

Abstract
The clinical manifestation of metastasis in a vital organ is the final stage of cancer progression and the main culprit of cancer related mortality. Once established, metastasis is devastating, yet only a small proportion of the cancer cells that leave a tumor succeed at infiltrating, surviving, and ultimately overtaking a distant organ. The bottlenecks that challenge cancer cells in newly invaded microenvironments are organ specific and consequently demand distinct mechanisms for metastatic colonization. Here we review the metastatic traits that allow cancer cells to colonize distinct organ sites.
Keywords: Cancer, metastasis, organ specific metastasis, metastatic colonization
Organ tropism of metastatic cells
Metastasis results from disseminated cancer cells that initiate new tumors at distant organ sites. The metastatic cascade involves multiple steps, including invasion, entry into the circulation from the primary tumor, systemic dissemination, arrest and extravasation in secondary organs, settlement into latency, reactivation, outgrowth, and potential seeding of tertiary metastasis (see Box 1 for information on the early steps of the metastatic cascade) [1–3]. The pattern of affected organs is remarkably variable depending on the cancer type [1, 2, 4, 5] (Figure 1, Key Figure). Some cancer types predominantly spread to one organ (e.g. prostate cancer to bone, pancreatic cancer and uveal melanoma to liver), or show sequential organ specific colonization (e.g. colorectal cancer, CRC, frequently metastasizes first to the liver, later to lungs and brain). Other cancer types, such as breast cancer, lung cancer, or melanoma, are able to colonize many different organ sites, either sequentially or synchronously [1, 5, 6]. While defined organ tropisms are not rigid phenomena, the organ-specific patterns of metastasis are clear (Figure 1). Beyond lymph node spread, the liver, lung, bone and brain are frequently colonized by a variety of cancer types. The skin, ovaries and spleen are less common sites of metastasis. Skin metastases generally occur in melanoma and breast cancer, ovarian metastases in breast and gastric cancers, and spleen metastases almost exclusively in melanoma [5].
Box 1. The metastatic cascade.
Overt metastasis is the final manifestation of a series of stochastic events that are collectively called the “metastatic cascade”. The cascade can be parsed into distinct steps: (1) local invasion and intravasation, (2) dissemination in the circulation, (3) arrest at the distant site, (4) extravasation, (5) survival as micrometastasis, and (6) colonization of target organs. These steps have been extensively reviewed in [1, 2, 122].
Step 1. To invade from the confined primary tumor to the adjacent parenchyma, tumor cells utilize the action of a variety of extracellular proteases, including matrix metalloproteinases (MMPs) and cathepsins, which break down extracellular matrix (ECM) and trigger the release growth factors that influence tumor growth and invasion [79, 123]. The invasive front of a tumor is an important interface at which cancer and stromal cells interact closely [124]. Myeloid cells accumulate at the invasive front, generating an immunosuppressive environment. Tumor-associated macrophages and fibroblasts promote the invasion of cancer cells by producing pro-migratory factors or depositing fibrillar collagen [125–128]. Departure from a primary tumor is favored by the epithelial-to-mesenchymal transition (EMT) of cancer cells. EMT involves a loss of intercellular adhesion, epithelial polarization and the gain of mesenchymal traits [122]. In cancer cells, EMT supports self-renewal, motility and invasiveness, traits that favor metastatic dissemination [122, 129, 130]. A leaky neovasculature generated by the primary tumor contributes to easier access to the circulation.
Step 2. Cancer cells may invade and intravasate as single cells or as multi-cellular clusters and associate with non-neoplastic cells, which may enhance their survival during dissemination [120, 125, 131]. At distant organ sites, circulating tumor cells arrest in narrow capillary beds and extravasate. Rapid physical trapping due to vasculature size likely plays a major role in this process [132]. The capacity to arrest at distant organs may also be determined by specific functions of the cancer cells, e.g. by forming adhesive connections in specific organs as it has been described for breast cancer in the lung vasculature [133].
Step 3, 4. Cancer cells lodged in the microvasculature may initiate intraluminal growth and form an embolus that eventually ruptures the blood vessel or, more frequently, cancer cells may extravasate directly into the tissue parenchyma by penetrating the microvascular wall. In the bone marrow or the liver, the vasculature is fenestrated and poses a lower physical barrier than in other organs such as the lungs or the brain [1, 2]. There, the vasculature is surrounded by a tight basement membrane and additionally reinforced by pericytes and astrocytes, which requires specialized functions by the cancer cells to extravasate into the parenchyma [14, 64].
Step 5, 6. The vast majority of cancer cells that extravasate into the parenchyma will die, but a minority of these cells may enter a period of dormancy and survive for months to decades. From such disseminated tumor cell populations a few cancer cells may re-initiate growth and establish a full-fledged tumor at the distant site.
Figure I for Text Box 1.
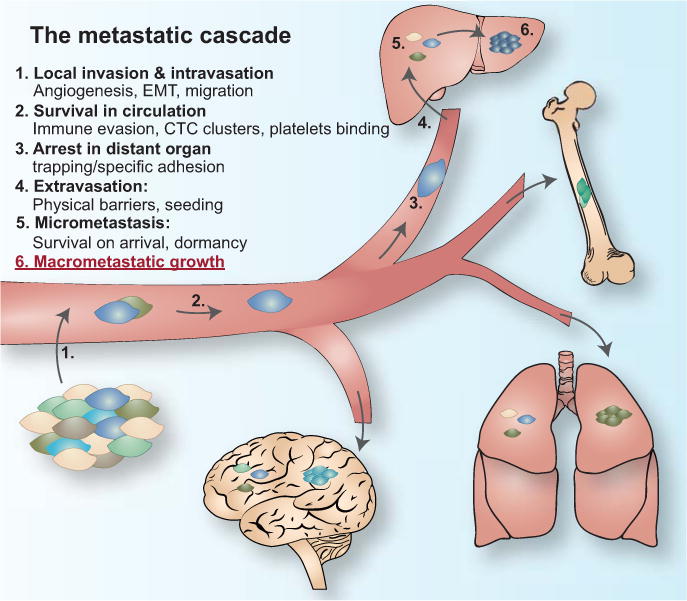
The metastatic cascade
Figure 1, Key Figure. Patterns of metastatic spread of solid tumors.
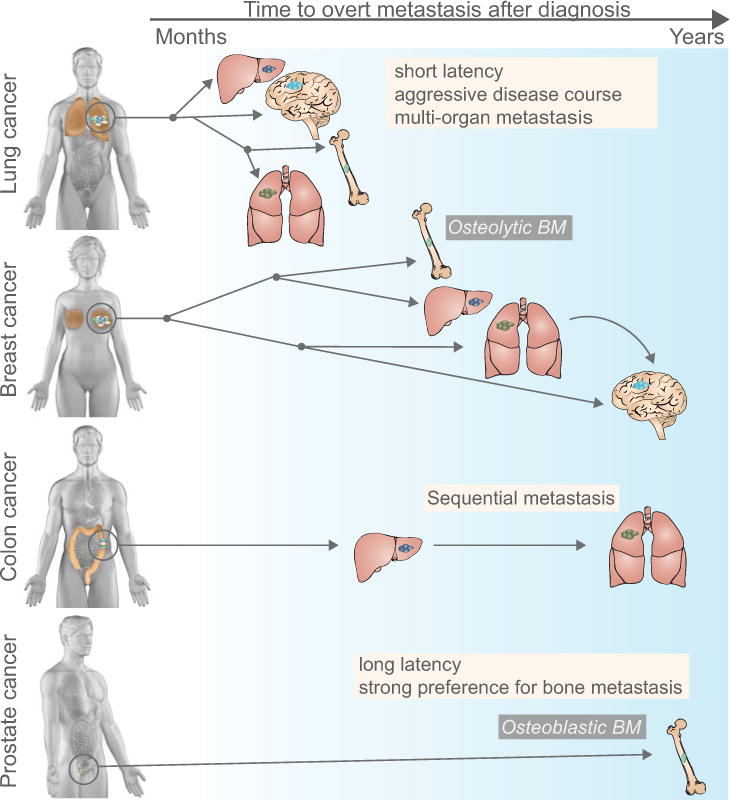
Different cancer types exhibit remarkable variability in their metastatic course, reflected in the length of the latency period (months to years), the affected organs (most commonly the liver, lung, bone, and brain) and the type of metastasis (e.g. osteolytic or osteoblastic bone metastasis). Latency period (denoted by the arrow on top of the figure, left: months, right: years after diagnosis): Lung cancer metastasis typically occurs within months after initial diagnosis, whereas prostate cancer and certain subtypes of breast cancer can produce distant relapse up to decades after initial diagnosis. Lung cancer is the main contributor to brain metastasis, a late occurrence in breast cancer. Organ pattern (most frequently affected organ is located on the top of each cancer type): Lung and breast cancers metastasize to different organs (with a different propensity), whereas colon cancer most frequently metastasizes to liver, and from established liver metastasis secondarily to lung. Prostate cancer typically, though not exclusively metastasizes to bone. Different cancer types also vary in the type of metastatic lesions they induce, well illustrated by the development of osteolytic bone metastasis in breast and lung cancer and osteoblastic bone metastasis in prostate cancer.
