

Abstract
Background
Thyroid-derived cell models are commonly used to investigate the characteristics of thyroid cancers. It is noteworthy that each in vitro single cell model system imitates only a few characteristics of thyroid cancer depending on e.g. source of cells or oncogene used to transform the cells.
Methods
In the current work we utilized rat thyroid cancer cell models to determine their clinical relevance in redox gene studies by comparing in vitro expression data to thyroid Oncomine microarray database. To survey the cell lines we analyzed mRNA expression of genes that produce superoxide anion (nox family), genes that catalyze destruction of superoxide anion to hydrogen peroxide (sod family), and genes that remove hydrogen peroxide from cellular environment (catalase, gpx family and prdx family).
Results
Based on the current results, rat thyroid PC Cl3, PC PTC1, PC E1A, or FRLT5 cell models can be used to study NOX2, NOX4, SOD2, SOD3, CATALASE, GPX1, GPX2, GPX5, PRDX2, and PRDX3 gene expression and function.
Conclusions
Redox gene expression in rat originated single cell model systems used to study human thyroid carcinogenesis corresponds only partly with human redox gene expression, which may be caused by differences in redox gene activation stimulus. The data suggest careful estimation of the data observed in rat thyroid in vitro models.
Electronic supplementary material
The online version of this article (doi:10.1186/s12935-015-0264-3) contains supplementary material, which is available to authorized users.
Keywords: Thyroid cancer, Redox gene expression, Cell line, Cell model, Rat, PC Cl3, FRTL5, SOD3, Extracellular superoxide dismutase
Background
Tumors of thyroid gland, which include e.g. differentiated papillary thyroid cancer and poorly differentiated anaplastic thyroid cancer, are among the foremost-characterized cancers. A number of cell models have been created to study signal transduction and function of oncogenes in thyroid carcinogenesis [1–4]. These model systems, however, may not reflect all aspects of thyroid cancers emphasizing the need to compare the gene expression between the cell model system and human tissues.
Although the function of reactive oxygen species (ROS) is unspecific they are important mediators of tumor formation inducing mutations and regulating signal transduction, thus affecting a large range of cellular molecules. Reduction/oxidation (redox) system is highly sensitive to cellular changes, such as ageing and transformation, and therefore can function as a sensor in cell differentiation and carcinogenesis. The importance of ROS for primary cell immortalization and transformation was emphasized by a study demonstrating that increased nox1 expression is essential for cellular transformation by ras oncogene [5]. NOX proteins are catalytic subunits in NADPH complexes inducing superoxide anion (O2−) production and activating signal transduction by interacting with other cellular proteins. Superoxide dismutase (SOD) gene family catalyzes dismutation of superoxide anion into hydrogen peroxide (H2O2), which is then further metabolized by catalase, glutathione peroxidase (GPX) family, and peroxiredoxin (PRDX) family.
To characterize thyroid cancer models derived from rat primary thyroid cells we analyzed the expression of different nox genes representing superoxide anion radical source and sod, catalase, gpx, and prdx gene families that dismutase superoxide to hydrogen peroxide and further to less reactive derivatives, such as oxygen and water molecules. The obtained expression data was compared to microarray data from thyroid cancer patients to validate the use of cell models in redox studies.
Results
Expression of NOX1-4, SOD1-3, GPX1-8, and PRDX1-6 genes in normal human thyroid, papillary thyroid cancer, and in anaplastic thyroid cancer
Although ROS are important second messengers in normal cellular functions, in pathological conditions, such as cancer, redox enzyme expression is unbalanced causing oxidative stress. Thyroid carcinogenesis models consist of a number of different in vitro models that are derived from rat origin by transforming the cells with oncogenes. Since rat and human thyroid models may have different characteristics, especially in highly sensitive redox system, in the current work we analyzed redox gene expression in PC Cl3 and FRLT5 derived thyroid cancer models. For the survey we selected redox enzymes producing superoxide anion (O2−) and on enzymes neutralizing it to hydrogen peroxide (H2O2) and further to e.g. water (H2O) and oxygen (O2). To compare the observed redox gene expression in rat thyroid cell models with human thyroid tissue and thyroid cancers we first extracted microarray data from Oncomine database (http://www.oncomine.org) that contains a number of patients and hence moderate the differences observed between individuals. NOX1, NOX3, and NOX5 expression suggested minor changes in thyroid cancers (Fig. 1a, c, e), whereas the expression of NOX2 and NOX4 showed increased mRNA synthesis in papillary thyroid and anaplastic thyroid cancers (Fig. 1b, d). SOD1 stayed at similar levels, SOD2 expression increased in cancer, and SOD3 mRNA synthesis decreased correlating to reduced differentiation degree of thyroid tissue (Fig. 1f–h). CATALASE expression was markedly decreased in anaplastic thyroid cancer as compared to normal thyroid tissue and papillary thyroid cancer (Fig. 1i). Glutathione peroxidase family showed variable gene expression: GPX1 and GPX2 expression analysis suggested minor increase in mRNA synthesis (Fig. 1j, k), GPX3, GPX5, and GPX7 expression was downregulated in thyroid cancers (Fig. 1l, n, o), whereas GPX4 expression did not change (Fig. 1m). PRDX1, PRDX2, and PRDX3 mRNA synthesis decreased in papillary and anaplastic thyroid cancers (Fig. 1p–r), PRDX4 mRNA expression was moderately increased in anaplastic thyroid cancer tissues (Fig. 1s), and PRDX6 expression status was at similar levels in normal thyroid and thyroid cancers (Fig. 1t).
Fig. 1.
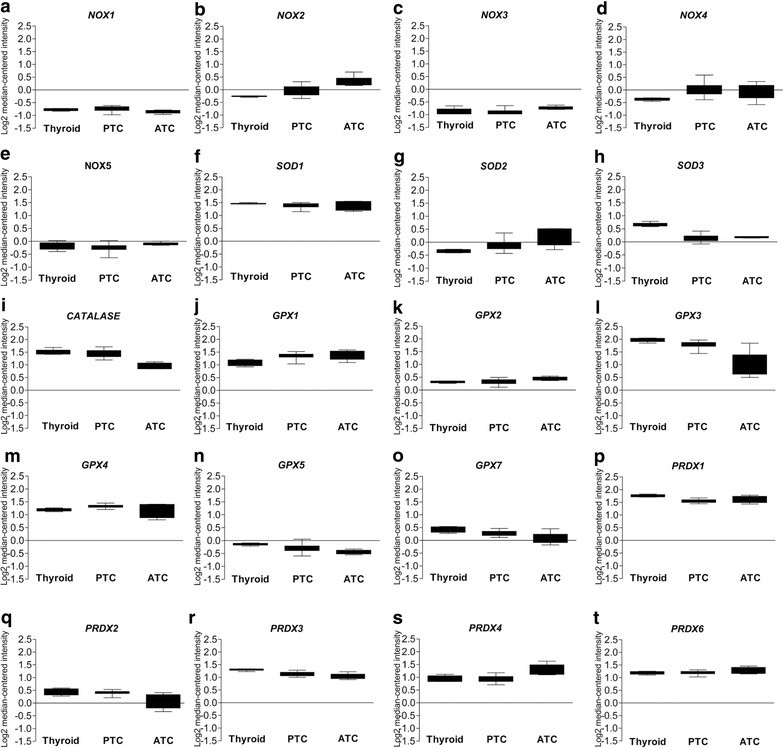
Microarray data extracted from Oncomine database. The average expression of redox genes obtained from the database is shown as a histograms. a expression of nox1, b expression of nox2, c expression of nox3, d expression of nox4, e expression of nox5, f expression of sod1, g expression of sod2, h expression of sod3, i expression of catalase, j expression of gpx1, k expression of gpx2, l expression of gpx3, m expression of gpx4, n expression of gpx5, o expression of gpx7, p expression of prdx1, q expression of prdx2, r expression of prdx3, s expression of prdx4, t expression of prdx6
TSH increases redox gene expression in rat FRLT5 cells
Rat thyroid cells immortalized and transformed with oncogenes are widely used to study the development of thyroid cancer. Previous studies have suggested increased ROS production in cells treated with thyroid stimulating hormone (TSH) [6, 7], which binds to G-protein couple TSH receptor (TSH-R) and activates downstream signal cascades, such as cyclic AMP, PKA, PKC, ERK1/2, PI3K-AKT. Hence, we dissected nox1-4, sod1-3, catalase,gpx1-8, and prdx1-6 expression status in rat thyroid FRLT5 stimulated with TSH.
