MA) between two platinum electrodes for electrical field stimulation (EFS) and data was charted with Labscribe (iWorx, Dover, NH) as described previously elsewhere.18 Responses were normalized to an initial bethanechol (BCH, 10 μM, cholinergic muscarinic agonist)-induced contraction. Neurogenic contractions and relaxations were induced by application of EFS (20V, 0.1 milliseconds, 2–30 Hz). Neurogenic relaxations were studied in tissues precontracted with 5 μM prostaglandin F2-α (PGF2α). Relaxations were induced when the contractile response to PGF2α was stable for at least 5 minutes. Tetrodotoxin (TTX, 0.3 μM, voltage-gated sodium channel inhibitor) was applied to block neurogenic responses.
Colonic Migrating Motor Complexes
Colonic migrating motor complexes (CMMCs) were recorded from intact colons ex vivo as previously described elsewhere.27 Colons were collected in warmed media and luminal contents were gently flushed. A stainless-steel rod was inserted into the lumen, and the tissue was secured at both ends with surgical silk. Force transducers (Grass Instruments) were placed 2 cm apart and attached to the oral and aboral ends by surgical silk. Tissue was placed into a bath containing DMEM-F12 media (37°C) and adjusted to an initial tension of 0.5 g. CMMCs were recorded with LabChart 8 (ADInstruments, Colorado Springs, CO) for 20 minutes following an acclimation period and the initial 6-minute interval was used as baseline. Agonists were bath applied and CMMCs recorded for an additional 6-minute interval. CMMCs were defined as a complex in which contraction occurs first at the oral site followed by a contraction at the aboral site. Amplitude, integral, frequency, and propagation velocity were calculated as percent of baseline.
Colon Bead Assay
Distal colonic transit was assessed by measuring the latency to expel a small (2 mm diameter) plastic bead inserted 3 cm into the colon.18
Endogenous Pellet Production
Mice were individually housed, and fecal pellet output was measured on 2 consecutive days. Pellets were collected for 1 hour beginning at 9:00 AM (Zeitgeber +3). The wet weight of fecal matter was measured immediately, and the dry weight was obtained the next day after dehydration.18 Data from the 2 days was averaged.
Whole Gut Transit
Total intestinal transit time was defined as the latency from gavage of 0.2 mL of a 6% carmine red solution in H2O with 0.5% methylcellulose to the appearance of red dye in fecal pellets.18
Upper Gastrointestinal Transit
Upper gastrointestinal transit was assessed as described previously.28 Briefly, mice received a gavage of 0.2 mL of a 6% carmine red solution in H2O with 0.5% methylcellulose. Mice were euthanized 15 minutes later, and the distance travelled was measured to calculate upper gastrointestinal velocity.
Solutions
Calcium imaging experiments were performed in modified Krebs buffer consisting of (in mmol/L): 121 NaCl, 5.9 KCl, 2.5 CaCl2, 1.2 MgCl2, 1.2 NaH2PO4, 10 HEPES, 21.2 NaHCO3, 1 pyruvic acid, 8 glucose (pH adjusted to 7.4 with NaOH) with 3 μmol/L nicardipine and 1 μmol/L scopolamine to inhibit muscle contractions. Muscle contractility studies were performed in normal Krebs buffer consisting of (in mmol/L): 117 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgCl2, 1.2 NaH2PO4, 25 NaHCO3 and 11 glucose. CMMC studies were conducted in DMEM/Nutrient Mixture F-12 (Life Technologies) supplemented with l-glutamine and HEPES.
Chemicals and Reagents
Unless otherwise listed, all chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO). Clozapine-N-oxide (CNO) was obtained from the National Institute on Drug Abuse Drug Supply Program at the National Institutes of Health and bath applied at 10 μM for isolated preparations or administered via an intraperitoneal injection at 0.25 mg kg−1 for in vivo experiments. Drugs were bath applied via a gravity-fed perfusion system in Ca2+ imaging experiments and directly added to organ baths for isometric muscle tension recordings and CMMC recordings. Thus, there is a lag between drug application and cellular response in Ca2+ imaging experiments that is not present in organ baths because of the time required for the drug to reach the tissue through the perfusion system.
Statistical Analysis
Digital images were analyzed offline using ImageJ software (National Institutes of Health, Bethesda, MD). Data were analyzed using Prism 5 (GraphPad Software, La Jolia, CA) and are shown as mean ± standard error of the mean (SEM). Contractility studies were analyzed by two-way analysis of variance (ANOVA) with a Bonferroni post-test. Remaining data were analyzed by Student t test. P < .05 was considered statistically significant.
Results
Enteric glia respond to neurotransmitters6, 11 and physiologic patterns of neural activity in the ENS9 through GPCR pathways that evoke intracellular Ca2+ responses.6, 8 Yet what role, if any, glial signaling downstream of intracellular Ca2+ responses plays in the neural control of gut functions is unknown. We addressed this issue directly by using a chemogenetic approach22 to selectively trigger glial Ca2+ responses and examining the effect on neuromuscular transmission in the gut measured with in vivo and ex vivo motility assays (Figure 1A).
Figure 1.

