

Abstract
For the treatment of monogenetic hematological disorders, restoration of transgene expression in affected cell populations is generally considered to have beneficial effects. However, X-linked chronic granulomatous disease (XCGD) is unique since the appearance of functional neutrophils in the peripheral blood following hematopoietic stem cell gene therapy is transient only. One contributing factor could be the occurrence of detrimental effects secondary to ectopic gp91phox expression in neutrophils, which has not been formally demonstrated previously. This study uses iPSCs to model XCGD, which allows the process of differentiation to be studied intensely in vitro. Alpharetroviral vectors carrying a ubiquitous promoter were used to drive the “ectopic” expression of codon optimized gp91phox cDNA. In the mature fraction of neutrophils differentiated from transduced XCGD-iPSCs, cellular recovery in terms of gp91phox expression and reactive oxygen species production was abruptly lost before cells had fully differentiated. Most critically, ectopic gp91phox expression could be identified clearly in the developing fraction of the transduced groups, which appeared to correspond with reduced cell viability. It is possible that this impedes further differentiation of developing neutrophils. Therefore, affording cellular protection from the detrimental effects of ectopic gp91phox expression may improve XCGD clinical outcomes.
Introduction
Neutrophils are professional phagocytes with critical functions in eliminating invading pathogens. This is reliant upon the catalytic production of reactive oxygen species (ROS) by the enzyme nicotinamide adenine dinucleotide (NADPH) oxidase. One of the most well characterized disorders of neutrophil functionality is X-linked chronic granulomatous disease (XCGD). It is due to a deficiency in expression of the gp91phox protein,1 which is a critical component of NADPH oxidase thus compromising the catalytic production of ROS.2 Patients are susceptible to potentially life-threatening fungal and bacterial infections. The definitive treatment for primary immune deficiencies (PIDs) such as XCGD is hematopoietic stem cell gene therapy (SCGT) involving the transplantation of genetically modified patient autologous hematopoietic stem cells (HSCs).3 The therapeutic concept behind it is that stable transgene expression at the stem cell level will be transmitted to mature progenies thus leading to the recovery of cell functionality. This approach has been demonstrated to be effective for treating other PIDs such as adenosine deaminase-severe combined immunodeficiency disease (ADA-SCID),4 SCID-X1,5 and Wiskott–Aldrich syndrome.6 However, despite adopting similar vector designs and patient conditioning regimens, outcomes from XCGD clinical trials have witnessed only the transient appearance of functionally reconstituted neutrophils in the peripheral blood (PB) following transplantation. Initially, these unfavorable outcomes were attributed to the primitiveness of vector designs and the avoidance of myelosuppression,7 which is particularly undesirable especially in treating young children. Thereafter, clinical protocols were amended with the inclusion of myelosuppresion and the use of strong ubiquitous promoters such as that derived from the spleen focus-forming virus (SFFV) to drive gp91phox expression.8 Much higher levels of transduction efficiency could be achieved with significantly higher levels of gene marking. Unfortunately, this came at the cost of undesired insertional activation of the EVI1-MDS1 proto-oncogene leading to myelodysplasia with monosomy 7 (ref. 9). Taken together, it is possible to speculate that there may exist disease characteristics unique to XCGD such that direct restoration of gp91phox expression may not be entirely favorable for neutrophils.