The data shown in Fig. 2 suggested that TSH stimulation of cells after three-day hormone starvation induced mRNA production of nox1, nox2, and nox4 genes (Fig. 2a–c), whereas nox3 was not expressed or the expression level was extremely low and thus not detectable in FRLT5 thyroid cells. The expression analysis of superoxide dismutase family members, which dismutase O2− to H2O2, suggested decreased cytoplasmic sod1 and extracellular sod3 mRNA synthesis, whereas the expression of mitochondrial sod2 was increased (Fig. 2d–f). Interestingly, there was a significant upregulation of genes responsible of H2O2 removal; the expression of catalase, gpx1, gpx4, gpx5, gpx6, gpx8, prdx1, prdx2, prdx3, prdx4, and prdx6 were all upregulated (Fig. 2g, h, j–q, s). Only gpx2 and prdx5 (Fig. 2i, r) did not respond to TSH starvation, and FRTL5 cells did not express gpx3 and gpx7, or their expression level was extremely low.
Fig. 2.
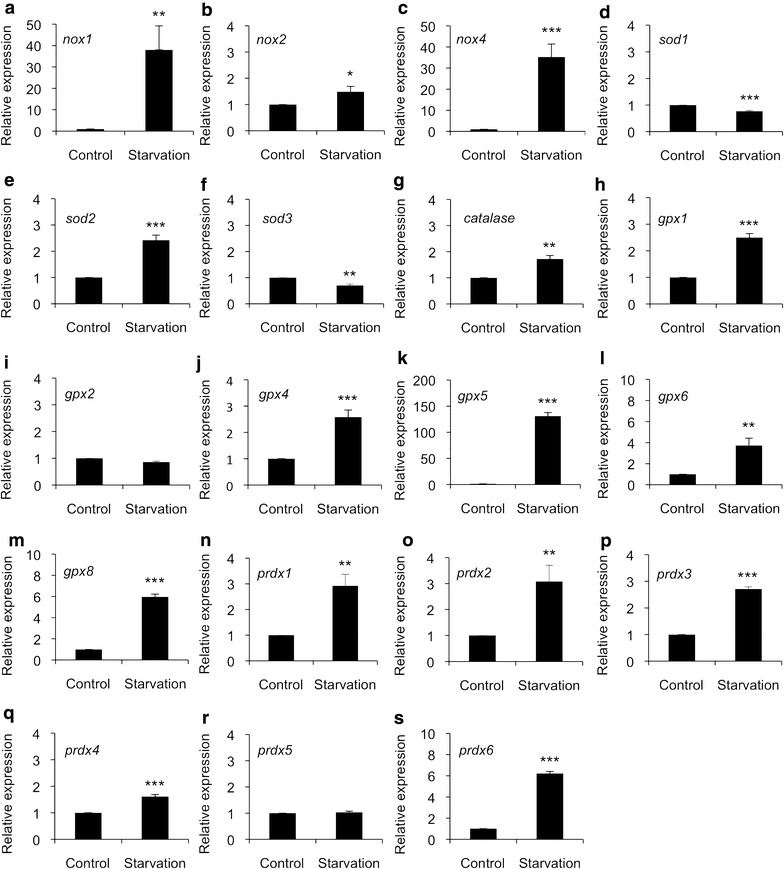
The effect of TSH on redox gene expression. FRLT5 cells were treated with TSH after 3-day hormone starvation. a expression of nox1, b expression of nox2, c expression of nox4, d expression of sod1, e expression of sod2, f expression of sod3, g expression of catalase, h expression of gpx1, i expression of gpx2, j expression of gpx4, k expression of gpx5, l expression of gpx6, m expression of gpx8, n expression of prdx1, o expression of prdx2, p expression of gpx3, q expression of prdx4, r expression of prdx5, s expression of prdx6. The data are expressed as the mean ± SD. The p values are represented (*p < 0.05, **p < 0.01, ***p < 0.001)
Effect of PTC1 and e1a oncogenes on redox gene expression
In cancer ROS, especially superoxide anion, mediates primary cell transformation and cell proliferation [5]. We thus analyzed the effect of PTC1 and e1a oncogene expression on redox gene mRNA levels in rat PC Cl3, PC PTC1 and PC E1A cells. As shown in Fig. 3, the mRNA levels of nox1-4 were upregulated by the oncogenes, although in PC PTC1 cells there was only a tendency for increased nox1 and nox4 mRNA expression. Interestingly, while O−.2 production was upregulated, there was a downregulation of genes dismutating O−.2 to H2O2 and of genes removing H2O2 from the cellular environment. The expression of sod1-3, cat, gpx1, gpx2, gpx4, gpx5,gpx7, and gpx8, and prdx1-6 genes was mostly decreased by PTC1 and e1a oncogenes thus suggesting redox unbalance towards increased O2− cellular concentration. The expression analysis suggested reduced, or tendency towards reduced catalase and gpx6 expression in PTC1 transformed PC Cl3 cells but not clear effect in e1a transformed cells. Only the expression of gpx3 was upregulated by PTC1 and e1a oncogenes. Based on the data there is increased expression of nox genes forming NADPH complex that mainly produces O2−, whereas expression of genes coding for enzymes responsible for removing ROS was mostly downregulated.
Fig. 3.