What determines the organ tropism of metastases? Each organ varies in its physical accessibility, vascular and nutrient supply, and stromal composition, thus placing different demands on infiltrating cancer cells [1]. The organ-specific circulation pattern and the anatomy of vessels certainly influence metastatic spread. However, this does not fully explain the organ-specific pattern of metastasis clinically observed in most cancers. For example, kidneys, liver and brain equally receive approximately 10-20% of blood volume, but each shows a very different pattern of metastasis [5]. This discrepancy between anatomy and metastasis in different organs has long been observed and forms the basis for the ‘seed and soil’ hypothesis, according to which, cancer cell seeds have intrinsic compatibilities with certain, welcoming organ microenvironment soils [7, 8]. This view is supported by recent observations of distinct cancer subtypes displaying significant variations in their organ specificity. For instance, adenocarcinoma of the lung spreads more frequently to the brain and adrenal gland than does squamous carcinoma of the lung [5]. Among different breast cancer subtypes luminal breast tumors have a higher propensity to form bone metastasis, and HER2+ breast cancer is associated with a higher frequency of liver metastases [9–11]. Nonetheless, the proportion of disseminated cancer cells that survive to achieve metastatic colonization is vanishingly low [2, 12, 13], meaning that most seeds are poorly endowed and no soil is really very welcoming.
These clinical observations are complemented by a wealth of data from experimental mouse models. These models have revealed tumor intrinsic and extrinsic mechanisms dictating organ specific metastatic progression, against a background of massive attrition of the disseminated cancer cells [13–31]. These studies support the notion that organ-specific metastasis depends not only on extrinsic factors enabling cancer cell access to organs, such as circulation patterns and vascular wall accessibility, but also on the intrinsic abilities of the metastatic cancer cells themselves. For example, intrinsic capabilities to interact with the host microenvironment allow cancer cells to cross physical barriers, survive in distant sites, engage with a distinct organ specific cell types, and eventually overtake the host tissue (Box 1).
Metastasis is above all a Darwinian selection process in which cancer cells with distinct metastatic traits that enable them to overcome metastatic bottlenecks, are being selected from a genetically and epigenetically heterogeneous tumor cell population [32, 33]. The bottlenecks that exert selective pressures are distinct at each step of the metastatic cascade. Cancer cell clones expand as a function of their ability to surpass the specific demands of each step of the metastatic cascade and continue to evolve thereafter [33–36].
General mediators of metastasis, such as those supporting invasion, ability to amplify survival pathways, or immune evasion increase the probability of cancer cells to adapt and, consequently, survive through multiple specific challenges in multiple organs. In contrast, certain genes and pathways enable passage through critical organ-specific barriers, such as crossing the blood-brain barrier, or mediate beneficial interactions with organ-specific cell types, such as the osteoclasts in the bone marrow. In addition to tumor cell autonomous traits that increase the probability of disseminated cancer cells to establish overt metastasis, the paracrine interaction with stromal cells and tumor-driven systemic processes can have a profound impact on metastasis; for instance, by stimulating the growth of distant tumor cells or by generating a pre-metastatic niche at a distant site [37]. During all stages of the metastatic cascade tumor cells enlist the help of non-neoplastic cells, extensively reviewed in [38–40]. Individually, all of these traits promote survival of individual cancer cells when facing the harsh encounter with a new organ microenvironment, and with that, these traits increase the probability of achieving clinically overt metastasis. In this review we will focus on the mechanisms that enable cancer cells to grow out and take over the distant organ.
The final stage of the metastatic cascade: organ colonization
A fine-tuned crosstalk between cancer cells and their microenvironment is required for successful colonization of a distant organ. This may be achieved by distinct mechanisms including, but not limited to, (1) evasion of the immune system or other detrimental signals that may threaten cancer cell survival; (2) interaction with stem cell niches and resident cell populations to promote survival signals in the local microenvironment; and, (3) recruitment of cell populations that modify the new host microenvironment to better match the growth requirements of the cancer cells. Mechanistic dissection of organ-specific metastatic colonization in experimental mouse models over the past decade has shed light on these organ-specific metastatic traits, the composition of permissive metastatic niches, and how complex interactions between cancer cells and their niche result in overt metastasis.
Bone metastasis
60–85% of patients with metastatic breast and prostate cancer harbor bone metastases, often resulting in pathological fractures, chronic pain, and neurological compression syndromes [41]. The small blood vessels in the bone marrow, called sinusoids, are lined with fenestrated endothelia to allow the traffic of hematopoietic cells. The bone marrow sinusoids are likely more permissive to circulating tumor cells (CTCs) than are other types of capillaries. In addition, bone matrix cells like osteoblasts secrete a variety of chemo-attracting factors (e.g. CXCL12, RANKL, OPN, or BMPs) that recruit cancer cells to the bone marrow [41, 42] (Figure 2, top).
Figure 2. Osteolytic metastatic colonization of the bone.
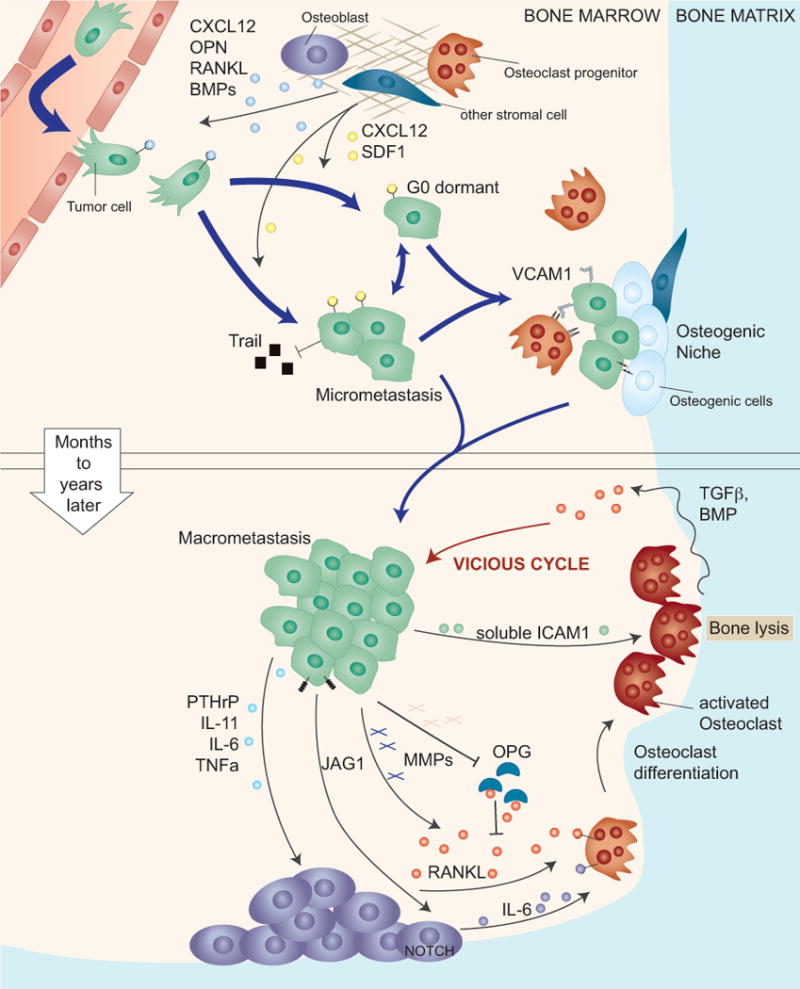
The capillaries in the bone, called sinusoids, are lined with fenestrated endothelia that facilitate the traffic of hematopoietic cells. Thus, the bone marrow sinusoids are likely permissive to cancer cell passage. Upper panel: Upon infiltrating the bone marrow cancer cells are exposed to a variety of growth and death promoting signals, which are thought to force cancer cells into a latent state until they acquire the necessary traits for overt metastasis. In this state cancer cells benefit from secreted survival signals (CXCL12, SDF1) from bone resident cells and by direct interaction with osteogenic cells and pre-osteoclasts. Lower panel: A critical step in the formation of overt osteolytic bone metastasis is the activation of osteoclasts. This process is locally facilitated by cancer cell-derived mediators including PTHrP, IL-11 and others that stimulate the secretion of RANKL by osteoblasts. Cleavage and release of membrane bound RANKL, or inactivation of the antagonist osteoprotegerin (OPG) can also contribute to increasing RANKL activity. Alternatively, cancer cells trigger the secretion of IL-6 by osteoblasts, which in turn induces osteoclast differentiation. Activated osteoclasts execute bone resorption, which releases TGF-β and other growth factors that are embedded in the mineralized bone matrix. TGF-β then further stimulates the expression of osteolytic factors in the cancer cells, resulting in a vicious cycle of bone metastasis.