One possibility that has not been considered previously is the consequence of ectopic gp91phox expression. In hematopoietic SCGT, integrating viral vectors containing a ubiquitous promoter to drive transgene expression are commonly used. In the context of XCGD, it means the potential expression of gp91phox in nontargeted cell populations even within the same path of myeloid differentiation. This nonphysiologically regulated expression of ectopic gp91phox could result in perturbations in ROS production leading to unknown detrimental effects. It has been reported that raised intracellular levels of ROS (mitochondria derived) could negatively compromise the functionality of hematopoietic stem cells HSCs.10 However, this may not be fully attributable to vector-mediated constitutive expression of gp91phox, which is stable only in the assembled form with other NADPH oxidase subunits.11 In HSCs, these subunits are produced in much lower amounts compared with mature phagocytes,12 which may be insufficient to fully support stable enzyme assembly through incorporating transgene-derived gp91phox protein. However, once differentiating cells become committed myeloid progenitor cells, there is greater possibility that NADPH oxidase-mediated perturbations in ROS production could result in the occurrence of detrimental effects secondary to ectopic gp91phox expression. Although the precise underlying mechanisms are unclear, it has been reported that dysregulation in NADPH-derived ROS production can lead to the induction of endoplasmic reticulum (ER) stress.13 It is also known that sustained ER stress can lead to the activation of apoptotic pathways and eventually cell death if stress levels remain elevated.14 Should this occur in developing neutrophils (myeloblast/myelocytes) then cell viability would likely become compromised.
Previously, it may not have been practical to investigate this theory further due to the absence of a suitable disease model. In terms of physiological relevance, the preferred in vitro modeling system would be to use CD34+ cells from XCGD patients. For reasons of practicality, however, these cells are difficult to obtain repeatedly, therefore making it challenging to study granulocyte differentiation into neutrophils intensely. However, the development of technology to generate induced pluripotent stem cells (iPSCs)15 from patients has allowed for the recapitulation of disease phenotypes and is particularly suited to modeling monogenetic disorders such as XCGD.16,17 It is known that most of hematopoiesis occurs within the bone marrow with only relatively mature blood cells released into the bloodstream. Therefore, an in vitro modeling system would allow possible impediments to differentiation to be identified that are caused by ectopic gp91phox expression in developing neutrophils.
In this study, it was found that cellular recovery in the mature fraction of neutrophils differentiated from transduced XCGD iPSCs was incomplete both in terms of gp91phox expression and ROS production. This appears to be related to an abrupt loss of transgene expression before cells had fully differentiated. Interestingly, “ectopic” expression of gp91phox could be found only in the developing fraction of neutrophils differentiated from transduced XCGD-iPSCs, which corresponded with nonphysiological ROS production. Most critically, developing neutrophils ectopically expressing gp91phox appeared to show an increased propensity for undergoing cell apoptosis irrespective of a XCGD or healthy genetic background. Therefore, future strategies targeted at the physiological regulation of gp91phox expression may improve the viability of neutrophils.
Results
Generation and characterization of XCGD iPSCs
PB CD34+ cells from an XCGD patient were reprogrammed into iPSCs (Figure 1a). Within 30 days following transduction, embryonic stem cell–like colonies could be identified. The following representative characterization data was generated using XCGD iPSC (clone #8), which was used for all subsequent transduction and differentiation experiments. These XCGD iPSCs exhibited alkaline phosphatase (ALP) activity and displayed the surface markers SSEA-4, Tra-1–60, and Tra-1–81 (Figure 1b). They could form teratomas consisting of tissues derived from the three germ layers (Figure 1c), confirming that these cells possess typical features of pluripotent stem cells. XCGD iPSCs expressed pluripotency genes as determined by reverse transcription polymerase chain reaction (Supplementary Figure S1) and exhibited a normal karyotype (Figure 1d). Most importantly, the same exact point mutation that had been identified in the patient in the CYBB gene (c.1448G>A p.Trp483X) was successfully retained (Figure 1e). Therefore, neutrophils differentiated from these unmodified XCGD iPSCs should display the characteristic phenotype of the disease.
Figure 1.

Reprogramming XCGD PB CD34+ cells into iPSCs. (a) Schematic overview of the reprogramming process using a Sendai virus (SeV) vector carrying the four Yamanaka factors to transduce peripheral blood (PB) CD34+ cells. XCGD iPSC (#8) was subjected to detailed characterization and used for subsequent experiments. Embryoid body medium (EBM). (b) XCGD iPSCs stained positive for ALP, SSEA-4, Tra-1–60, and Tra-1–81. (c) XCGD iPSCs could form teratomas consisting of tissue derived from the three germ layers endoderm, mesoderm, and ectoderm. (d) A normal karyotype was exhibited. (e) Confirmation of the point mutation CYBB: c.1448G>A (p.Trp483X). Bars = 200 μm.