Selective expression of hM3Dq receptors by enteric glia in the intestine of GFAP::hM3Dq mice. (A) Model of experimental paradigm used in this study. (B) Schematic of transgene containing hemagglutinin (HA)-tagged hM3Dq driven by the glial fibrillary acidic protein (GFAP) promoter. (C–G) Confocal images of dual-label immunohistochemistry for the HA-tagged hM3Dq protein (green in all panels at left) and enteric glia (C–E center; GFAP immunoreactivity and S100beta immunoreactivity shown in magenta), enteric neurons (F center; HuC/D immunoreactivity shown in magenta) or interstitial cells of Cajal (ICCs) (G center; c-Kit immunoreactivity shown in magenta) in the myenteric plexus of GFAP::hM3Dq mice. Overlays are shown in right-hand panels. The images in C and G are z-stacks and the images in D–F are a single optical slice (1 μm). The dashed line in G denotes the boundary of a myenteric ganglion that lies on top of ICCs and smooth muscle cells. Note that HA-immunoreactivity covers the surface of GFAP-immunoreactive (C, D) and S100beta-immunoreactive (E) enteric glial cells and does not localize with enteric neurons (F) or ICCs (G). Also note that no HA-immunoreactivity is present within the smooth muscle coats (G). Images are representative of labeling in a minimum of 9 animals. Scale bars in C, D: 10 μm; scale bar in C applies to panels C, E, and F; scale bar in G is 20 μm.
Functional Expression of hM3Dq “Designer Receptors Exclusively Activated by Designer Drugs” by Enteric Glia in GFAP::hM3Dq Mice
Our first goal was to establish a suitable model to selectively evoke reliable Ca2+ responses in enteric glial cells. To this end, we obtained several chemogenetic mouse lines (all transgenic) that express designer receptors exclusively activated by designer drugs (DREADDs) under transcriptional control of the glial fibrillary acidic protein (GFAP) promoter and tested their feasibility for use evoking glial Ca2+ responses in the gut. We chose lines expressing the hM3Dq variant of DREADD receptors because this mutant GPCR activates the same canonical Gq signaling pathway as endogenous glial GPCRs.6, 8
These mouse lines included one line where hM3Dq is constitutively expressed under the control of the GFAP promoter23 (Figure 1B) and three double transgenic lines that express the tetracycline-controlled transactivator protein (tTA) under regulatory control of the GFAP promoter24 and the tetracycline-responsive promoter element (TRE; tetO) upstream of the hM3Dq receptor29 (Figure 2A). In double mutant offspring of the latter “Tet-Off” system, hM3Dq transcription occurs in the absence of the tetracycline analog doxycycline. Specific details on mouse lines are provided in the Materials and Methods section. We reasoned that a suitable model should meet certain basic criteria including 1) confinement of hM3Dq receptor expression to GFAP-positive enteric glia and 2) reliable Ca2+ responses evoked in glia in response to the physiologically inert hM3Dq agonist CNO.
Figure 2.

Ectopic expression of hM3Dq receptors in the intestines of GFAP::tTA/tetO::hM3Dq transgenic mice. (A) Breeding scheme to generate double transgenic mice that express the tetracycline-controlled transactivator protein (tTA) driven by the glial fibrillary acidic protein (GFAP) promoter and the tetracycline-responsive cytomegalovirus promoter element (TRE; tetO CMV) upstream of the hM3Dq receptor. Note that glial cell hM3Dq transcription occurs in the absence of the tetracycline analog, doxycycline (dox) in these lines. (B–D) Representative epifluorescence microscopy images of immunoreactivity for GFAP (glia, green, left panels) and the hemagglutinin (HA)-tagged hM3Dq protein (grayscale, middle panels) in whole-mount preparations of myenteric plexus and the adherent longitudinal muscle from the colons of GFAP::tTA/tetO::hM3Dq transgenic mice (overlays of GFAP and HA shown in panels at right). Images are representative of labeling in a minimum of 3 animals from each of the following transgenic lines: (B) GFAP::tTA67/tetO::hM3Dq, (C) GFAP::tTA78/tetO::hM3Dq, and (D) GFAP::tTA110/tetO::hM3Dq. Scale bar in D: 60 μM, and applies to all panels.
All transgenic mouse lines tested express hM3Dq with a hemagglutinin (HA) protein tag, and we took HA immunoreactivity as acceptable evidence for hM3Dq protein expression and localization. In GFAP::hM3Dq transgenic mice, we observed robust immunoreactivity for HA-tagged hM3Dq that was confined to GFAP-immunoreactive and S100beta-immunoreactive enteric glia within the myenteric plexus of the ileum and colon (Figure 1C–E and Figure 3). HA-immunoreactivity was distributed across the surface of all observable GFAP-immunoreactive and S100beta-immunoreactive enteric glial cells and was never observed in non-GFAP-immunoreactive cells such as neurons (Figure 1F), interstitial cells of Cajal (Figure 1G), or smooth muscle cells (Figure 1G). Likewise, HA immunoreactivity was never observed in tissue from wild type (WT) littermates of GFAP::hM3Dq transgenic mice (Figure 3). In contrast to the GFAP::hM3Dq mice, we observed only weak HA-immunoreactivity within the myenteric plexus of the three lines of GFAP::tTA/tetO::hM3Dq double-transgenic mice tested, and the pattern of HA labeling more closely resembled that of nerve varicosities than glial cells (Figure 2). This likely reflects ectopic expression due to differing transgene insertion sites into the genome in these lines. These data are consistent with previous work showing that these specific lines of GFAP::tTA mice yield neuronal expression in the cerebellum, forebrain, and spinal cord.24
Figure 3.