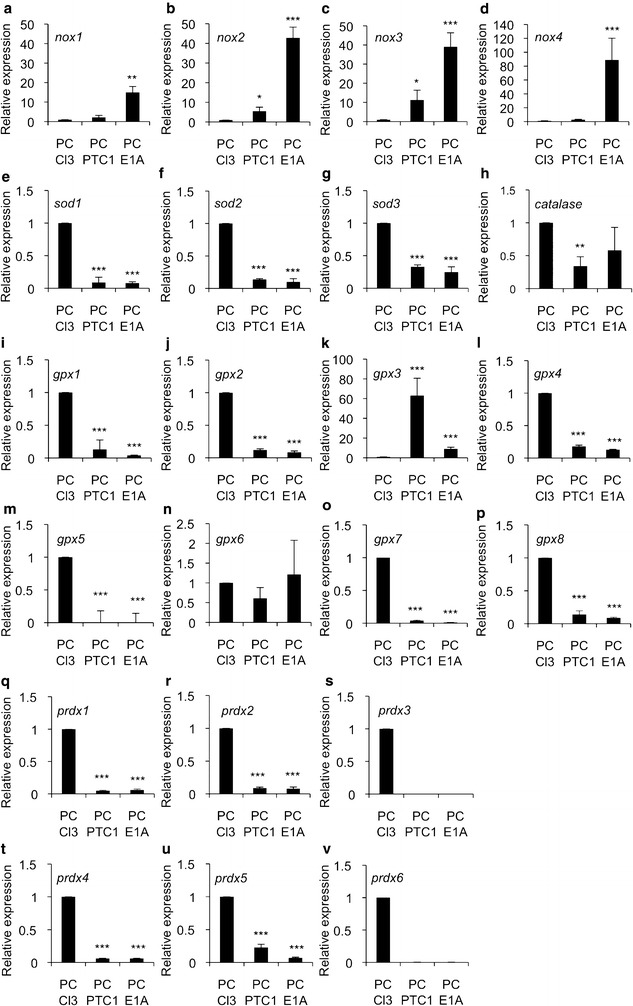
The effect of PTC1 and e1a oncogenes on redox gene expression in PC Cl3 cell derived cancer models. a expression of nox1, b expression of nox2, c expression of nox3, d expression of nox4, e expression of sod1, f expression of sod2, g expression of sod3, h expression of catalase, i expression of gpx1, j expression of gpx2, k expression of gpx3, l expression of gpx4, m expression of gpx5, n expression of gpx6, o expression of gpx7, p expression of gpx8, q expression of prdx1, r expression of prdx2, s expression of prdx3, t expression of prdx4, u expression of prdx5, v expression of prdx6. The data are expressed as the mean ± SD. The p values are represented (*p < 0.05, **p < 0.01, ***p < 0.001)
The effect of RAS on redox gene expression
Next we investigated the expression of redox genes in FRLT5 cell model that consists of cells harboring different levels of RAS: FRLT5 clone V13 has 1.4-fold increased RAS activation, clone V21 has tenfold increased RAS activation, and clone V39 has 35-fold increased RAS activation as compared to FRLT5 control cells [2]. The expression analysis of nox gene family suggested gradually increased nox1 mRNA synthesis and decreased nox2 expression correlating to increased RAS activation level (Fig. 4a, b), whereas nox4 expression was strongly upregulated in FRLT5 V13, FRLT5 V21, FRLT5 V21, and in FRLT5 V39 clones as compared to control FRLT5 cells (Fig. 4c). In line with previous data [8], sod1 and sod2 expression varies depending on the cell model used. Gradually increased RAS activation correlated to increased sod1 mRNA levels whereas sod2 synthesis suggested differential pattern (Fig. 4d, e). The expression analysis of sod3 showed increase in mRNA synthesis at low RAS activation levels and significantly decreased sod3 mRNA production at high RAS levels (Fig. 4f), as we have published recently [8]. Similarly with dismutase family, genes responsible of H2O2 removal showed variable expression. Catalase expression was gradually downregulated (Fig. 4g), whereas gene expression analysis of members of glutathione peroxidase family suggested increased mRNA synthesis. Gpx1 and gpx6 mRNA expression was upregulated in clone V21 (tenfold RAS activity) and in clone V13 (1.4-fold RAS activity), respectively (Fig. 4h, l). Other members of gpx gene family, gpx2, gpx3, gpx4, gpx7, and gpx8 showed increased mRNA production corresponding to increased RAS activity (Fig. 4i–k, m, n). The expression of peroxiredoxin family members was decreased already at moderate 1.4-fold RAS activation level excluding prdx1 and prdx6 that showed only a minor changes in mRNA synthesis levels in the cell model (Fig. 4o–t).
Fig. 4.
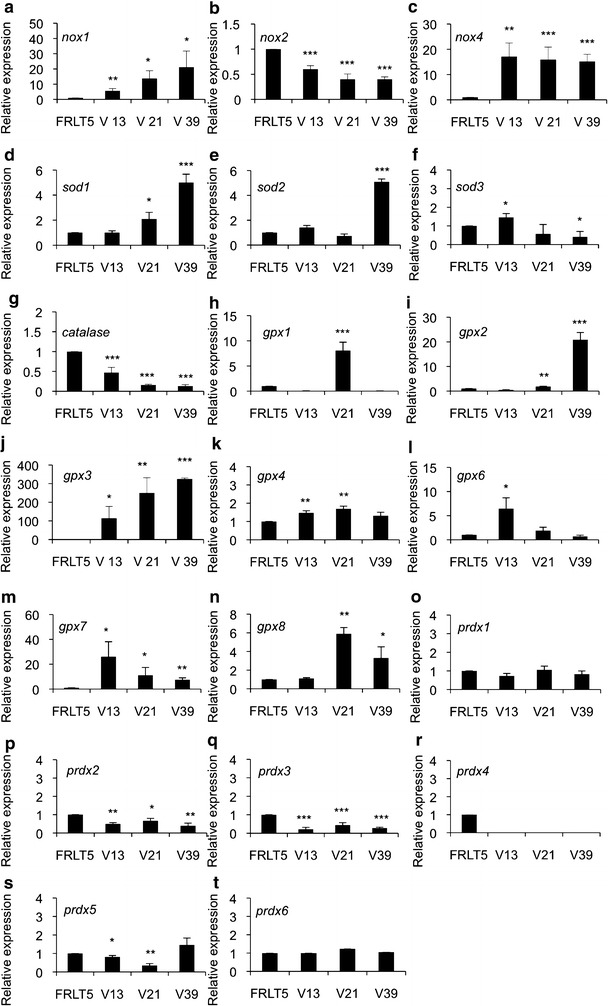
The effect of H-RasV12 oncogene on redox gene expression in FRLT5 cells derived cancer model. a expression of nox1, b expression of nox2, c expression of nox4, d expression of sod1, e expression of sod2, f expression of sod3, g expression of catalase, h expression of gpx1, i expression of gpx2, j expression of gpx3, k expression of gpx4, l expression of gpx6, m expression of gpx7, n expression of gpx8, o expression of prdx1, p expression of gpx2, q expression of prdx3, r expression of prdx4, s expression of prdx5, t expression of prdx6. The data are expressed as the mean ± SD. The p values are represented (*p < 0.05, **p < 0.01, ***p < 0.001)
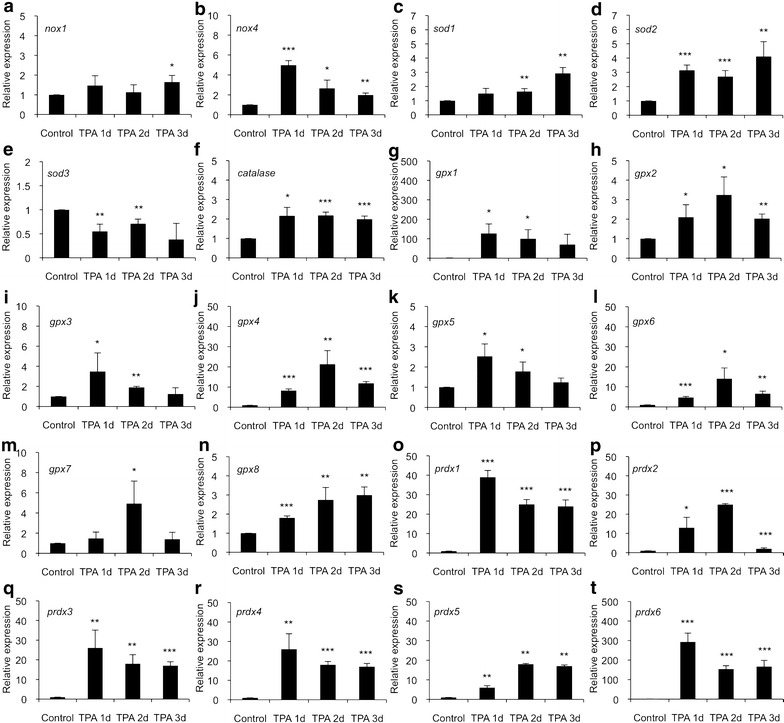
TPA has a time-dependent effect on redox gene expression
The gene expression analysis of PC Cl3 cells treated with 1.6 nM tetradecanoylphorbol acetate (TPA) on three consecutive days was studied to dissect the effect of general mitogenic pressure on redox gene expression. There was only a modest increase in nox1 expression and a significant long-term increase in nox4 expression (Fig. 5a, b). The expression of nox2 and nox3 mRNA synthesis was not detected suggesting that TPA treatment decreased their mRNA synthesis unlike PTC1 and e1a oncogenes (Fig. 3b, c). The expression of cytoplasmic sod1 and mitochondrial sod2 was significantly increased at all time points, whereas extracellular sod3 mRNA production showed modest reduction in gene expression during the treatment (Fig. 5c–e). Cytoplasmic catalase expression was significantly increased on all three consecutive days (Fig. 5f). The overall pattern of glutathione peroxidase gene family mRNA synthesis suggested initially increased expression that returned towards the baseline on day three. Gpx1 expression was significantly increased on day 1 and on day 2, whereas on day 3 there was only an insignificant tendency for increased mRNA synthesis (Fig. 5g). The gene expressions of gpx2, gpx3, gpx4, gpx5, gpx6, and gpx8 were significantly increased during the treatment (Fig. 5h–l, n), whereas the mRNA synthesis of gpx7 was increased only on day 2 and returned back to the base line on day 3 (Fig. 5m). Prdx 1-6 gene expressions were upregulated by TPA treatment on all consecutive days (Fig. 5o–t).