Neutrophil differentiation and recapitulation of the XCGD phenotype
The overall differentiation process from either healthy or XCGD iPSCs occurs over ~21 days (Figure 2a). A co-culture system with irradiated C3H10T1/2 cells was adopted throughout the procedure. No purification of hematopoietic progenitor cells (HPCs) was carried out following harvest on day 14 before inducing to undergo further differentiation into neutrophils for an additional 5 to 7 days. To begin with, healthy iPSCs were used to optimize this protocol. Differentiated neutrophils displayed the characteristic multi-lobed appearance of the nuclei following Wright-Giemsa staining. Cells contained myeloperoxidase (MPO) and ALP, which are enzymes found in cytoplasmic granules, the appearance of which corresponds with the increasing maturation status of neutrophils (Figure 2b). The ability of differentiated neutrophils to phagocytose infectious microorganisms was mimicked through the nitroblue tetrazolim (NBT)-coated yeast assay.18 Ingested NBT-coated yeast is purple/black in color due to NBT reduction by ROS (Figure 2c, black arrows). It has been reported that neutrophils can generate neutrophil extracellular traps, composing of nuclear constituents and granules for the extracellular killing of large pathogens.19 Here, neutrophil extracellular traps formed by iPSC-derived neutrophils can be visualized by staining for nucleic acids and MPO, a critical component of these structures20 (Figure 2d, white arrows). Within each cell culture, differentiating neutrophils may be defined as developing (CD64dullCD15dull) or mature (CD64highCD15high) (Supplementary Figure S2) according to surface marker expression as reflected in the morphology of cells sorted by FACS. These two populations were also clearly separable by determining CD66b expression, which is another marker that can be used to assess neutrophil maturation (Supplementary Figure S3). Clinically, the characteristic XCGD phenotype is defined by the absence of any oxidase activity dependent on the NADPH oxidase system in neutrophils. This could be assessed using the dihydrorhodamine123 assay. Following phorbol myristate acetate stimulation, differentiated neutrophils with a healthy genetic background produced ROS at levels comparable to that of PB neutrophils (Supplementary Figure S4). However, this was completely absent in neutrophils differentiated from XCGD-iPSCs, indicating that the XCGD phenotype has been successfully recapitulated in this modeling system (Figure 2e).
Figure 2.

Characteristic features of iPSC-derived neutrophils. (a) Detached iPSCs were co-cultured on a layer of C3H10T1/2 cells and induced to undergo hematopoietic differentiation. One week later, HPCs could be harvested from sac-like structures and then further induced to undergo neutrophil differentiation. (b) Histological appearance of differentiated neutrophils Wright Giemsa (WG) staining, myeloperoxidase (MPO) (black dots) and alkaline phosphatase (ALP) (purple dots). (c) NBT-coated yeast were added to a suspension of differentiated cells for 1 hour at 37 °C and then stained with 1% safranin-O. Phagocytosed yeast is black in color due to the reduction of NBT to formazan by ROS (black arrows). (d) Differentiated cells were seeded onto Poly-l-lysine coated cover slips and stimulated with PMA to induce the formation of neutrophil extracellular traps (white arrows). After permeabilization and fixation, cells were stained for SYTO 13 (green) and MPO (red). (e) Recapitulation of the XCGD phenotype. Neutrophils were differentiated from either healthy (Healthy) or XCGD iPSCs. Nonstimulated (gray) and stimulated cells with PMA (black line) were assessed for ROS production through the DHR assay. Bars = 200 μm. DHR, dihydrorhodamine123; PMA, phorbol myristate acetate.