After extravasation into the bone marrow, cancer cells may benefit from abundantly expressed soluble factors, such as CXCL12 and IGF1, that stimulate PI3K-AKT signaling – a pathway well known to enhance cancer cell survival in challenging environments [43] (Figure 2, top). Cancer cells with elevated Src signaling and high expression levels of CXCR4 (the receptor for CXCL12) are especially primed to utilize the physiological survival signals in the bone marrow, increasing the probability of establishing overt metastasis later on [21, 26]. In addition, high Src activity has been shown to counteract pro-apoptotic signaling of TRAIL, a cytokine also present in bone metastatic lesions [21, 44]. These findings from animal models are also reflected in clinical datasets in which CXCR4 expression and expression of the Src signature in tumor cells is associated with breast cancer bone relapse [21].
The need to find supportive niches within an organ is of importance for the survival of disseminated metastatic stem cells [45]. Cancer cells may take up residence in stem cell niches of the bone marrow. Prostate cancer cells compete with hematopoietic stem cells (HSCs) for occupancy of stem cell niches [46] and breast cancer cells can occupy osteogenic niches [47] (Figure 2, top). In these niches, cancer cells may benefit from heterotypic adherens junctions between E-cadherin on cancer cells and N-cadherin on osteogenic cells. E-cadherin expression correlates with bone metastasis in patient samples and early disruption of the adherens junctions reduces bone metastasis in mouse models [47]. Similarly, the expression of α4β1 integrin and its ligand, vascular cell adhesion molecule-1 (VCAM1), facilitates microenvironmental crosstalk in the bone marrow to promote the expansion of micrometastases in pre-clinical models [28, 48, 49].
During the final phase of overt colonization metastatic cancer cells can also actively modify the bone microenvironment in their favor, by disturbing the complex and tightly regulated network of signals that control bone homeostasis by regulating osteoblasts and osteoclasts (Figure 2, bottom). Depending on the signals released from cancer cells, bone metastases manifest as osteoblastic lesions, osteolytic lesions, or a combinations thereof [41, 50]. In osteoblastic lesions, which are typically of prostate cancer metastasis, tumor cells stimulate bone matrix deposition by osteoblasts, resulting in increased bone density and eventual displacement of the bone marrow [51]. Factors secreted by prostate cancer cells that promote osteoblastic bone metastasis include fibroblast (FGFs) insulin-like (IGFs), vascular endothelial (VEGF), and platelet derived growth factor (PDGF) as well as endothelin 1 (ET1), WNT family members, and bone morphogenetic proteins (BMPs) [41, 51–55].
In osteolytic lesions, which are caused most commonly by breast cancer and lung cancer, the metastatic cells activate bone resorbing osteoclasts, which produce collagenases and other proteases, that degrade ECM proteins and demineralize the bone matrix [41, 56]. Taking center stage in the formation of osteolytic bone metastasis is nuclear factor-kB ligand (RANKL) signaling [41]. Tumor cell-derived PTHrP, IL-11, IL-6, and TNF-α cue osteoblasts to release RANKL, which induces osteoclast formation and the subsequent resorption of the bone [41, 50]. Bone metastatic cancer cells also secrete MMPs, which increase local RANKL activity, either directly by cleaving and releasing membrane-bound RANKL [57], or indirectly, by reducing the level of the RANKL antagonist osteoprotegrin [58].
One consequence of bone resorption in osteolytic metastasis is the release of growth factors that are normally embedded in the mineralized bone matrix (Figure 2, bottom). The released growth factors then stimulate tumor growth, leading to the production of additional osteolytic and osteoblastic factors, and resulting in the ‘vicious cycle’ of bone metastasis [41, 50, 56]. TGF-β is abundant in the bone matrix and is released during osteoclastic bone resorption [41]. In breast and melanoma models, TGF-β signaling plays an essential role in the establishment of bone metastasis. TGF-β signaling is activated in bone metastasis of breast cancer patients and inhibition of the TGF-β pathway reduces bone metastasis formation in pre-clinical models [24, 59–61]. Recently, it has been shown that bone tropic prostate cancer cells also benefit from TGF-β signaling, which is further amplified by reduced levels of PMEPA1, a negative regulator of TGF-β signaling. In patients, PMEPA1 levels decreased in metastatic lesions compared with the primary tumor and low PMEPA1 levels correlated with worse prognosis [62].
Additional mechanisms that promote osteolytic bone metastasis involve the Notch ligand Jagged1 (JAG1), the expression of which is also regulated by TGF-β. JAG1 overexpression mediates bone metastasis in a human hormone receptor-negative (‘triple negative’) breast cancer cell line [24] and is associated with bone metastatic relapse in different patient cohorts [30]. In xenograft models, JAG1 promotes osteolytic bone metastasis by activating Notch signaling in osteoblasts, which induces the secretion of IL6 and directly activates osteoclast differentiation [30] (Figure 2, bottom). Osteoclast differentiation is also influenced by tumor-derived factors (e.g. soluble intracellular adhesion molecule-1, sICAM-1), which induce widespread changes in microRNA abundance [31]. In in vitro experiments osteoclast differentiation could be blocked by the ectopic expression of several microRNAs, which target osteoclast genes. In a xenograft model, the delivery of these micro RNAs inhibited osteoclast activity and reduced osteolytic bone metastasis from breast cancer cells. Clinically, serum levels of sICAM-1 and two microRNAs that were elevated during osteoclast differentiation, mir-16 and mir-378, are associated with bone metastasis burden [31]. These examples show that factors secreted by cancer cells can modulate the bone metastatic microenvironment and determine the type of bone metastases formed.
Lung metastasis
Lung metastasis is frequent in different types of cancer, including breast cancer, gastrointestinal tumors, renal carcinomas, melanoma, different types of sarcomas, and lung cancer itself [5]. Lung capillaries are lined with endothelial cells that are surrounded by a basement membrane and adjacent alveolar cells [1]. To cross these structural obstacles, breast cancer and melanoma cells express specific mediators such as SPARC and the TGF-β inducible factor angiopoietin-like 4 (ANGPTL4) and the secreted C-terminal fibrinogen-like domain of angiopoietin-like 4 (cANGPTL4) [63–65]. The expression of these mediators enhances the extravasation of tumor cells in the lung by dissociating the cell-cell junctions between endothelial cells (Figure 3). Other factors expressed by cancer cells are the EGF receptor ligand epiregulin, the prostaglandin synthase COX2, and the metalloproteinases MMP1 and MMP2, which foster the breaching of lung capillaries to seed metastasis [66]. All these mediators are up-regulated in breast tumors and their expression predicts relapse to the lungs [16, 63, 66], reinforcing the concept that metastatic traits required in early steps of the metastatic cascade are already selected for in the primary tumor, where they may play a different role in processes such as tumor angiogenesis.
Figure 3. Metastatic colonization of the lung.