Expression of hM3Dq receptors is confined to enteric glia in the intestines of GFAP::hM3Dq mice. (A–D) Epifluorescence images of immunoreactivity for enteric glia (glial fibrillary acidic protein [GFAP] shown in green) and the hemagglutinin (HA)-tagged hM3Dq protein (grayscale) in whole-mount longitudinal muscle myenteric plexus (LMMP) preparations from the colon (A, B) and ileum (C, D) of GFAP::hM3Dq transgenic mice (A, C) and wild-type (WT) littermates (B, D). Note that HA-immunoreactivity is confined to GFAP-immunoreactive glia in GFAP::hM3Dq mice. Images are representative of labeling in a minimum of 3 animals. Scale bar in D: 60 μM, and applies to all panels.
Next, we tested the functionality of hM3Dq receptors expressed by enteric glia by imaging intracellular Ca2+ transients evoked by the hM3Dq agonist CNO (Figure 4). Our results show that stimulation of hM3Dq with CNO (10 μM)23 in tissue from GFAP::hM3Dq mice selectively induces reliable intracellular Ca2+ responses within enteric glia (Figure 4A, C, and D and Video 1) but has no effect on cellular Ca2+ levels in tissue from WT littermates (Figure 4B). Enteric glia from GFAP::hM3Dq mice and WT littermates exhibited comparable purine-evoked Ca2+ responses (adenosine diphosphate [ADP], 100 μM, data not shown), suggesting that the expression of endogenous receptor pathways is not significantly altered in this transgenic line.
Figure 4.

Activation of glial hM3Dq receptors reliably evokes intracellular calcium (Ca2+) transients. (A, B) Representative traces of intracellular Ca2+ levels in myenteric glia in ganglia from (A) GFAP::hM3Dq transgenic mice and (B) wild-type (WT) littermates in response to the hM3Dq agonist clozapine-N-oxide (CNO, 10 μM). Each trace represents an individual glial cell and all glial cells within an individual ganglion are shown. (C) Representative traces showing that activation of glial hM3Dq receptors with CNO (blue shaded area) in GFAP::hM3Dq mice elicits intracellular Ca2+ transients in glial processes (green lines, top) and cell bodies (black lines, bottom). (D) Representative images of Fluo-4 fluorescence in a myenteric ganglion from a GFAP::hM3Dq mouse at rest (baseline, top image) and after stimulation with CNO (peak, bottom image). Scale bar: 10 μM. Traces are representative of recordings in n = 6 ganglia from at least three mice of each genotype.
Glial responses to CNO in GFAP::hM3Dq mice are characterized by large Ca2+ transients in glial cell bodies (mean peak ΔF/F = 1.35 ± 0.18, n = 60 glial cells from six ganglia, three animals) and many oscillating Ca2+ transients throughout glial processes (Figure 4C). In contrast to GFAP::hM3Dq mice, we never observed glial Ca2+ transients in response to CNO in tissue from any of the three GFAP::tTA/tetO::hM3Dq lines of mice tested (Figure 5). However, enteric glia in tissue from GFAP::tTA/tetO::hM3Dq mice still exhibited robust purine-evoked intracellular Ca2+ responses (ADP, Figure 5). Together, these results show that GFAP::hM3Dq mice are a suitable model system to study the effects of selectively evoking glial Ca2+ responses and we proceeded to use this line for all additional studies.
Figure 5.
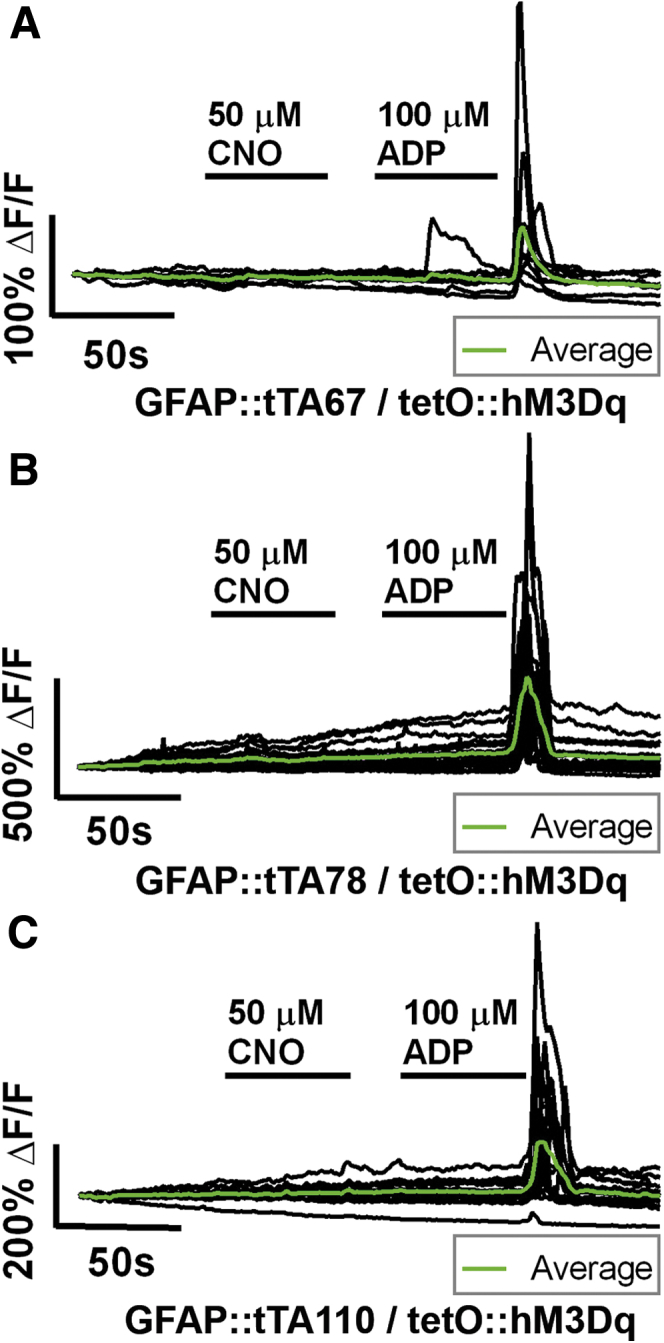
Representative calcium (Ca2+) imaging of myenteric glia in experimental lines (A) GFAP::tTA67/tetO::hM3Dq, (B) GFAP::tTA78/tetO::hM3Dq, and (C) GFAP::tTA110/tetO::hM3Dq. Each panel shows the activity of all individual glial cells (black traces) within a myenteric ganglion (averaged response of all glia within ganglion overlaid in green). In all cases, application of clozapine-N-oxide (CNO; 30 seconds, 50 μM) failed to elicit Ca2+ activity in glial cells. These same cells exhibited robust elevations in intracellular Ca2+ in response to adenosine diphosphate (ADP; 30 seconds, 100 μM).