Fig. 5.
Dose-dependent effect of TPA on redox gene expression in PC Cl3 cells. a expression of nox1, b expression of nox4, c expression of sod1, d expression of sod2, e expression of sod3, f expression of catalase, g expression of gpx1, h expression of gpx2, i expression of gpx3, j expression of gpx4, k expression of gpx5, l expression of gpx6, m expression of gpx7, n expression of gpx8, o expression of prdx1, p expression of gpx2, q expression of prdx3, r expression of prdx4, s expression of prdx5, t expression of prdx6. The data are expressed as the mean ± SD. The p values are represented (*p < 0.05, **p < 0.01, ***p < 0.001)
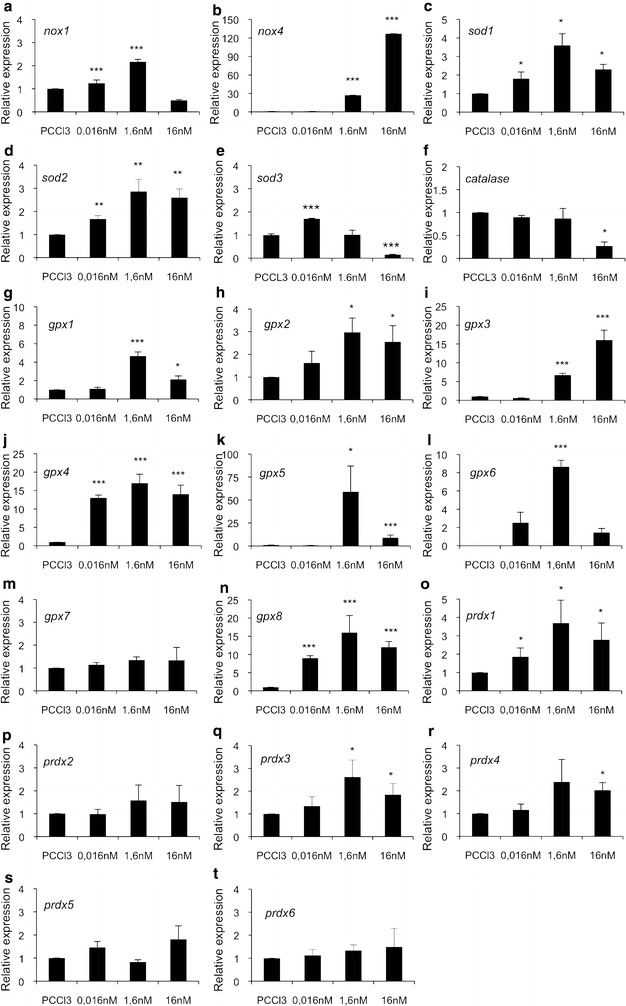
TPA has a concentration dependent effect on redox gene expression
To further dissect the effect of mitogenic pressure on redox gene expression we treated rat thyroid PC Cl3 cells with 0.016, 1.6, and 16 nM TPA. We observed increased nox1 expression at 0.016 nM dose that was further strengthened at 1.6 nM concentration and returned back to baseline at high 16 nM TPA (Fig. 6a), whereas nox4 expression initiated to increase at 1.6 nM concentration and suggested robust mRNA production at 16 nM TPA (Fig. 6b). The expression of nox2 and nox3 was not detectable supporting the data shown in Fig. 4. Sod1 and sod2 mRNA expression analysis suggested significantly increased mRNA synthesis at all TPA doses used (Fig. 6c, d). Sod3 expression analysis suggested significantly increased mRNA synthesis at low 0.016 nM TPA concentration, baseline expression at 1.6 nM TPA, and significantly decreased expression at high 16 nM TPA (Fig. 6e). Catalase expression was at similar levels with control cells at 0.016 nM and at 1.6 nM TPA doses, and decreased at 16 nM TPA (Fig. 6f). The expression analysis of glutathione peroxidases family members gpx1, gpx2, gpx3, gpx4, gpx5, gpx6, gpx7, and gpx8 suggested increased mRNA synthesis correlating to increased TPA concentration (Fig. 6g–n). The expression analysis of peroxiredoxins suggested increased mRNA synthesis of prdx1 at all TPA doses (Fig. 5o), of prdx3 at 1.6 nM and at 16 nM TPA concentrations (Fig. 6q), and of prdx4 at 16 nM TPA (Fig. 6r). The expressions of prdx2, prdx5, and prdx6 were unaffected at all TPA concentrations (Fig. 6p, s, t).
Fig. 6.
Time-dependent effect of TPA on redox gene expression in PC Cl3 cells. a expression of nox1, b expression of nox4, c expression of sod1, d expression of sod2, e expression of sod3, f expression of catalase, g expression of gpx1, h expression of gpx2, i expression of gpx3, j expression of gpx4, k expression of gpx5, l expression of gpx6, m expression of gpx7, n expression of gpx8, o expression of prdx1, p expression of gpx2, q expression of prdx3, r expression of prdx4, s expression of prdx5, t expression of prdx6. The data are expressed as the mean ± SD. The p values are represented (*p < 0.05, **p < 0.01, ***p < 0.001)
SOD3 enhances the TPA derived increased cell proliferation and loss of normal cellular phenotype
Based on the current data sod3 gene expression showed RAS activation-dependent and TPA concentration-dependent initial increase that was then decreased at high RAS activation and TPA concentration levels (Figs. 4f, 6e). Thus, we studied the effect of SOD3 over-expression on the growth of PC Cl3 cells during TPA treatment. BrdU DNA incorporation and cell count analysis suggested that SOD3 over-expression significantly enhanced TPA stimulated DNA proliferation and cell growth as compared to control cells (Fig. 7a, b). PC Cl3 cells characteristically grow in follicular clusters in the growth medium supplemented with thyroid hormones and growth factors. TPA treatment had a concentration and a time-dependent effect on control PC Cl3 cells causing the loss of the characteristic follicular cluster conformation phenotype (Fig. 7a–h). At 1.6 nM TPA concentration (Fig. 7a–c) the loss of the follicular growth formation was slower than at high 16 nM TPA concentration (Fig. 7f–h) that disrupted the normal growth phenotype already at 12-h time point after the initiation of TPA supplementation. PC Cl3 cells over-expressing human SOD3 showed earlier loss of the follicular cluster conformation phenotype that initiated at 12-h time point and suggested spindle like phenotype at 24-h time point (Fig. 7i–k).