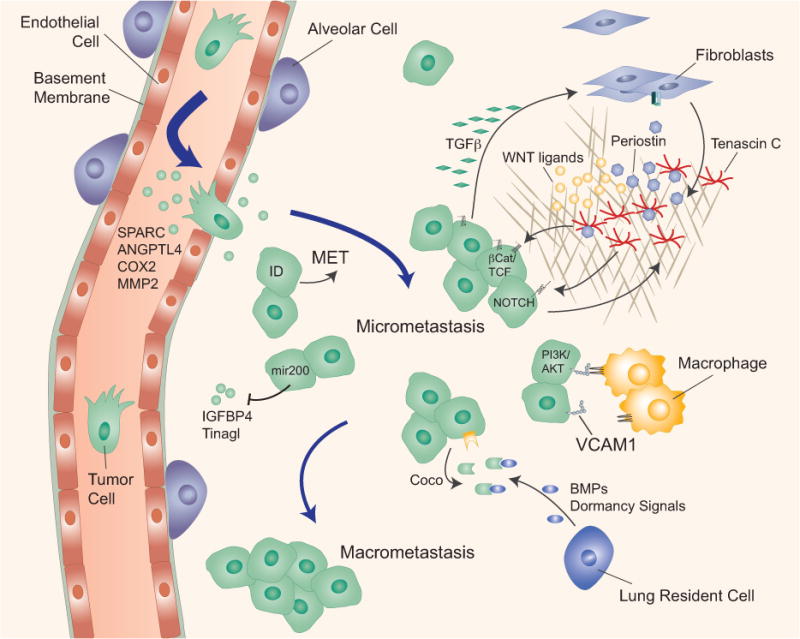
The lung capillaries are lined with a basement membrane and mediators of cancer cell extravasation in the lung have been identified (e.g. SPARC, ANGPTL4, COX2, MMP2). The expression of ID proteins in breast cancer cells induces mesenchymal-epithelial transition (MET) at the metastatic site and supports in breast cancer cells in bypassing senescence. The cancer cell-and myofibroblast-derived extracellular matrix proteins periostin (POSTN) and tenascin C (TNC) stimulate cancer cell survival by enhancing Wnt access to their receptors (periostin) or by amplifying Wnt and Notch signaling (TNC). Cancer cells express high levels of vascular cell adhesion molecule 1 (VCAM1), which tethers macrophages to cancer cells and triggers activation of the cell survival AKT pathway in cancer cells. Cancer cells overcome dormancy signals (BMPs) from lung resident cells by overexpressing Coco, which increases formation of macrometastasis.
After extravasation in the lung parenchyma, tumor-stroma interactions play a critical role in amplifying the output of survival and stemness pathways in cancer cells, consequently increasing their chances of surviving (Figure 3). In an MMTV-driven polyoma middle T (PyMT) mouse breast cancer model, lung metastatic cancer stem cells stimulate the expression of the extracellular matrix (ECM) protein periostin in lung fibroblasts via secretion of TGF-β3 [67]. Increased periostin levels recruit WNT ligands and stimulate WNT signaling preferentially in cancer stem cells, ultimately promoting lung colonization [67]. In the ECM, periostin tightly interacts with the hexameric glycoprotein tenascin C (TNC) [68]. TNC is expressed at the invasive front of tumors where it also binds to fibronectin, integrins and syndecan membrane proteoglycan, and is associated with poor prognosis and lung relapse in breast cancer patients [69]. TNC expression by fibroblasts or the tumor cells amplifies the Notch signaling output, and promotes survival of the tumor cells and their colonization of the lung [69, 70]. As the metastatic lesion grows and recruits cancer-associated fibroblasts, tumor cell derived TNC is joined by TNC from the stroma in this supportive role [69]. Lung tropic human breast cancer cells express high levels of VCAM-1, which is engaged by α4β1-integrins on tumor-associated macrophages. In xenograft models this interaction triggers VCAM-1 activation of ezrin, which subsequently enhances PI3K–Akt signaling in the cancer cells and increases their survival [71].
In addition to these interactions with the metastatic niche, cell intrinsic mechanisms are also essential for outgrowth of disseminated tumor cells. For example, the expression of inhibitor of differentiation 1 (ID1) and ID3 in breast cancer cells supports metastasis initiation after infiltration of the target parenchyma [72]. ID1 is under the control of TGF-β signaling and induces mesenchymal-epithelial transition (MET) by antagonizing the transcription factor Twist 1 at the metastatic site. Loss of ID1 dramatically reduced lung colonization in a xenograft model [73]. The microRNA mir-200 is intricately linked to the EMT-MET program and its overexpression promotes metastatic colonization of the lung. In addition to regulating E-cadherin, expression of mir-200 promotes metastatic colonization by targeting Sec23a, which regulates the secretion of metastasis suppressive proteins IGFBP4 and Tinagl1 [74]. Metastatic cells in the lungs may also have to overcome antagonistic signals from the stroma [75]. For example, BMP signals can promote differentiation of allograft breast cancer cells in the lungs. In this model, the BMP-sequestering protein Coco promotes metastatic outgrowth [76] (Figure 3).
Brain metastasis
Metastasis to the CNS principally involves the brain parenchyma and the leptomeninges, and it has a particularly poor prognosis with high morbidity and mortality. The median survival of patients with brain metastasis is in the order of months and few effective treatments are currently available [77]. More than half of brain metastasis derives from lung adenocarcinoma, followed by breast cancer and melanoma [5]. To enter the brain parenchyma, cancer cells must traverse micro-capillary walls that constitute the blood-brain barrier, which consists of tightly adjoined endothelial cells that are lined by a basement membrane, pericytes, and astrocyte foot processes [78]. To cross this barrier and access the brain parenchyma, cancer cells require specialized mechanisms. Some of the molecular mediators of this process have been identified, including the sialyltransferase ST6GalNac5, COX2, HB-EGF, MMP-2, mir-105, and the protease cathepsin S [14, 79, 80] (Figure 4).
Figure 4. Metastatic colonization of the brain.
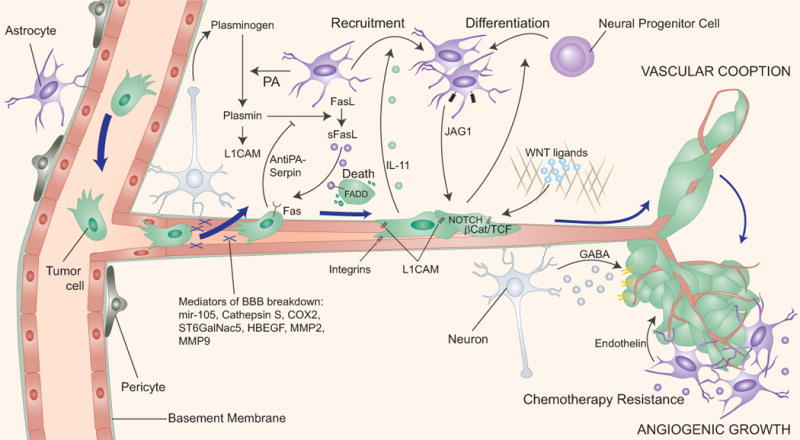
To establish parenchymal brain metastasis cancer cells have to cross the vascular walls that constitute the blood brain barrier (BBB), which consists of tightly connected endothelial cells lined with a basement membrane and contacting astrocyte and pericytes. Several classes of mediators of cancer cell passage through the BBB have been identified (mir-105, Cathepsin S, COX2, ST6GalNac5, HBEGF, MMP2, MMP9). Cancer cells express high levels of anti-PA serpins which prevent the release of cytotoxic soluble Fas-ligand (sFasl) from reactive astrocytes and the inactivation of the L1CAM adhesion molecule that mediates vascular cooption by the cancer cells. Once cancer cells evade astrocyte-mediated killing they can take advantage of astrocyte-derived survival and chemo-protective functions of largely unknown nature. Cancer cells may stimulate the accumulation of astrocytes in metastatic lesions. Cancer cells may also utilize neuron-secreted GABA as support for metastasis.
Metastatic colonization of the brain proceeds with a close apposition of cancer cells at the abluminal side of the microcapillaries [81, 82]. Small lesions often develop without establishing new vasculature [83]. Recently, lung cancer and breast cancer metastatic cells were shown to express the cell adhesion molecule L1CAM for spreading on the basement membrane of brain capillaries after extravasation into the brain parenchyma (Figure 4). Brain metastatic cells also produce specific serpin protease inhibitors to prevent L1CAM cleavage by astrocyte-derived plasminogen activator (PA) [84]. High expression of SerpinB2 and Neuroserpin correlates with lower brain metastasis-free survival in patients with lung adenocarcinoma [84]. Integrins also play a critical role in mediating brain metastatic cell spreading and angiogenesis [85, 86]. Human lung adenocarcinoma and murine myeloma cells that reached mouse brain tissue and failed to establish required β1 integrin mediated adhesion to the vascular basement membrane were less efficient at forming overt brain metastasis [87, 88].