Enteric Glial Cell Calcium Signaling Driven by Gq-G Protein-Coupled Receptors Triggers Neurogenic Smooth Muscle Contractions
Calcium responses in enteric glia are mainly reported in myenteric circuits controlling gut contractility. Thus, we initially tested whether the activation of glial Ca2+ signaling alone has any effect on intestinal contractility. To this end, we conducted isometric muscle tension recordings in longitudinally oriented segments of intestine and selectively triggered glial Ca2+ responses with CNO in tissue from GFAP::hM3Dq mice (Figure 6).
Figure 6.

Selective activation of glial calcium (Ca2+) responses elicits neurogenic contractions of intestinal smooth muscle. (A–D) Representative isometric muscle tension recordings of segments of ileum and colon from (A, B) GFAP::hM3Dq transgenic mice and (C, D) WT littermates. Note that clozapine-N-oxide (CNO; 10 μM, blue traces), bethanechol (BCH; 10 μM, black traces), and electrical field stimulation (EFS; 20 V, 0.1 ms, 20 Hz, purple traces) (A, B) elicit comparable contractions in the intestines of GFAP::hM3Dq transgenic mice and (C, D) that CNO has no effect on the contractility of tissue from wild-type (WT) littermates. Contractions elicited by CNO in GFAP::hM3Dq mice are abolished in the presence of tetrodotoxin (TTX, 0.3 μM, red dashed trace in A, B). (E, F) Summary data of contractions measured in response to CNO, BCH, and EFS in tissue from GFAP::hM3Dq transgenic mice and WT littermates (n = 5–7, *P < .05, **P < .01, two-way analysis of variance).
Application of CNO (10 μM) elicited large contractions in segments of ileum and colon from GFAP::hM3Dq mice (Figure 6A and B) but had no effect on tissue from WT littermates (Figure 6C and D). Contractions elicited by CNO were equal in magnitude to those driven by the direct stimulation of smooth muscle with bethanechol (Figure 6E and F, BCH, 10 μM) and equal in magnitude to those driven by maximal stimulation of ENS neurons with EFS (20 Hz, 20 V; Figure 6E and F). Contractions driven by CNO were entirely dependent upon neuronal activation because they were absent in the presence of tetrodotoxin (Figure 6A and B, TTX). BCH and EFS elicited equal contractions in bowel segments from GFAP::hM3Dq and WT mice, indicating that the transgenic mice do not have any gross abnormalities in the excitability of enteric neurons and/or the ability of intestinal smooth muscle to contract (Figure 6E and F).
EFS induced neurogenic relaxations in segments of ileum and colon precontracted with PGF2α (Figure 7). CNO had no effect on neurogenic relaxations in either WT or GFAP::hM3Dq mice (Figure 7). In addition, CNO did not stimulate neurogenic relaxations in the absence of EFS (Figure 7A–D). Together, these results show that glial stimulation predominantly affects excitatory motor circuits controlling gut contractions and that glial stimulation has little or no effect on inhibitory circuits regulating gut relaxations.
Figure 7.

Glial stimulation has no effect on neurogenic relaxations. (A, B) Representative traces showing neurogenic relaxations elicited by electrical field stimulation (EFS) (20 V; 0.1 ms; 20 Hz, purple traces) in prostaglandin F2α-precontracted, longitudinally oriented segments of ileum (A, C) and colon (B, D) from GFAP::hM3Dq transgenic mice (A, B) and wild-type (WT) littermates (C, D). All relaxations are abolished in the presence of tetrodotoxin (TTX, 0.3 μM, red dashed traces in A-B). Clozapine-N-oxide (CNO) alone (10 μm; blue traces in A–D) does not stimulate relaxations in precontracted segments of intestine and has no effect on neurogenic relaxations driven by EFS in WT or GFAP::hM3Dq mice (gray traces in A–D). (E, F) Summary data of relaxations measured in response to EFS in the presence and absence of CNO in tissue from GFAP::hM3Dq transgenic mice and WT littermates (n = 5–7, P > .05, two-way analysis of variance).
Stimulation of Enteric Glial Cell Calcium Signaling Enhances Colonic Migrating Motor Complexes
Fecal pellet propulsion in mice is driven by an enteric neural reflex that manifests as a rhythmic migrating motor pattern in the isolated large intestine called the colonic migrating motor complex (CMMC).30, 31 Enteric glial cell Ca2+ responses are entrained with neuronal activity during the CMMC,9 but their significance remains unclear. Therefore, we tested how the activation of glial Ca2+ signaling affects the ongoing pattern of the CMMC in the isolated colon (Figure 8).