Fig. 7.
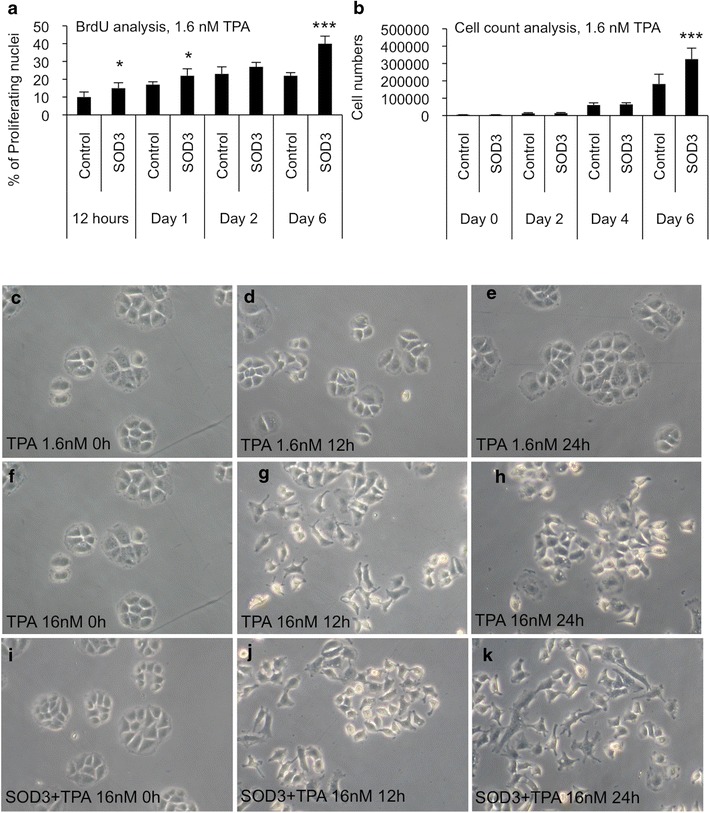
The effect of SOD3 over-expression on TPA treated PC Cl3 cells. a BrdU incorporation analysis suggested SOD3-enhanced DNA replication. b Growth curve analysis supported BrdU assay suggesting increased growth in the presence of SOD3. c–e The effect of 1.6 nM TPA on PC Cl3 cell follicular growth formation. f–h The effect 16 nM TPA on PC Cl3 cell follicular growth formation. i–k The combined effect of 16 nM TPA and SOD3 over-expression on PC Cl3 cell follicular growth formation. The data are expressed as the mean ± SD. The p values are represented (*p < 0.05, **p < 0.01, ***p < 0.001)
Discussion
Primary cell transformation and malignant cancer development is frequently characterized by increased oxidative stress that induces mutations and increases growth signaling. The data extracted from Oncomine database suggested increased NOX1-5 gene expression that, as part of NADPH oxidase complex, induces O2− production (Fig. 1). In line with previously published reports, Oncomine data showed reduced expression of genes coding for enzymes that remove oxygen radicals from the tissue environment. The only exceptions were increased expression of mitochondrial SOD2, GPX1, and GPX2 [9–11] thus highlighting the importance of mitochondrial activity in tumorigenesis [12].
Thyroid stimulating hormone is an important mediator of normal thyroid physiology whereas transformed thyroid cells develop the ability to grow independently of hormone stimulation [13]. Several thyroid pathologies are characterized by abnormal TSH production thus compromising hormone related signal transduction and thyroid function. TSH acts through TSH receptor, a member of small G-protein coupled receptor family, regulating thyroid hormone T3 and T4 production, thyroid growth, and cell differentiation through adenylyl cyclase and cAMP signaling pathways [14]. TSH concentration is markedly increased in Hashimoto’s disease, which is also characterized by increased oxidative stress [15], whereas Graves’s disease is characterized by low levels of TSH in compensation of high T3 and T4 hormone concentrations in circulation. Grave’s disease is an autoimmune disease associated with increased oxidative stress, mainly originating from migrating inflammatory cells [16]. The current data showed TSH-stimulated signal transduction regulating redox gene expression in FRLT5 cells (Fig. 2) suggesting feasibility of the cells for Hashimoto’s disease and Grave’s disease related oxidative stress studies. In support of the current data, we have previously shown in line with the current work that TSH stimulation decreases sod3 expression in PC Cl3 cells [6].
Thyroid cancers arise from epithelial cell mutations, such as RET/PTC genetic aberration formed by chromosomal rearrangement of RET tyrosine kinase domain to N-terminal of H4 (D10S170) [17] and frequently associated to radiation derived papillary thyroid cancer [18]. A number of cell lines expressing oncogenes, e.g. adenovirus adenovirus Ad12early region 1A (e1a), are used to study the development and characteristics of thyroid cancer. The oncogenic nature of e1a has been linked to virus dose, host genetic constitution, age of the host at the time of infection, and immune status of the host. The viral e1a sequence drives primary cells towards transformation by affecting signal transduction pathways involved in immune escape, p53 response, and protein stability [19]. Rat thyroid PC Cl3 cells transfected with e1a lack the differentiation ability and show characteristics of TSH independent growth and function [20]. The frequency of ras mutations in thyroid tumors vary from 0 % up to 85 % showing increased occurrence in poorly differentiated and undifferentiated thyroid cancers [21, 22]. The aberrant activation of RAS-BRAF-MEK1/2-ERK1/2 kinase signaling pathway, which mediates the extracellular signals into the nucleus, contributes to thyroid carcinoma by increasing cell proliferation, survival, and metastasis.
In the current work redox gene expression PC Cl3 cells transformed with PTC1 and e1a oncogenes suggested partial similarity between human microarray data and cell models. The expression of nox2, nox3, and nox4 in cell models showed similar increase as in thyroid cancers tissues whereas the decreased expression of sod3, catalase, gpx5, gpx7, prdx1, prdx2, and prdx3 corresponded to decreased expression in microarray data. It is important to note that the expression of nox1, sod1, sod2, gpx1, gpx3, gpx4, gpx8, prdx4, and prdx6 in cell models was opposite to human tissues. Further, gpx6 and prdx5 that showed variable expressions in cell models were not expressed in human cancers (Fig. 3). Thus, out of 22 redox genes 10 showed similar expression pattern in thyroid tissues and PTC1 or e1a derived cell models. In FRLT5 based H-RasV12 cell model the expression of nox4, sod2, sod3, catalase, gpx2, gpx4, and prdx6 corresponded to human tissue mRNA production. The expression of gpx1 was variable and the expression of nox1, nox2, sod1, gpx3, gpx7, prdx1, prdx2, prdx3, and prdx4 in cell model was opposite to expression in thyroid tissue. In line with PTC1 and e1a models, gpx6, gpx8, and prdx5, which were absent in human microarray data, showed mRNA production in FRLT5 cell model indicating that seven redox genes out of 22 had similar expression pattern in cells and tissues (Fig. 4).
The differences between tissue microarray database and in vitro gene expression assays may be caused by presence of stromal paracrine effect influencing tissue redox balance or presence of multiple genetic aberrations in vivo. In human tissues increased expression of NOX genes is mainly caused by small RAS GTPase protein activation [23–25] or cytokines stimulus [26–28], thus suggesting direct involvement of ras family genes in nox mRNA production in FRLT5 clones V13, V21, and V39 (Fig. 4). Similarly with NOX genes ras oncogene signaling has been suggested to be involved in SOD1, SOD2, and SOD3 gene expression through PI3K-AKT [29, 30] and RAS-RAF-MEK-ERK pathways [31] CATALASE mRNA expression, which was decreased in vitro models by oncogenes, is stimulated by PI3K-AKT and AMPK signaling [32, 33], whereas glutathione peroxidase family gene expression, another H2O2 metabolizing enzyme group, is activated by proto-oncogenes, such as ets-1 [34], by antioxidative Keap1 signaling [35], by peroxisome proliferator-activated receptor γ signaling [36], by ROS [37], and by cytokines [35]. The third H2O2 metabolizing enzyme group, peroxiredoxin gene family, is activated by immune response inducing proteins, such as lipopolysaccharide through ROS-p38 MAPK [38, 39] by proto-oncogene SRC-PI3 K-JNK pathway [40], by PI3K-AKT [41], by cytokines [42, 43] and by various kinds of antioxidative molecules [35, 44].