Metastatic cancer cells encounter a variety of resident cell types in the brain parenchyma. Astrocytes can provide a growth-permissive microenvironment to infiltrated cancer cells, first, however, cancer cells must evade astrocyte induced cell death. In xenograft models of brain metastasis, activated astrocytes overexpress the pro-apoptotic cytokine Fas ligand (FasL) and release it from a membrane-anchored form by the action of PA to kill infiltrated metastatic cells. Brain metastatic cells express anti-PA serpins that shield cancer cells from PA-released FasL [84]. The surviving cancer cells can induce astrocytes to establish Notch signaling [25] and endothelin production, which favor metastasis in experimental systems [89, 90]. Conversely, cancer cells can increase the density of astrocytes by promoting the differentiation of neural progenitor cells towards the astrocyte lineage [91] (Figure 4).
The contribution of other brain cells, including oligodendrocytes, pericytes, microglia, or neurons, is less defined. Although brain metastatic cells need to neutralize cytotoxic microglia signals, microglia infiltration correlates with metastatic progression [81, 92]. Brain metastatic cells may up-regulate GABA transporters and utilize neuron-released GABA neurotransmitter as a metabolite, supporting outgrowth in the brain [93] (Figure 4).
The WNT pathway was identified to support colonization of brain and bone by KRAS-mutant and EGFR-mutant human lung adenocarcinoma cells [20]. Clinically, a specific WNT responsive gene signature is associated with metastatic relapse in lung adenocarcinoma patients [20]. Two WNT regulated genes, Lef1 and HoxB9, were specifically implicated in metastatic cell invasiveness and colony formation [20]. Brain metastases in patients show up-regulation of WNT target genes [94].
Liver metastasis
The liver is the most common site of distant metastasis in solid tumors. Gastrointestinal cancers such as CRC, pancreatic cancer and tumors of the gallbladder, which are drained by the enterohepatic circulation, reach the liver first. As such, the liver provides a large number of cancer cells with ample opportunity to arrest, extravasate, and colonize the hepatic parenchyma [22]. Indeed, a recent study found a higher number of circulating CRC cells in the portal venous blood than in the peripheral blood, suggesting that a significant percentage of tumor cells are trapped in the liver [95]. Other primary tumors that metastasize to the liver include lung and breast cancers [4, 5]. Uveal (ocular) melanoma almost exclusively relapses to the liver, providing a clear indication that, beyond circulation patterns, certain compatibilities of metastatic cells with the host stroma also count in organ-specific metastasis [96].
In contrast to the vessels in the brain or the lungs, the hepatic vasculature is fenestrated (sinusoidal endothelium) and lacks an organized sub-endothelial basement membrane. Therefore, cancer cell extravasation is less restricted in the liver than it is in the brain or the lungs, as shown in quantitative cell-tracking studies in mice [12]. However, the liver parenchyma is rich in cells of the innate immune system, potentially posing a obstacle to cancer cells. Indeed, the neutralization of pro-apoptotic TRAIL on resident natural killer cells in the liver increases experimental metastasis [97] (Figure 5).
Figure 5. Metastatic colonization of the liver.
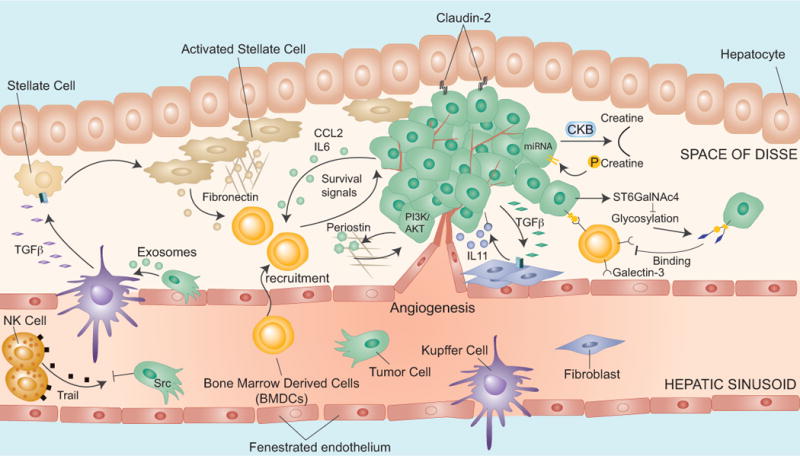
The extravasation into the liver is facilitated by the hepatic vascular endothelium, which is fenestrated and lacks an organized basement membrane. High Src signaling protects cancer cells from TRAIL-mediated apoptosis. Cancer cells release MIF containing exosomes that trigger TGF-β production for the activation of stellate cells, leading to the recruitment of bone marrow derived cells (BMDCs). BMDCs can also be attracted by secretion of CCL2 and IL6. Other survival signals may be provided by galectin-3. In the liver, colon cancer cells secrete periostin, which induces PI3K/AKT signaling. Cancer cells also interact with hepatocytes via claudin-2, stimulating overt metastasis. The secretion of creatine kinase B (CKB) by cancer cells contributes to metastatic outgrowth by generating phosphocreatine as a metabolite to regenerate ATP in cancer cells.
Certain liver parenchyma cell types favor metastatic outgrowth (Figure 5). In experimental models, claudin-2-mediated cell-cell interactions between breast cancer cells and hepatocytes led to induction of c-Met and stimulated metastasis to the liver [98]. In an allograft model, exosome vesicles released by murine pancreatic ductal adenocarcinoma cells caused TGF-β secretion, stimulated fibronectin production by hepatic stellate cells, and triggered a recruitment of bone-marrow derived macrophages [99]. The macrophage migration inhibitory factor (MIF) was highly enriched in murine and human pancreatic cancer exosomes, and its blockade inhibited metastasis in the mice [99] (Figure 5). CRC and lung cancer cells mobilize myeloid cell populations through soluble factors, such as CCL2 or IL6, that promote liver metastasis [100, 101] (Figure 5).
Certain cancer cells express the glycosyltransferases St6GalnAc4 and C2GnT2, which alter the glycosylation of a Galectin-3 ligand on tumor cells and thereby increase interaction with galectin-3 expressed on myeloid cells [101] (Figure 5). Clinically, aberrant glycosylation and high galectin-3 levels are associated with metastatic progression [101, 102]. Another significant case of metastatic interaction with the hepatic microenvironment is provided by CRC stem cells. The cells are often unresponsive to TGF-β owing to mutations that disable the TGF-β receptors or the SMAD signal transducers proteins. However, these cells abundantly secrete TGF-β, which enhances metastasis formation in the liver by activating a paracrine loop with production of IL-11 from stromal fibroblasts. IL-11 then activates STAT3 signaling in CRC stem cells to support their survival in the liver [27] (Figure 5).
Proliferating cancer cells in the liver have high biosynthetic demands and compete with hepatocytes for glycolytic substrates. In vivo screens identified two microRNAs (miR-551a and miR-483) that were down regulated in liver-tropic CRC cells leading to increased expression of the creatine kinase brain-type (CKB) [103]. The cancer cells benefit from high levels of creatine in the liver, which CKB coverts into phosphocreatine that the CRC import for their bioenergetic needs [103] (Figure 5).
Concluding remarks
Genomics and other systems level approaches combined with extensive work in experimental models have started to shed light on the traits of cancer cells, the composition of stromal niches, and the interaction between cancer cells and these niches that increase the probability of overt colonization of a specific organ by cancer cells from different tumors of origin. However, many questions remain open (Outstanding questions).
The manifestation of organ specific metastasis can take months to decades and is the result of multiple different traits that each provide a small advantage to individual cancer cells to survive and thrive. Despite physical barriers that need to be overcome, the arrival in a distant organ does not seem to be the most limiting factor. Tumor cells can be found in the blood in early-stage cancer [104], in some cases even before tumors are overtly invasive [105–107]. Notably, even non-transformed epithelial stem cells are able to infiltrate and survive in the lung when injected in large numbers in the circulation [108]. It remains unknown whether cancer cells that leave the primary site very early during tumor progression are able to initiate clinically manifest metastasis or whether cancer cells that leave the primary tumor later during tumor progression stand a better chance.
Disseminated tumor cells can survive for decades after surgical resection of a tumor. Latent cancer cell populations may reside in specialized protective niches in the bone marrow or in other organs, as suggested by cases in which recipients of liver, kidney or heart transplant developed metastasis from dormant cancer cells carried by the donor’s organ [109–111]. The identification of mediators of cancer cell survival during metastatic dormancy is of interest as targeting such mediators with adjuvant therapy could prevent overt metastasis. However, the most limiting step of the metastatic cascade appears to be the transition from infiltration of an organ to overt colonization, which involves the evasion of organ-derived detrimental signals and the exploitation of organ-derived survival signals.