Figure 8.

Selective activation of glial calcium (Ca2+) transients enhances the colonic migrating motor complex (CMMC) in GFAP::hM3Dq transgenic mice. Representative (A) oral and (B) aboral recordings of CMMCs in GFAP::hM3Dq mice (black traces) and WT littermates (gray traces) under basal conditions (solid lines) and after glial activation with clozapine-N-oxide (CNO) (dashed lines). (C) Model of experimental setup for CMMC recordings. (D) Frequency distribution of CMMC propagation initiation point in wild-type and GFAP::hM3Dq transgenic mice in the presence or absence of CNO (n = 3–5, two-way analysis of variance).
We observed spontaneous, regular CMMCs in both GFAP::hM3Dq transgenic mice and WT littermates as contractions migrating from proximal to distal colon that were separated by periods of quiescence. Baseline CMMC activity was comparable between GFAP::hM3Dq mice and WT littermates, and we did not observe any differences in contractile activity or overall propagation characteristics of CMMCs between genotypes in the absence of CNO. Upon application of CNO (10 μM), we observed a significant alteration in the properties of the CMMCs in GFAP::hM3Dq mice but no significant changes in the CMMCs of WT littermates (Figure 8A, B, and D). Overall, we observed a significant enhancement of all contractile parameters of the CMMC after glial activation including increased CMMC frequency (by 25.8%), propagation velocity (by 55.5%), amplitude (oral by 12.6%, aboral by 13.5%), and integral (oral by 21.4%, aboral by 17.7%) (Figure 9). These results show that glial Ca2+ signaling functions to enhance excitatory neural circuits that control the CMMC in mice.
Figure 9.

Activation of glial calcium (Ca2+) signaling enhances key aspects of colonic migrating motor complexes (CMMCs). (A–F) Summary data of CMMC characteristics including oral contraction (A) amplitude and (B) integral, (C) CMMC frequency, aboral contraction (D) amplitude and (E) integral, and (F) propagation velocity. All data are expressed as percentage change from baseline after the addition of clozapine-N-oxide. (n = 3–5, *P < .05, **P < .01, ***P < .001, two-tailed t-test).
Stimulation of Enteric Glial Cell Ca2+ Signaling Augments Gastrointestinal Motility in Vivo
Our results show that glial mechanisms downstream of Ca2+ signaling can activate neural circuits that drive neurogenic contractions of intestinal smooth muscle and that glial Ca2+ signaling enhances neural circuits that drive CMMCs. We hypothesized these effects observed in ex vivo preparations would correspond to an overall enhancement of gut motility in vivo. To this end, we triggered glial Ca2+ signaling in vivo with a single injection of CNO in GFAP::hM3Dq mice and assessed several measures of gut motility (Figure 10). After administration of CNO, we observed a nearly threefold increase in the number of fecal pellets produced per hour by GFAP::hM3Dq mice as compared to their WT littermates (Figure 10A). Despite enhancing the number of pellets produced, we found that the composition of water and fecal matter in the pellets produced remained comparable (Figure 10B).
Figure 10.

Selective activation of enteric glial calcium (Ca2+) signaling in vivo enhances gastrointestinal motility. Effect of the hM3Dq agonist clozapine-N-oxide (0.25 mg kg−1, intraperitoneal) on (A) endogenous pellet production, (B) fecal pellet composition, (C) colonic transit time, and (D) whole-gastrointestinal transit time and (E) upper gastrointestinal transit velocity in GFAP::hM3Dq transgenic mice and WT littermates (n = 4–5, **P < .01, ***P < .001, two-tailed t-test).
To more accurately measure colonic transit, we measured the latency to expel a small plastic bead inserted into the distal colon. Similar to the enhancement of endogenous pellet production, the administration of CNO decreased the time required to expel the bead by nearly two-thirds in GFAP::hM3Dq mice as compared to their WT littermates (Figure 10C). However, the activation of glial Ca2+ signaling did not significantly alter whole intestinal transit time or small intestinal transit velocity in GFAP::hM3Dq mice (Figure 10D and E). Together, these results indicate that glial excitation triggering Ca2+ signaling activates excitatory neural programs in the colon.
Discussion
Enteric glial cells have been recognized as the sole companions of neurons within enteric ganglia since the earliest studies of the ENS,32 and our current understanding of the neural control of gut reflexes stems from the assumption that there is a clear division of labor between neurons and glia. Indeed, glial cells are widely considered silent in terms of the synaptic physiology underlying gut reflexes33 and are thought to play more significant roles in the metabolic support of these neuronal circuits.2 However, recent experimental findings showing that enteric glia display a form of excitability encoded by elevations in intracellular Ca2+ suggest that this division many not be as straightforward as once thought.6, 7, 8, 9, 11 Indeed, results showing that glial Ca2+ responses are evoked by a variety of neuromodulators11 including those released by neurons during physiologic pattern of ENS activity9 have raised the controversial possibility that glial cells actively participate in information transfer in enteric circuits. Yet the outcomes of glial Ca2+ signaling have thus far remained unclear despite intense interest in observing glial Ca2+ signaling in response to various mediators.10
Our goal in the present study was to clarify the significance of evoked glial Ca2+ signaling in the regulation of intestinal motility. To this end, we used a novel transgenic chemogenetic mouse model to selectively evoke glial Gq-GPCR signaling leading to intracellular Ca2+ responses.23 Our results show that evoked glial Ca2+ signaling has a strong, excitatory effect on enteric motor circuits. Glial excitation enhanced ongoing patterns of ENS activity such as the CMMC and glial excitation per se was sufficient to drive neurogenic contractions of the intestine The latter outcome is quite astounding because it suggests that glial excitation may play an important role in the initiation of specific motor programs in the gut. Given the present results, it is conceivable that glial excitation initiated by neuronal signaling functions to ‘call up’ certain motor programs and distributes this message through the glial network. Whether the motor programs of digestion are stored in glial networks is certainly still a hypothetical question, but it will be an intriguing issue addressed in future work.