TPA is a tumor promoter that induces high ROS, especially O2−, production [28] and therefore it is often used to characterize the effect of superoxide dismutases in vitro [45] and in vivo models [46]. In the current study TPA administration time-dependently increased redox gene expressions (Fig. 5) but dose-dependently showed differential response in redox gene expression (Fig. 6). Because sod3 mRNA dose-dependent response to TPA (Fig. 6e) suggested similar expression pattern as observed in FRLT5 H-RasV12 clones V13, V21, and V39 (Fig. 4f), we characterized in more detail the effect of sod3 over-expression in TPA treated PC Cl3 cells (Fig. 7). Our previous observations have suggested a coordinated action of SOD3 and RAS small GPTase signal transduction thus indicating that SOD3 supports growth by increasing RAS-ERK1/2 and PI3 K-AKT signal transduction pathway activation [8, 31, 47, 48]. The current data suggesting increased cell proliferation in TPA treated cells in the presence of SOD3 as compared to control cells (Fig. 7) supports previous observations suggesting that SOD3 may be a growth promoter contributing to cell proliferation at early phase of tumorigenesis.
Conclusions
As a conclusion, according to our data PC Cl3, PC PTC1, PC E1A cell model systems corresponded to human NOX2, NOX4, SOD3, CATALASE, GPX5, and GPX7 gene expressions in thyroid cancer. Used FRLT5 clones V13, V21, and V39 modeled human NOX2, NOX4, SOD2, SOD3, CATALASE, GPX1, GPX2, PRDX2, and PRDX3 gene expressions. TPA administration to rat thyroid PC Cl3 cells could be used to study the effect of NOX2, NOX4, SOD2, SOD3, CATALASE, GPX1, GPX2, and PRDX4 genes.
Methods
Cells
Rat thyroid PC Cl3, PC PTC1, PC E1A, FRLT5, and FRTL5-related cell clones V13, V21, and V39 stably transfected with H-RasV12 expression plasmid [2] were cultured in Ham’s F12 medium Coon’s modified (Sigma, St Louis, MO, USA) supplemented with 5 % calf serum (Life Technologies, Inc., Carlsbad, CA, USA), penicillin (100 U/mL) (Sigma), and streptomycin (100 mg/L) (Sigma). PC Cl3 and FRLT5 cells (modeling normal thyroid cells) were additionally supplemented with 10 nM thyroid stimulating hormone (TSH), 10 nM hydrocortisone, 100 nM insulin, 5 mg/mL transferrin, 5 nM somatostatin, and 20 mg/mL glycyl-histidyl-lysine.
Treatment of cells
To study effect of TSH on redox gene expression, FRLT5 cells were cultured 3 days without hormone supplementation, stimulated for 6 h with 10 nM TSH, and collected for RNA isolation. To study dose-dependent effect of tetradecanoylphorbol acetate (TPA) (Sigma) on redox gene expression and cellular growth, PC Cl3 cells were treated with 0.016, 1.6 or 16 nM TPA for 8 h and pelleted for RNA isolation. To study time-dependent effect of TPA on redox gene expression, PC Cl3 cells were supplemented with fresh medium containing 1.6 nM TPA at day 0, day 1, and day 2. Cells were collected on day 1, day 2, and day 3 for RNA isolation. To study effect of TPA on cell growth, PC Cl3 control cells and PC Cl3 cells over-exressing SOD3 (kindly provided by professor Stefan L. Marklund from the University of Umeå, Sweden) were treated with 1.6 and 16 nM TPA and prepared for BrdU DNA replication analysis, growth curve analysis, and microscopy. SOD3 over-expressing and control plasmid PC Cl3 cells were prepared by transfecting cells with Fugene 6 (Roche, Mannheim, Germany) followed by geneticin antibiotic (Sigma) selection to create stable cells lines.
Real time PCR for mRNA expression analysis
For mRNA expression total RNA was extracted using an RNeasy mini kit (Qiagen, Hilden, Germany). RNA (1 μg) was reverse transcribed with Quantitect Reverse® Transcription Kit (Qiagen). Primers were designed with (http://www-genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi). cDNA was done by GeneAmp RNA PCR Core Kit system (Qiagen). Reactions were performed in triplicate using SYBR Green PCR Master mix (Applied Biosystems, Foster City, CA, USA) and iCycler (BioRad, Hercules, CA, USA). Fluorescent threshold values were calculated using formula 2−(sample 1 ΔCt − sample 2 ΔCt), where ΔCt is difference between amplification fluorescent threshold (Ct) of mRNA of interest, and Ct of ß-actin mRNA used as housekeeping gene. Primers are listed in Additional file 1: Table S1.
Growth analysis
For BrdU DNA replication analysis PC Cl3 cell cultures grown on coverslips were supplemented with 10 mM bromodeoxyuridine (BrdU; Roche, Basel, Switzerland) for 15 min. Subsequently, cells were fixed in 3 % paraformaldehyde (Sigma) and permeabilized with 0.2 % Triton X-100 (Sigma). The coverslips were incubated with anti-BrdU antibody followed by FITC–conjugated secondary antibody (Jackson ImmunoResearch Laboratories Inc., West Grove, PA, USA). Nuclei were counterstained with HOECST (Sigma). For growth curve analysis PC Cl3 cells were grown in 6-well dishes. The number of cells was counted in triplicate from control and SOD3 wells in each analysis date by burker chamber using formula: number of cells/ml = average of cells in each square × 104 × dilution factor.
Statistical analyses
The experiments were repeated at least three times. All results are expressed as the mean ± SD. The p values (*p < 0.05, **p < 0.01, ***p < 0.001) were determined by two-tailed independent samples t test.
Authors’ contributions
FC, FF, and TZ carried out cell culture experiments, real time PCR experiments, data analysis, and interpretation of data. GdV and MS carried out experimental design, interpretation of data, provided cell lines, material and reagents, and helped to draft the manuscript. MOL carried out coordination of the study, experimental design, cell culture experiments, real time PCR experiments, data analysis, interpretation of data, and manuscript preparation. All authors read and approved the final manuscript.
Acknowledgements
Work has been supported by Ministry of Health, SDN Funds 5 per mille 2012 and by Fondazione SDN decision number RC2010-M-0001.
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- cAMP
cyclic adenosine monophosphate
- BRAF
serine threonine protein kinase
- ERK
extracellular signal regulated kinase
- GPX
glutathione peroxidase
- H2O2
hydrogen peroxide
- H-RasV12
Harvey sarcoma virus mutated
- JNK
c-Jun terminal kinase
- MAPK
mitogen activated protein kinase
- MEK
mitogen activated protein kinase kinase
- NOX
nicotinamide adenine dinucleotide phosphate-oxidase
- PI3 K-AKT
phosphatidylinositol-4,5-bisphosphate 3-kinase-protein kinase B
- PKA
protein kinase A
- PKC
protein kinase C
- PRDX
peroxiredoxin
- REDOX
reduction oxidation
- RET
ret proto-oncogene
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- O2−
superoxide anion
- SRC
proto-oncogene tyrosine-protein kinase Src
- TPA
tetradecanoylphorbol acetate
- TSH
thyroid stimulating hormone
- TSH-R
thyroid stimulating hormone receptor
Additional file
Contributor Information
Francesca Cammarota, Email: francesca.cammarota88@gmail.com.