Several of the mediators of organ specific colonization, such as those involved in cancer cell interactions with osteoblasts and osteoclasts, are only relevant to metastasis in that particular organ site. However, many mediators of metastasis that were identified in studies on one or another organ are not necessarily restricted to that particular organ. For example, periostin was originally implicated in lung metastasis by breast cancer cells [67] but is also utilized by CRC cells for liver metastasis [112]. Similarly, VCAM1 gives tumor cells in the lung a distinct survival advantage by fostering interactions with macrophages [71], whereas in the bone marrow VCAM1 mediates the interaction of tumor cells with myeloid osteoclast progenitors promoting their osteolytic expansion [28]. Other examples are COX2 and MMP1, which were initially shown to be mediators of breast cancer cell extravasation in the lungs but also play a role in extravasation through the blood-brain barrier [14, 16, 22, 113, 114].
Tumor heterogeneity, cancer cell plasticity, and complex cooperations between different cancer clones provide additional challenges in the modeling, interpretation and therapeutic intervention of metastatic cancer [115, 116]. Already at the primary site different cancer cell clones may cooperate to sustain the growth of the tumor [117, 118], and crosstalk between tumor cells stimulates metastasis [119]. Collective invasion of multicellular clusters increases survival and metastatic efficiency of disseminated tumor cells in preclinical models [120], and multiclonal seeding has been detected in prostate cancer patients [35, 36].
Each of the steps of the metastatic cascade poses natural vulnerabilities of the cancer cells that could be targeted to prevent overt metastasis and to improve the outcome of patients with metastatic cancer. In a therapeutic setting, signals released by cancer cells under the stress of targeted kinase therapy stimulate the proliferation and dissemination of drug-resistant cancer cell minorities [121]. It is possible that the mechanisms that provide a survival benefit during the crucial steps of metastasis may also increase the survival of cancer cells during drug treatment, thus contributing to therapy resistance and disease progression. Future research must be directed to identifying the most critical mediators of metastatic colonization as therapeutic targets. The most valuable of these targets might well be those that mediate not organ-specific metastasis, but multi-organ metastasis.
Trends Box.
The pattern of affected organs in metastasis is variable depending on the tumor of origin, indicating that intrinsic cancer cell traits, the physical accessibility of target organs, and the composition of host organ microenvironments are important determinants of distant metastasis.
Metastasis is an inefficient process whereby few cells succeed at reestablishing a tumor at a distant organ.
Organ specific metastasis involves cancer cell interactions with the host microenvironment including activation of paracrine cytokine loops, modification of the host cellular composition, and alteration of extracellular matrix structures.
Immune cell evasion, association with a supportive niche, and the ability to amplify survival pathways, often achieved through interaction with the stroma, are essential for successful metastatic colonization.
Outstanding questions.
When and where do the traits for organ specific metastasis arise - in the primary tumor or at the distant organ site?
What is the origin of these metastatic traits - genetic or epigenetic?
Do metastatic cells utilize different niches for their initial survival on arrival, for dormancy, and for aggressive outgrowth?
What gives cancer cells the ability to enter a dormant state up to several years, while retaining tumor-initiating capacity?
What are the signals that allow cancer cells to exit dormancy and reactivate their proliferative programs?
How do cancer cells acquire metastatic traits for organ colonization during the dormant state?
Are organs that serve as sanctuary sites for dormant metastatic cells the same organs in which overt metastasis eventually emerges?
Are the mechanisms that support the survival of cancer cells after extravasation related to those that support the survival of residual cancer cells under anti-cancer therapy?
What is the basis for the notorious drug resistance of metastatic cells in distant organ microenvironments, such as the brain?
Would therapeutic targeting of the mechanisms that specifically support the survival of dormant metastatic cells prove an efficient strategy to prevent metastasis?
Acknowledgments
We thank T. Wiesner, D. Macalinao, K. Ganesh, A. Laughney and S. Malladi for useful input. A.O. was an Erwin Schroedinger Fellowship awardee (J3013, FWF, Austrian Science Fund). J.M. was supported by the NIH (CA163167 and CA129243), the Congressionally Directed Medical Research Program of the Department of Defense DOD W81XWH-12-1-0074, the Cancer Center Support Grant P30 CA008748 and the MSKCC Metastasis Research Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature
- 1.Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9(4):274–84. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 2.Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147(2):275–92. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12(8):895–904. doi: 10.1038/nm1469. [DOI] [PubMed] [Google Scholar]
- 4.Disibio G, French SW. Metastatic patterns of cancers: results from a large autopsy study. Arch Pathol Lab Med. 2008;132(6):931–9. doi: 10.5858/2008-132-931-MPOCRF. [DOI] [PubMed] [Google Scholar]
- 5.Budczies J, et al. The landscape of metastatic progression patterns across major human cancers. Oncotarget. 2015;6(1):570–83. doi: 10.18632/oncotarget.2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Urosevic J, et al. Colon cancer cells colonize the lung from established liver metastases through p38 MAPK signalling and PTHLH. Nat Cell Biol. 2014;16(7):685–94. doi: 10.1038/ncb2977. [DOI] [PubMed] [Google Scholar]
- 7.Paget S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1989;8(2):98–101. 1889. [PubMed] [Google Scholar]
- 8.Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3(6):453–8. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 9.Kennecke H, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol. 2010;28(20):3271–7. doi: 10.1200/JCO.2009.25.9820. [DOI] [PubMed] [Google Scholar]
- 10.Soni A, et al. Breast cancer subtypes predispose the site of distant metastases. Am J Clin Pathol. 2015;143(4):471–8. doi: 10.1309/AJCPYO5FSV3UPEXS. [DOI] [PubMed] [Google Scholar]
- 11.Smid M, et al. Subtypes of breast cancer show preferential site of relapse. Cancer Res. 2008;68(9):3108–14. doi: 10.1158/0008-5472.CAN-07-5644. [DOI] [PubMed] [Google Scholar]
- 12.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2(8):563–72. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 13.Kienast Y, et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med. 2010;16(1):116–22. doi: 10.1038/nm.2072. [DOI] [PubMed] [Google Scholar]
- 14.Bos PD, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459(7249):1005–9. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tavazoie SF, et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008;451(7175):147–52. doi: 10.1038/nature06487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Minn AJ, et al. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J Clin Invest. 2005;115(1):44–55. doi: 10.1172/JCI22320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vanharanta S, et al. Loss of the multifunctional RNA-binding protein RBM47 as a source of selectable metastatic traits in breast cancer. Elife. 2014;3 doi: 10.7554/eLife.02734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vanharanta S, et al. Epigenetic expansion of VHL-HIF signal output drives multiorgan metastasis in renal cancer. Nat Med. 2013;19(1):50–6. doi: 10.1038/nm.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim MY, et al. Tumor self-seeding by circulating cancer cells. Cell. 2009;139(7):1315–26. doi: 10.1016/j.cell.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nguyen DX, et al. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell. 2009;138(1):51–62. doi: 10.1016/j.cell.2009.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang XH, et al. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell. 2009;16(1):67–78. doi: 10.1016/j.ccr.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gupta GP, et al. Identifying site-specific metastasis genes and functions. Cold Spring Harb Symp Quant Biol. 2005;70:149–58. doi: 10.1101/sqb.2005.70.018. [DOI] [PubMed] [Google Scholar]
- 23.Cheung WK, et al. Control of alveolar differentiation by the lineage transcription factors GATA6 and HOPX inhibits lung adenocarcinoma metastasis. Cancer Cell. 2013;23(6):725–38. doi: 10.1016/j.ccr.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang Y, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3(6):537–49. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 25.Xing F, et al. Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating Notch signalling in brain. EMBO Mol Med. 2013;5(3):384–96. doi: 10.1002/emmm.201201623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Myoui A, et al. C-SRC tyrosine kinase activity is associated with tumor colonization in bone and lung in an animal model of human breast cancer metastasis. Cancer Res. 2003;63(16):5028–33. [PubMed] [Google Scholar]