Precisely how glial Ca2+ signaling is translated into neuronal excitation in the ENS is unknown and will likely remain an active area of research for quite some time. Likewise, the downstream effects of astrocytic Ca2+ signaling remain a matter of great controversy.19 Several models have been put forth to account for the effects of glia on central networks. Given that enteric glia share many similarities with astrocytes, we speculate that enteric glia modulate enteric neuronal networks using similar mechanisms. The most promising of these include the release of “gliotransmitters”34 and the active control of extracellular potassium (K+) ion concentration.35
Either mechanism, or both, could contribute to the effects we observed upon glial activation in the gut by directly activating excitatory circuits with excitatory gliotransmitters36 or by decreasing the inhibiting tone by decreasing extracellular K+37 and hyperpolarizing inhibitory neurons. We feel that the most likely explanation for our current data is that glial stimulation drives the release of an excitatory mediator that stimulates one, or all of the following classes of myenteric neurons: excitatory ascending interneurons, excitatory motorneurons, or intrinsic primary afferent neurons. Excitation of any one or combination of these neuron classes could produce the observed result on neurogenic contractions. Importantly, we did not observe any noticeable effect of glial excitation on neurogenic relaxations. This outcome strongly suggests that glia regulate neurotransmission on a synapse-by-synapse basis and not by the diffuse release of neuroactive compounds that broadly affect all types of neurons. Thus, mechanisms downstream of glial Ca2+ signaling appear to not necessarily set the tone of neurotransmission but rather to play an active role in the modulation of specific neuronal circuits controlling the contractile aspect of gut motility. However, more work is clearly needed to decipher the exact mechanisms involved.
Interestingly, our in vivo data suggest that the role of glial Ca2+ signaling could differ between gut regions. For example, we observed a marked increase in colonic transit but no change in whole gut transit or small intestinal transit. However, our ex vivo data show that the activation of glial Ca2+ signaling evokes similar contractions in segments of ileum and colon. One explanation for these results is that there is significant glial heterogeneity along the length of the gastrointestinal tract.38, 39 Thus, it is conceivable that the different populations of enteric glia have different roles in synaptic transmission. However, a more likely explanation lies in the fact that in vivo motility patterns and neural innervation differ significantly between the ileum and colon.40 The fact that glial excitation elicited similar contractions in segments of ileum and colon in vitro strongly suggests that glial excitation has a similar effect on enteric circuits in both organs. In vivo, motility patterns in the small intestine are heavily influenced by central pathways, and the concomitant activation of enteric and central glia could confound clear results in this region of gut. In contrast, extrinsic innervation plays less of a role in colonic motility, and the effects we observe in this organ likely reflect a more pure stimulation of enteric glial cells.
In addition to differing between gut regions, the role of glial Ca2+ signaling appears to differ significantly between the myenteric and submucosal plexuses. In our hands, we did not observe a major effect of glial stimulation on the fluid content of fecal matter. In agreement, MacEachern et al41 found that the gliotoxin fluoroacetate had no effect on electrogenic ion transport in the colon. Based on their findings, these investigators concluded that enteric glia do not play a role in the regulation of electrogenic ion transport in the gut under physiologic conditions. Our current data would support this conclusion in that we show that glial Ca2+ signaling does not play a major role in the regulation of fluid exchange in the colon on an acute time scale. However, many other glial signaling mechanisms that do not rely on fluxes of intracellular Ca2+ may play important roles in the regulation of fluid exchange, and investigating these alternate signaling pathways will be important to understand the integrated function of glia in the intestine.
Together, our results provide a framework for understanding the consequences of glial excitation in the form of Ca2+ signaling in past and future work. Importantly, our results show that the activation of glia can have profound effects on gut physiology. This is particularly important when considering disorders such as chronic constipation and other functional gastrointestinal disorders where the glial network is disrupted.42 In these conditions, modifying glial activity could prove to be an extremely effective and novel therapeutic strategy to restore gut motility.
Acknowledgments
The authors thank Drs. Ken McCarthy (University of North Carolina at Chapel Hill) and Brian Popko (University of Chicago) for their generous donations of GFAP::hM3Dq and GFAP::tTA transgenic mice, respectively.
Footnotes
Conflicts of interest The authors disclose no conflicts.
Funding This study was funded by start-up funds from Michigan State University (to B.G.) and grants from the American Neurogastroenterology and Motility Society (to B.G.), the National Institutes of Health (grants HD065879 and RO1DK103723) (to B.G.), and a Senior Research Award from the Crohn’s and Colitis Foundation of America (to B.G.).
Supplementary Material
A myenteric ganglia from a GFAP::hM3Dq transgenic mouse containing enteric glia (loaded with the Ca2+ indicator Fluo-4) is challenged with the hM3Dq agonist CNO. Note the transient responses, which are limited to enteric glia within the myenteric ganglion.