Francesco Fiscardi, Email: fiscardi.f@gmail.com.
Tiziana Esposito, Email: tiziana.espo.89@gmail.com.
Gabriella de Vita, Email: gdevita@unina.it.
Marco Salvatore, Email: marcosalvatore2.segreteria@gmail.com.
Mikko O. Laukkanen, Email: mlaukkanen@sdn-napoli.it
References
- 1.Ambesi-Impiombato FS, Parks LA, Coon HG. Culture of hormone-dependent functional epithelial cells from rat thyroids. Proc Natl Acad Sci USA. 1980;77(6):3455–3459. doi: 10.1073/pnas.77.6.3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Vita G, Bauer L, da Costa VM, De Felice M, Baratta MG, et al. Dose-dependent inhibition of thyroid differentiation by RAS oncogenes. Mol Endocrinol. 2005;19(1):76–89. doi: 10.1210/me.2004-0172. [DOI] [PubMed] [Google Scholar]
- 3.Fusco A, Berlingieri MT, Di Fiore PP, Portella G, Grieco M, Vecchio G. One- and two-step transformations of rat thyroid epithelial cells by retroviral oncogenes. Mol Cell Biol. 1987;7(9):3365–3370. doi: 10.1128/MCB.7.9.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grieco M, Santoro M, Berlingieri MT, Donghi R, Pierotti MA, Della Porta G, et al. Molecular cloning of PTC, a new oncogene found activated in human thyroid papillary carcinomas and their lymph node metastases. Ann N Y Acad Sci. 1988;551:380–381. doi: 10.1111/j.1749-6632.1988.tb22370.x. [DOI] [PubMed] [Google Scholar]
- 5.Mitsushita J, Lambeth JD, Kamata T. The superoxide-generating oxidase Nox1 is functionally required for Ras oncogene transformation. Cancer Res. 2004;64(10):3580–3585. doi: 10.1158/0008-5472.CAN-03-3909. [DOI] [PubMed] [Google Scholar]
- 6.Laatikainen LE, Castellone MD, Hebrant A, Hoste C, Cantisani MC, Laurila JP, et al. Extracellular superoxide dismutase is a thyroid differentiation marker down-regulated in cancer. Endocr Relat Cancer. 2010;17(3):785–796. doi: 10.1677/ERC-10-0021. [DOI] [PubMed] [Google Scholar]
- 7.Song Y, Driessens N, Costa M, De Deken X, Detours V, Corvilain B, et al. Roles of hydrogen peroxide in thyroid physiology and disease. J Clin Endocrinol Metab. 2007;92(10):3764–3773. doi: 10.1210/jc.2007-0660. [DOI] [PubMed] [Google Scholar]
- 8.Castellone MD, Langella A, Cantara S, Laurila JP, Laatikainen LE, Bellelli R, et al. Extracellular superoxide dismutase induces mouse embryonic fibroblast proliferative burst, growth arrest, immortalization, and consequent in vivo tumorigenesis. Antioxid Redox Signal. 2014;21(10):1460–1474. doi: 10.1089/ars.2013.5475. [DOI] [PubMed] [Google Scholar]
- 9.Esworthy RS, Ho YS, Chu FF. The Gpx1 gene encodes mitochondrial glutathione peroxidase in the mouse liver. Arch Biochem Biophys. 1997;340(1):59–63. doi: 10.1006/abbi.1997.9901. [DOI] [PubMed] [Google Scholar]
- 10.Ukai Y, Kishimoto T, Ohdate T, Izawa S, Inoue Y. Glutathione peroxidase 2 in Saccharomyces cerevisiae is distributed in mitochondria and involved in sporulation. Biochem Biophys Res Commun. 2011;411(3):580–585. doi: 10.1016/j.bbrc.2011.06.189. [DOI] [PubMed] [Google Scholar]
- 11.Zimmermann R, Flohe L, Weser U, Hartman HJ. Inhibition of lipid peroxidation in isolated inner membrane of rat liver mitochondria by superoxide dismutase. FEBS Lett. 1973;29(2):117–120. doi: 10.1016/0014-5793(73)80539-3. [DOI] [PubMed] [Google Scholar]
- 12.Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015;11(1):9–15. doi: 10.1038/nchembio.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goretzki PE, Koob R, Koller T, Simon R, Branscheid D, Clark OH, et al. The effect of thyrotropin and cAMP on DNA synthesis and cell growth of human thyrocytes in monolayer culture. Surgery. 1986;100(6):1053–1061. [PubMed] [Google Scholar]
- 14.Corda D, Bizzarri C, Di Girolamo M, Valitutti S, Luini A. G protein-linked receptors in the thyroid. Adv Exp Med Biol. 1989;261:245–269. doi: 10.1007/978-1-4757-2058-7_9. [DOI] [PubMed] [Google Scholar]
- 15.Yi JW, Park JY, Sung JY, Kwak SH, Yu J, Chang JH, et al. Genomic evidence of reactive oxygen species elevation in papillary thyroid carcinoma with Hashimoto thyroiditis. Endocr J. 2015 (Epub ahead of print). [DOI] [PubMed]
- 16.Zarkovic M. The role of oxidative stress on the pathogenesis of graves’ disease. J Thyroid Res. 2012;2012:302537. doi: 10.1155/2012/302537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grieco M, Santoro M, Berlingieri MT, Melillo RM, Donghi R, Bongarzone I, et al. PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell. 1990;60(4):557–563. doi: 10.1016/0092-8674(90)90659-3. [DOI] [PubMed] [Google Scholar]
- 18.Bounacer A, Wicker R, Caillou B, Cailleux AF, Sarasin A, Schlumberger M, et al. High prevalence of activating ret proto-oncogene rearrangements, in thyroid tumors from patients who had received external radiation. Oncogene. 1997;15(11):1263–1273. doi: 10.1038/sj.onc.1200206. [DOI] [PubMed] [Google Scholar]
- 19.Gallimore PH, Turnell AS. Adenovirus E1A: remodelling the host cell, a life or death experience. Oncogene. 2001;20(54):7824–7835. doi: 10.1038/sj.onc.1204913. [DOI] [PubMed] [Google Scholar]
- 20.Berlingieri MT, Akamizu T, Fusco A, Crieco M, Colletta G, Cirafici AM, et al. Thyrotropin receptor gene expression in oncogene-transfected rat thyroid cells: correlation between transformation, loss of thyrotropin-dependent growth, and loss of thyrotropin receptor gene expression. Biochem Biophys Res Commun. 1990;173(1):172–178. doi: 10.1016/S0006-291X(05)81037-X. [DOI] [PubMed] [Google Scholar]
- 21.Fukushima T, Takenoshita S. Roles of RAS and BRAF mutations in thyroid carcinogenesis. Fukushima J Med Sci. 2005;51:67–75. doi: 10.5387/fms.51.67. [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Rostan G, Zhao H, Camp RL, Pollan M, Herrero A, Pardo J, et al. Ras mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer. J Clin Oncol. 2003;21(17):3226–3235. doi: 10.1200/JCO.2003.10.130. [DOI] [PubMed] [Google Scholar]
- 23.Adachi Y, Shibai Y, Mitsushita J, Shang WH, Hirose K, Kamata T. Oncogenic Ras upregulates NADPH oxidase 1 gene expression through MEK-ERK-dependent phosphorylation of GATA-6. Oncogene. 2008;27(36):4921–4932. doi: 10.1038/onc.2008.133. [DOI] [PubMed] [Google Scholar]
- 24.Lee J, Choi KJ, Lim MJ, Hong F, Choi TG, Tak E, et al. Proto-oncogenic H-Ras, K-Ras, and N-Ras are involved in muscle differentiation via phosphatidylinositol 3-kinase. Cell Res. 2010;20(8):919–934. doi: 10.1038/cr.2010.92. [DOI] [PubMed] [Google Scholar]