- 27.Calon A, et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22(5):571–84. doi: 10.1016/j.ccr.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu X, et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging alpha4beta1-positive osteoclast progenitors. Cancer Cell. 2011;20(6):701–14. doi: 10.1016/j.ccr.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cox TR, et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature. 2015 doi: 10.1038/nature14492. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Sethi N, et al. Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell. 2011;19(2):192–205. doi: 10.1016/j.ccr.2010.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ell B, et al. Tumor-induced osteoclast miRNA changes as regulators and biomarkers of osteolytic bone metastasis. Cancer Cell. 2013;24(4):542–56. doi: 10.1016/j.ccr.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481(7381):306–13. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Naxerova K, Jain RK. Using tumour phylogenetics to identify the roots of metastasis in humans. Nat Rev Clin Oncol. 2015;12(5):258–272. doi: 10.1038/nrclinonc.2014.238. [DOI] [PubMed] [Google Scholar]
- 34.Klein CA. Selection and adaptation during metastatic cancer progression. Nature. 2013;501(7467):365–72. doi: 10.1038/nature12628. [DOI] [PubMed] [Google Scholar]
- 35.Hong MK, et al. Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat Commun. 2015;6:6605. doi: 10.1038/ncomms7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gundem G, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520(7547):353–7. doi: 10.1038/nature14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McAllister SS, Weinberg RA. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol. 2014;16(8):717–27. doi: 10.1038/ncb3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kitamura T, Qian BZ, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol. 2015;15(2):73–86. doi: 10.1038/nri3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–37. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–22. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 41.Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nat Rev Cancer. 2011;11(6):411–25. doi: 10.1038/nrc3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones DH, et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature. 2006;440(7084):692–6. doi: 10.1038/nature04524. [DOI] [PubMed] [Google Scholar]
- 43.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 44.Xu J, et al. Regulation of the Src-PP2A interaction in tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-induced apoptosis. J Biol Chem. 2013;288(46):33263–71. doi: 10.1074/jbc.M113.508093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oskarsson T, Batlle E, Massague J. Metastatic stem cells: sources, niches, and vital pathways. Cell Stem Cell. 2014;14(3):306–21. doi: 10.1016/j.stem.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shiozawa Y, et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest. 2011;121(4):1298–312. doi: 10.1172/JCI43414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang H, et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell. 2015;27(2):193–210. doi: 10.1016/j.ccell.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Michigami T, et al. Cell–cell contact between marrow stromal cells and myeloma cells via VCAM-1 and alpha(4)beta(1)-integrin enhances production of osteoclast-stimulating activity. Blood. 2000;96(5):1953–60. [PubMed] [Google Scholar]
- 49.Matsuura N, et al. Induction of experimental bone metastasis in mice by transfection of integrin alpha 4 beta 1 into tumor cells. Am J Pathol. 1996;148(1):55–61. [PMC free article] [PubMed] [Google Scholar]
- 50.Guise TA, et al. Basic mechanisms responsible for osteolytic and osteoblastic bone metastases. Clin Cancer Res. 2006;12(20 Pt 2):6213s–6216s. doi: 10.1158/1078-0432.CCR-06-1007. [DOI] [PubMed] [Google Scholar]
- 51.Logothetis CJ, Lin SH. Osteoblasts in prostate cancer metastasis to bone. Nat Rev Cancer. 2005;5(1):21–8. doi: 10.1038/nrc1528. [DOI] [PubMed] [Google Scholar]
- 52.Chiang AC, Massague J. Molecular basis of metastasis. N Engl J Med. 2008;359(26):2814–23. doi: 10.1056/NEJMra0805239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guise TA, Yin JJ, Mohammad KS. Role of endothelin-1 in osteoblastic bone metastases. Cancer. 2003;97(3 Suppl):779–84. doi: 10.1002/cncr.11129. [DOI] [PubMed] [Google Scholar]
- 54.Hall CL, et al. Prostate cancer cells promote osteoblastic bone metastases through Wnts. Cancer Res. 2005;65(17):7554–60. doi: 10.1158/0008-5472.CAN-05-1317. [DOI] [PubMed] [Google Scholar]
- 55.Dai J, et al. Vascular endothelial growth factor contributes to the prostate cancer-induced osteoblast differentiation mediated by bone morphogenetic protein. Cancer Res. 2004;64(3):994–9. doi: 10.1158/0008-5472.can-03-1382. [DOI] [PubMed] [Google Scholar]
- 56.Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2(8):584–93. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 57.Lynch CC, et al. MMP-7 promotes prostate cancer-induced osteolysis via the solubilization of RANKL. Cancer Cell. 2005;7(5):485–96. doi: 10.1016/j.ccr.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 58.Lu X, et al. ADAMTS1 and MMP1 proteolytically engage EGF-like ligands in an osteolytic signaling cascade for bone metastasis. Genes Dev. 2009;23(16):1882–94. doi: 10.1101/gad.1824809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Juarez P, Guise TA. TGF-beta in cancer and bone: implications for treatment of bone metastases. Bone. 2011;48(1):23–9. doi: 10.1016/j.bone.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 60.Yin JJ, et al. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest. 1999;103(2):197–206. doi: 10.1172/JCI3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Javelaud D, et al. Stable overexpression of Smad7 in human melanoma cells impairs bone metastasis. Cancer Res. 2007;67(5):2317–24. doi: 10.1158/0008-5472.CAN-06-3950. [DOI] [PubMed] [Google Scholar]
- 62.Fournier PG, et al. The TGF-beta Signaling Regulator PMEPA1 Suppresses Prostate Cancer Metastases to Bone. Cancer Cell. 2015 doi: 10.1016/j.ccell.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Padua D, et al. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell. 2008;133(1):66–77. doi: 10.1016/j.cell.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tichet M, et al. Tumour-derived SPARC drives vascular permeability and extravasation through endothelial VCAM1 signalling to promote metastasis. Nat Commun. 2015;6:6993. doi: 10.1038/ncomms7993. [DOI] [PubMed] [Google Scholar]
- 65.Huang RL, et al. ANGPTL4 modulates vascular junction integrity by integrin signaling and disruption of intercellular VE-cadherin and claudin-5 clusters. Blood. 2011;118(14):3990–4002. doi: 10.1182/blood-2011-01-328716. [DOI] [PubMed] [Google Scholar]
- 66.Gupta GP, et al. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature. 2007;446(7137):765–70. doi: 10.1038/nature05760. [DOI] [PubMed] [Google Scholar]
- 67.Malanchi I, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2012;481(7379):85–9. doi: 10.1038/nature10694. [DOI] [PubMed] [Google Scholar]
- 68.Kii I, et al. Incorporation of tenascin-C into the extracellular matrix by periostin underlies an extracellular meshwork architecture. J Biol Chem. 2010;285(3):2028–39. doi: 10.1074/jbc.M109.051961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oskarsson T, et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med. 2011;17(7):867–74. doi: 10.1038/nm.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.O’Connell JT, et al. VEGF-A and Tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proc Natl Acad Sci U S A. 2011;108(38):16002–7. doi: 10.1073/pnas.1109493108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen Q, Zhang XH, Massague J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell. 2011;20(4):538–49. doi: 10.1016/j.ccr.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gupta GP, et al. ID genes mediate tumor reinitiation during breast cancer lung metastasis. Proc Natl Acad Sci U S A. 2007;104(49):19506–11. doi: 10.1073/pnas.0709185104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stankic M, et al. TGF-beta-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep. 2013;5(5):1228–42. doi: 10.1016/j.celrep.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Korpal M, et al. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med. 2011;17(9):1101–8. doi: 10.1038/nm.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer. 2014;14(9):611–22. doi: 10.1038/nrc3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gao H, et al. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell. 2012;150(4):764–79. doi: 10.1016/j.cell.2012.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Eichler AF, et al. The biology of brain metastases-translation to new therapies. Nat Rev Clin Oncol. 2011;8(6):344–56. doi: 10.1038/nrclinonc.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7(1):41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 79.Sevenich L, Joyce JA. Pericellular proteolysis in cancer. Genes Dev. 2014;28(21):2331–47. doi: 10.1101/gad.250647.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhou W, et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell. 2014;25(4):501–15. doi: 10.1016/j.ccr.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fitzgerald DP, et al. Reactive glia are recruited by highly proliferative brain metastases of breast cancer and promote tumor cell colonization. Clin Exp Metastasis. 2008;25(7):799–810. doi: 10.1007/s10585-008-9193-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Leenders WP, Kusters B, de Waal RM. Vessel co-option: how tumors obtain blood supply in the absence of sprouting angiogenesis. Endothelium. 2002;9(2):83–7. doi: 10.1080/10623320212006. [DOI] [PubMed] [Google Scholar]