{kind=link}
References
- 1.Wood J.D., Alpers D.H., Andrews P.L. Fundamentals of neurogastroenterology. Gut. 1999;45(Suppl 2):II6–II16. doi: 10.1136/gut.45.2008.ii6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Furness J.B. The enteric nervous system and neurogastroenterology. Nat Rev Gastroenterol Hepatol. 2012;9:294. doi: 10.1038/nrgastro.2012.32. [DOI] [PubMed] [Google Scholar]
- 3.Keating D.J., Spencer N.J. Release of 5-hydroxytryptamine from the mucosa is not required for the generation or propagation of colonic migrating motor complexes. Gastroenterology. 2010;138:659–670. doi: 10.1053/j.gastro.2009.09.020. 670.e1–2. [DOI] [PubMed] [Google Scholar]
- 4.Kurahashi M., Mutafova-Yambolieva V., Koh S.D. Platelet-derived growth factor receptor-α-positive cells and not smooth muscle cells mediate purinergic hyperpolarization in murine colonic muscles. Am J Physiol Cell Physiol. 2014;307:C561–C570. doi: 10.1152/ajpcell.00080.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neunlist M., Rolli-Derkinderen M., Latorre R. Enteric glial cells: recent developments and future directions. Gastroenterology. 2014;147:1230–1237. doi: 10.1053/j.gastro.2014.09.040. [DOI] [PubMed] [Google Scholar]
- 6.Gulbransen B.D., Sharkey K.A. Purinergic neuron-to-glia signaling in the enteric nervous system. Gastroenterology. 2009;136:1349–1358. doi: 10.1053/j.gastro.2008.12.058. [DOI] [PubMed] [Google Scholar]
- 7.Gomes P., Chevalier J., Boesmans W. ATP-dependent paracrine communication between enteric neurons and glia in a primary cell culture derived from embryonic mice. Neurogastroenterol Motil. 2009;21 doi: 10.1111/j.1365-2982.2009.01302.x. 870–e62. [DOI] [PubMed] [Google Scholar]
- 8.Kimball B.C., Mulholland M.W. Enteric glia exhibit P2U receptors that increase cytosolic calcium by a phospholipase C-dependent mechanism. J Neurochem. 1996;66:604–612. doi: 10.1046/j.1471-4159.1996.66020604.x. [DOI] [PubMed] [Google Scholar]
- 9.Broadhead M.J., Bayguinov P.O., Okamoto T. Ca2+ transients in myenteric glial cells during the colonic migrating motor complex in the isolated murine large intestine. J Physiol. 2012;590:335–350. doi: 10.1113/jphysiol.2011.219519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boesmans W., Martens M.A., Weltens N. Imaging neuron-glia interactions in the enteric nervous system. Front Cell Neurosci. 2013;7:183. doi: 10.3389/fncel.2013.00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boesmans W., Cirillo C., van den Abbeel V. Neurotransmitters involved in fast excitatory neurotransmission directly activate enteric glial cells. Neurogastroenterol Motil. 2013;25:e151–e160. doi: 10.1111/nmo.12065. [DOI] [PubMed] [Google Scholar]
- 12.Gulbransen B.D. Morgan & Claypool Life Sciences; San Rafael, CA: 2014. Enteric Glia; pp. 1–70. [Google Scholar]
- 13.Gulbransen B.D., Bains J.S., Sharkey K.A. Enteric glia are targets of the sympathetic innervation of the myenteric plexus in the guinea pig distal colon. J Neurosci. 2010;30:6801–6809. doi: 10.1523/JNEUROSCI.0603-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aube A.-C., Cabarrocas J., Bauer J. Changes in enteric neurone phenotype and intestinal functions in a transgenic mouse model of enteric glia disruption. Gut. 2006;55:630–637. [Google Scholar]
- 15.Abdo H.H., Derkinderen P., Gomes P. Enteric glial cells protect neurons from oxidative stress in part via reduced glutathione. FASEB J. 2010;24:1082–1094. doi: 10.1096/fj.09-139519. [DOI] [PubMed] [Google Scholar]
- 16.Abdo H.H., Mahé M.M., Derkinderen P. The omega-6 fatty acid derivative 15-deoxy-Δ12,1-prostaglandin J2 is involved in neuroprotection by enteric glial cells against oxidative stress. J Physiol. 2012;590:2739–2750. doi: 10.1113/jphysiol.2011.222935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nasser Y., Fernandez E., Keenan C.M. Role of enteric glia in intestinal physiology: effects of the gliotoxin fluorocitrate on motor and secretory function. Am J Physiol Gastrointest Liver Physiol. 2006;291:912–927. doi: 10.1152/ajpgi.00067.2006. [DOI] [PubMed] [Google Scholar]
- 18.McClain J.L., Grubišić V., Fried D. Ca2+ responses in enteric glia are mediated by connexin-43 hemichannels and modulate colonic transit in mice. Gastroenterology. 2014;146:497–507.e1. doi: 10.1053/j.gastro.2013.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Volterra A., Liaudet N., Savtchouk I. Astrocyte Ca2+ signalling: an unexpected complexity. Nat Rev Neurosci. 2014;15:327–335. doi: 10.1038/nrn3725. [DOI] [PubMed] [Google Scholar]
- 20.Sternson S.M., Roth B.L. Chemogenetic tools to interrogate brain functions. Neuroscience. 2014;37:387–407. doi: 10.1146/annurev-neuro-071013-014048. [DOI] [PubMed] [Google Scholar]