- 25.Ogrunc M, Di Micco R, Liontos M, Liontos M, Bombardelli L, Mione M, et al. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ. 2014;21(6):998–1012. doi: 10.1038/cdd.2014.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manea A, Tanase LI, Raicu M, Simionescu M. Jak/STAT signaling pathway regulates nox1 and nox4-based NADPH oxidase in human aortic smooth muscle cells. Arterioscler Thromb Vasc Biol. 2010;30(1):105–112. doi: 10.1161/ATVBAHA.109.193896. [DOI] [PubMed] [Google Scholar]
- 27.Lim H, Kim D, Lee SJ. Toll-like receptor 2 mediates peripheral nerve injury-induced NADPH oxidase 2 expression in spinal cord microglia. J Biol Chem. 2013;288(11):7572–7579. doi: 10.1074/jbc.M112.414904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li L, He Q, Huang X, Man Y, Zhou Y, Wang S, et al. NOX3-derived reactive oxygen species promote TNF-alpha-induced reductions in hepatocyte glycogen levels via a JNK pathway. FEBS Lett. 2010;584(5):995–1000. doi: 10.1016/j.febslet.2010.01.044. [DOI] [PubMed] [Google Scholar]
- 29.Rojo AI, Salinas M, Martin D, Perona R, Cuadrado A. Regulation of Cu/Zn-superoxide dismutase expression via the phosphatidylinositol 3 kinase/Akt pathway and nuclear factor-kappaB. J Neurosci. 2004;24(33):7324–7334. doi: 10.1523/JNEUROSCI.2111-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Connor KM, Subbaram S, Regan KJ, Nelson KK, Mazurkiewicz JE, Bartholomew PJ, et al. Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation. J Biol Chem. 2005;280(17):16916–16924. doi: 10.1074/jbc.M410690200. [DOI] [PubMed] [Google Scholar]
- 31.Laurila JP, Castellone MD, Curcio A, Laatikainen LE, Haaparanta-Solin M, Gronroos TJ, et al. Extracellular superoxide dismutase is a growth regulatory mediator of tissue injury recovery. Mol Ther. 2009;17(3):448–454. doi: 10.1038/mt.2008.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Evans PJ. The regulation of hepatic tyrosine aminotransferase. Biochim Biophys Acta. 1981;677:433–444. doi: 10.1016/0304-4165(81)90257-9. [DOI] [PubMed] [Google Scholar]
- 33.Awad H, Nolette N, Hinton M, Dakshinamurti S. AMPK and FoxO1 regulate catalase expression in hypoxic pulmonary arterial smooth muscle. Pediatr Pulmonol. 2014;49(9):885–897. doi: 10.1002/ppul.22919. [DOI] [PubMed] [Google Scholar]
- 34.Verschoor ML, Verschoor CP, Singh G. Ets-1 global gene expression profile reveals associations with metabolism and oxidative stress in ovarian and breast cancers. Cancer Metab. 2013;1(1):17. doi: 10.1186/2049-3002-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu KC, Cui JY, Klaassen CD. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol Sci. 2011;123(2):590–600. doi: 10.1093/toxsci/kfr183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chung SS, Kim M, Youn BS, Lee NS, Park JW, Lee IK, et al. Glutathione peroxidase 3 mediates the antioxidant effect of peroxisome proliferator-activated receptor gamma in human skeletal muscle cells. Mol Cell Biol. 2009;29(1):20–30. doi: 10.1128/MCB.00544-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang Z, Rose AH, Hoffmann PR. The role of selenium in inflammation and immunity: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2012;16(7):705–743. doi: 10.1089/ars.2011.4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim SU, Park YH, Min JS, Sun HN, Han YH, Hua JM, et al. Peroxiredoxin I is a ROS/p38 MAPK-dependent inducible antioxidant that regulates NF-kappaB-mediated iNOS induction and microglial activation. J Neuroimmunol. 2013;259(1–2):26–36. doi: 10.1016/j.jneuroim.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 39.Sun HN, Kim SU, Huang SM, Kim JM, Park Yh, Kim SH, et al. Microglial peroxiredoxin V acts as an inducible anti-inflammatory antioxidant through cooperation with redox signaling cascades. J Neurochem. 2010;114(1):39–50. doi: 10.1111/j.1471-4159.2010.06691.x. [DOI] [PubMed] [Google Scholar]
- 40.Bast A, Fischer K, Erttmann SF, Walther R. Induction of peroxiredoxin I gene expression by LPS involves the Src/PI3K/JNK signalling pathway. Biochim Biophys Acta. 2010;1799(5–6):402–410. doi: 10.1016/j.bbagrm.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 41.Zha X, Wu G, Zhao X, Zhou L, Zhang H, Li J, et al. PRDX6 protects ARPE-19 cells from oxidative damage via PI3K/AKT signaling. Cell Physiol Biochem. 2015;36(6):2217–2228. doi: 10.1159/000430186. [DOI] [PubMed] [Google Scholar]
- 42.Yun SJ, Seo JJ, Chae JY, Lee CC. Peroxiredoxin I and II are up-regulated during differentiation of epidermal keratinocytes. Arch Dermatol Res. 2005;296(12):555–559. doi: 10.1007/s00403-005-0561-0. [DOI] [PubMed] [Google Scholar]
- 43.Wang HQ, Du ZX, Liu BQ, Gao YY, Meng X, Guan Y, et al. TNF-related apoptosis-inducing ligand suppresses PRDX4 expression. FEBS Lett. 2009;583(9):1511–1515. doi: 10.1016/j.febslet.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 44.Park HJ, Carr JR, Wang Z, Noguera V, Hay N, Tyner AL, et al. FoxM1, a critical regulator of oxidative stress during oncogenesis. EMBO J. 2009;28(19):2908–2918. doi: 10.1038/emboj.2009.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Makino J, Kamiya T, Hara H, Adachi T. TPA induces the expression of EC-SOD in human monocytic THP-1 cells: involvement of PKC, MEK/ERK and NOX-derived ROS. Free Radic Res. 2012;46(5):637–644. doi: 10.3109/10715762.2012.664841. [DOI] [PubMed] [Google Scholar]
- 46.Kim SH, Kim MO, Gao P, Youm CA, Park HR, Lee TS, et al. Overexpression of extracellular superoxide dismutase (EC-SOD) in mouse skin plays a protective role in DMBA/TPA-induced tumor formation. Oncol Res. 2005;15(7–8):333–341. doi: 10.3727/096504005776449725. [DOI] [PubMed] [Google Scholar]
- 47.Laatikainen LE, Incoronato M, Castellone MD, Laurila JP, Santoro M, Laukkanen MO. SOD3 decreases ischemic injury derived apoptosis through phosphorylation of Erk1/2, Akt, and FoxO3a. PLoS One. 2011;6(8):e24456. doi: 10.1371/journal.pone.0024456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laukkanen MO, Cammarota F, Esposito T, Salvatore M, Laukkanen MO. Extracellular superoxide dismutase regulates the expression of small gtpase regulatory proteins GEFs, GAPs, and GDI. PLoS One. 2015;10(3):e0121441. doi: 10.1371/journal.pone.0121441. [DOI] [PMC free article] [PubMed] [Google Scholar]