- 83.Kusters B, et al. The pattern of metastasis of human melanoma to the central nervous system is not influenced by integrin alpha(v)beta(3) expression. Int J Cancer. 2001;92(2):176–80. doi: 10.1002/1097-0215(200102)9999:9999<::aid-ijc1173>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 84.Valiente M, et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell. 2014;156(5):1002–16. doi: 10.1016/j.cell.2014.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rahmathulla G, Toms SA, Weil RJ. The molecular biology of brain metastasis. J Oncol. 2012;2012:723541. doi: 10.1155/2012/723541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lorger M, et al. Activation of tumor cell integrin alphavbeta3 controls angiogenesis and metastatic growth in the brain. Proc Natl Acad Sci U S A. 2009;106(26):10666–71. doi: 10.1073/pnas.0903035106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yoshimasu T, et al. Increased expression of integrin alpha3beta1 in highly brain metastatic subclone of a human non-small cell lung cancer cell line. Cancer Sci. 2004;95(2):142–8. doi: 10.1111/j.1349-7006.2004.tb03195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Carbonell WS, et al. The vascular basement membrane as “soil” in brain metastasis. PLoS One. 2009;4(6):e5857. doi: 10.1371/journal.pone.0005857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kim SJ, et al. Astrocytes upregulate survival genes in tumor cells and induce protection from chemotherapy. Neoplasia. 2011;13(3):286–98. doi: 10.1593/neo.11112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kim SW, et al. Role of the endothelin axis in astrocyte- and endothelial cell-mediated chemoprotection of cancer cells. Neuro Oncol. 2014;16(12):1585–98. doi: 10.1093/neuonc/nou128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Neman J, et al. Co-evolution of breast-to-brain metastasis and neural progenitor cells. Clin Exp Metastasis. 2013;30(6):753–68. doi: 10.1007/s10585-013-9576-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Louie E, et al. Neurotrophin-3 modulates breast cancer cells and the microenvironment to promote the growth of breast cancer brain metastasis. Oncogene. 2013;32(35):4064–77. doi: 10.1038/onc.2012.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Neman J, et al. Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc Natl Acad Sci U S A. 2014;111(3):984–9. doi: 10.1073/pnas.1322098111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kafka A, et al. Brain metastases from lung cancer show increased expression of DVL1, DVL3 and beta-catenin and down-regulation of E-cadherin. Int J Mol Sci. 2014;15(6):10635–51. doi: 10.3390/ijms150610635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Deneve E, et al. Capture of viable circulating tumor cells in the liver of colorectal cancer patients. Clin Chem. 2013;59(9):1384–92. doi: 10.1373/clinchem.2013.202846. [DOI] [PubMed] [Google Scholar]
- 96.Singh AD, Bergman L, Seregard S. Uveal melanoma: epidemiologic aspects. Ophthalmol Clin North Am. 2005;18(1):75–84. viii. doi: 10.1016/j.ohc.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 97.Takeda K, et al. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat Med. 2001;7(1):94–100. doi: 10.1038/83416. [DOI] [PubMed] [Google Scholar]
- 98.Tabaries S, et al. Claudin-2 promotes breast cancer liver metastasis by facilitating tumor cell interactions with hepatocytes. Mol Cell Biol. 2012;32(15):2979–91. doi: 10.1128/MCB.00299-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Costa-Silva B, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17(6):816–26. doi: 10.1038/ncb3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhao L, et al. Recruitment of a myeloid cell subset (CD11b/Gr1 mid) via CCL2/CCR2 promotes the development of colorectal cancer liver metastasis. Hepatology. 2013;57(2):829–39. doi: 10.1002/hep.26094. [DOI] [PubMed] [Google Scholar]
- 101.Reticker-Flynn NE, Bhatia SN. Aberrant glycosylation promotes lung cancer metastasis through adhesion to galectins in the metastatic niche. Cancer Discov. 2015;5(2):168–81. doi: 10.1158/2159-8290.CD-13-0760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu FT, Rabinovich GA. Galectins as modulators of tumour progression. Nat Rev Cancer. 2005;5(1):29–41. doi: 10.1038/nrc1527. [DOI] [PubMed] [Google Scholar]
- 103.Loo JM, et al. Extracellular metabolic energetics can promote cancer progression. Cell. 2015;160(3):393–406. doi: 10.1016/j.cell.2014.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nagrath S, et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature. 2007;450(7173):1235–9. doi: 10.1038/nature06385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Husemann Y, et al. Systemic spread is an early step in breast cancer. Cancer Cell. 2008;13(1):58–68. doi: 10.1016/j.ccr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 106.Schardt JA, et al. Genomic analysis of single cytokeratin-positive cells from bone marrow reveals early mutational events in breast cancer. Cancer Cell. 2005;8(3):227–39. doi: 10.1016/j.ccr.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 107.Rhim AD, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148(1–2):349–61. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Podsypanina K, et al. Seeding and propagation of untransformed mouse mammary cells in the lung. Science. 2008;321(5897):1841–4. doi: 10.1126/science.1161621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Stephens JK, et al. Fatal transfer of malignant melanoma from multiorgan donor to four allograft recipients. Transplantation. 2000;70(1):232–6. [PubMed] [Google Scholar]
- 110.Strauss DC, Thomas JM. Transmission of donor melanoma by organ transplantation. Lancet Oncol. 2010;11(8):790–6. doi: 10.1016/S1470-2045(10)70024-3. [DOI] [PubMed] [Google Scholar]
- 111.Riethmuller G, Klein CA. Early cancer cell dissemination and late metastatic relapse: clinical reflections and biological approaches to the dormancy problem in patients. Semin Cancer Biol. 2001;11(4):307–11. doi: 10.1006/scbi.2001.0386. [DOI] [PubMed] [Google Scholar]
- 112.Bao S, et al. Periostin potently promotes metastatic growth of colon cancer by augmenting cell survival via the Akt/PKB pathway. Cancer Cell. 2004;5(4):329–39. doi: 10.1016/s1535-6108(04)00081-9. [DOI] [PubMed] [Google Scholar]
- 113.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2(3):161–74. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 114.Wu K, et al. Roles of the cyclooxygenase 2 matrix metalloproteinase 1 pathway in brain metastasis of breast cancer. J Biol Chem. 2015;290(15):9842–54. doi: 10.1074/jbc.M114.602185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell. 2015;27(1):15–26. doi: 10.1016/j.ccell.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 116.Tabassum DP, Polyak K. Tumorigenesis: it takes a village. Nat Rev Cancer. 2015;15(8):473–83. doi: 10.1038/nrc3971. [DOI] [PubMed] [Google Scholar]
- 117.Cleary AS, et al. Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature. 2014;508(7494):113–7. doi: 10.1038/nature13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Marusyk A, et al. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature. 2014;514(7520):54–8. doi: 10.1038/nature13556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Calbo J, et al. A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer Cell. 2011;19(2):244–56. doi: 10.1016/j.ccr.2010.12.021. [DOI] [PubMed] [Google Scholar]
- 120.Aceto N, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158(5):1110–22. doi: 10.1016/j.cell.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Obenauf AC, et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature. 2015;520(7547):368–72. doi: 10.1038/nature14336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wan L, Pantel K, Kang Y. Tumor metastasis: moving new biological insights into the clinic. Nat Med. 2013;19(11):1450–64. doi: 10.1038/nm.3391. [DOI] [PubMed] [Google Scholar]
- 123.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9(4):239–52. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nguyen-Ngoc KV, et al. ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc Natl Acad Sci U S A. 2012;109(39):E2595–604. doi: 10.1073/pnas.1212834109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wyckoff JB, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67(6):2649–56. doi: 10.1158/0008-5472.CAN-06-1823. [DOI] [PubMed] [Google Scholar]
- 127.Condeelis J, Segall JE. Intravital imaging of cell movement in tumours. Nat Rev Cancer. 2003;3(12):921–30. doi: 10.1038/nrc1231. [DOI] [PubMed] [Google Scholar]
- 128.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(5):392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 129.Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013;19(11):1438–49. doi: 10.1038/nm.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Thiery JP, et al. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 131.Nash GF, et al. Platelets and cancer. Lancet Oncol. 2002;3(7):425–30. doi: 10.1016/s1470-2045(02)00789-1. [DOI] [PubMed] [Google Scholar]
- 132.Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127(4):679–95. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 133.Wang H, et al. Tumor cell alpha3beta1 integrin and vascular laminin-5 mediate pulmonary arrest and metastasis. J Cell Biol. 2004;164(6):935–41. doi: 10.1083/jcb.200309112. [DOI] [PMC free article] [PubMed] [Google Scholar]