- 21.Fenno L., Yizhar O., Deisseroth K. The development and application of optogenetics. Annu Rev Neurosci. 2011;34:389–412. doi: 10.1146/annurev-neuro-061010-113817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Armbruster B.N., Li X., Pausch M.H. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci USA. 2007;104:5163–5168. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Agulhon C., Boyt K.M., Xie A.X. Modulation of the autonomic nervous system and behaviour by acute glial cell Gq protein-coupled receptor activation in vivo. J Physiol. 2013;591:5599–5609. doi: 10.1113/jphysiol.2013.261289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin W., Kemper A., McCarthy K.D. Interferon-gamma induced medulloblastoma in the developing cerebellum. J Neurosci. 2004;24:10074–10083. doi: 10.1523/JNEUROSCI.2604-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gulbransen B.D., Bashashati M.M., Hirota S.A.S. Activation of neuronal P2X7 receptor-pannexin-1 mediates death of enteric neurons during colitis. Nat Med. 2012;18:600–604. doi: 10.1038/nm.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fried D.E., Gulbransen B.D. In situ Ca2+ imaging of the enteric nervous system. J Vis Exp. 2015 doi: 10.3791/52506. e52506–e52506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Devries M.P., Vessalo M., Galligan J.J. Deletion of P2X2 and P2X3 receptor subunits does not alter motility of the mouse colon. Front Neurosci. 2010;4:22. doi: 10.3389/fnent.2010.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grubišić V., Kennedy A.J., Sweatt J.D. Pitt-Hopkins mouse model has altered particular gastrointestinal transits in vivo. Autism Res. 2015 doi: 10.1002/aur.1467. http://dx.doi.org/10.1002/aur.1467 Published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alexander G.M., Rogan S.C., Abbas A.I. Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron. 2009;63:27–39. doi: 10.1016/j.neuron.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heredia D.J., Dickson E.J., Bayguinov P.O. Colonic elongation inhibits pellet propulsion and migrating motor complexes in the murine large bowel. J Physiol. 2010;588:2919–2934. doi: 10.1113/jphysiol.2010.191445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heredia D.J., Dickson E.J., Bayguinov P.O. Localized release of serotonin (5-hydroxytryptamine) by a fecal pellet regulates migrating motor complexes in murine colon. Gastroenterology. 2009;136:1328–1338. doi: 10.1053/j.gastro.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dogiel A.S. Über den Bau der Ganglien in den Geflechten des Darmes und der Gallenblase des Menschen und der Säugetiere [article in German] Arch Anat Physiol Leipz. 1899;5:130–158. [Google Scholar]
- 33.Hanani M., Francke M., Hartig W. Patch-clamp study of neurons and glial cells in isolated myenteric ganglia. Am J Physiol Gastrointest Liver Physiol. 2000;278:G644–G651. doi: 10.1152/ajpgi.2000.278.4.G644. [DOI] [PubMed] [Google Scholar]
- 34.Cao X., Li L.-P., Wang Q. Astrocyte-derived ATP modulates depressive-like behaviors. Nat Med. 2013;19:773–777. doi: 10.1038/nm.3162. [DOI] [PubMed] [Google Scholar]
- 35.Wang F., Smith N.A., Xu Q. Astrocytes modulate neural network activity by Ca2+-dependent uptake of extracellular K+ Sci Signal. 2012;5:ra26. doi: 10.1126/scisignal.2002334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang W., Segura B.J., Lin T.R. Intercellular calcium waves in cultured enteric glia from neonatal guinea pig. Glia. 2003;42:252–262. doi: 10.1002/glia.10215. [DOI] [PubMed] [Google Scholar]
- 37.Costagliola A., Van Nassauw L., Snyders D. Voltage-gated delayed rectifier Kv1-subunits may serve as distinctive markers for enteroglial cells with different phenotypes in the murine ileum. Neurosci Lett. 2009;461:80–84. doi: 10.1016/j.neulet.2009.06.053. [DOI] [PubMed] [Google Scholar]
- 38.Boesmans W., Lasrado R., Vanden Berghe P. Heterogeneity and phenotypic plasticity of glial cells in the mammalian enteric nervous system. Glia. 2015;63:229–241. doi: 10.1002/glia.22746. [DOI] [PubMed] [Google Scholar]
- 39.Bush T.G., Savidge T.C., Freeman T.C. Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic mice. Cell. 1998;93 doi: 10.1016/s0092-8674(00)81571-8. 13–13. [DOI] [PubMed] [Google Scholar]
- 40.Grundy D., Brookes S. Morgan & Claypool Life Sciences; San Rafael, CA: 2011. Neural Control of Gastrointestinal Function; pp. 1–134. [Google Scholar]
- 41.MacEachern S.J., Patel B.A., Keenan C.M. Inhibiting inducible nitric oxide synthase in enteric glia restores electrogenic ion transport in mice with colitis. Gastroenterology. 2015;149:445–455.e3. doi: 10.1053/j.gastro.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bassotti G., Villanacci V., Maurer C.A. The role of glial cells and apoptosis of enteric neurones in the neuropathology of intractable slow transit constipation. Gut. 2006;55:41–46. doi: 10.1136/gut.2005.073197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A myenteric ganglia from a GFAP::hM3Dq transgenic mouse containing enteric glia (loaded with the Ca2+ indicator Fluo-4) is challenged with the hM3Dq agonist CNO. Note the transient responses, which are limited to enteric glia within the myenteric ganglion.