Cellular recovery in XCGD iPSC-derived neutrophils was not restored to wild-type levels following transgene-derived gp91phox transfer
XCGD iPSCs were transduced using the Alpha.SIN.EFS.gp91.IRES.PURO vector (Figure 3a) for the purpose of restoring transgene-derived gp91phox expression and ROS production. Puromycin selection pressure was sustained throughout from the maintenance of transduced XCGD iPSCs to subsequent differentiation into polymorphonuclear neutrophils in order to ensure that only transgene-positive cells are analyzed. A multicolored flow cytometry analysis, optimized specifically for these objectives, allowed for the concomitant analysis of gp91phox expression or ROS production by focusing on the mature (CD64highCD15high) population (Figure 3b). In the untransduced XCGD group, differentiated neutrophils expressed neither gp91phox nor generated ROS (XCGD). In contrast, mature neutrophils from the healthy control group expressed gp91phox at normal levels on a per-cell basis (Healthy). It was discovered in transduced XCGD neutrophils that while increased recovery in gp91phox expression corresponded with increased vector copy number (VCN) from 1.01 to 9.25 (multiplicity of infection (MOI) 1 and 10; Table 1), the extent of recovery was considerably less compared with the healthy control group (Figure 3b, MOI 1 and MOI 10, top row). Similarly, the restoration in ROS-generating capacity did not reach wild-type levels (Figure 3b, MOI 1 and MOI 10, bottom row).
Figure 3.

Cellular recovery and expression of NOX2 subunits. XCGD-iPSCs were transduced with an alpharetroviral vector at MOIs 1 and 10. (a) Schema of the pAlpha.SIN.EFS.gp91s.IRES.PURO provirus. Control cells were those differentiated from untransduced XGCD or Healthy iPSCs. (b) Transduced XCGD-iPSCs were differentiated into neutrophils and assessed for gp91phox expression (top row) where cells were stained with the isotype control (gray) or anti-gp91phox antibody (black line). Nonstimulated (gray) and stimulated cells with phorbol myristate acetate (black line) were assessed for ROS production (bottom row) through the dihydrorhodamine123 assay. (c) In a separate experiment, differentiated neutrophils were assessed for gp91phox (top row), p47phox (middle row), and p67phox (bottom row) expression. Gray indicates staining with the isotype control and black lines indicate protein expression. All numbers indicate the mean fluorescence intensity (MFI). MOI, multiplicity of infection.
Table 1. Vector copy number in transduced iPSCs.
| Provirus | Cell source | MOI | VCN |
|---|---|---|---|
| pAlpha.SIN.EFS.gp91s.IRES.PURO.oPRE | XCGD iPSCs | 1 | 1.01 |
| pAlpha.SIN.EFS.gp91s.IRES.PURO.oPRE | XCGD iPSCs | 10 | 9.25 |
| pAlpha.SIN.SFFV.gp91s.IRES.PURO.oPRE | XCGD iPSCs | 10 | 14.15 |
| pAlpha.SIN.EFS.gp91s.F2A.PURO.T2A.NGFR.oPRE | XCGD iPSCs | 1 | 1.14 |
| pAlpha.SIN.EFS.PURO.T2A.NGFR.oPRE | XCGD iPSCs | 1 | 1.81 |
| pAlpha.SIN.EFS.gp91s.F2A.PURO.T2A.NGFR.oPRE | Healthy iPSCs | 1 | 6.78 |
| pAlpha.SIN.EFS.PURO.T2A.NGFR.oPRE | Healthy iPSCs | 1 | 8.11 |
MOI, multiplicity of infection; VCN, vector copy number.
Since assembly of NADPH oxidase requires additional intracellular components, the expression of gp91phox (Figure 3c, top row), p47phox (Figure 3c, middle row), and p67phox (Figure 3c, bottom row) was determined in mature neutrophils within the same experimental groups. Despite the absence of gp91phox expression, in the XCGD group, both p47phox and p67phox were still expressed albeit at a lower level (XCGD) compared with the healthy controls (Healthy). Unexpectedly, there was a further opposing decrease in the expression of these two subunits that appeared to correlate negatively with an increase in gp91phox expression (MOI 1 and MOI 10). This data may suggest that transgene-derived gp91phox expression could induce a detrimental cellular response since an opposite trend would be expected given that these subunits are known to be critical to gp91phox stability.
Ectopic gp91phox expression leads to nonphysiological expression of ROS
In this study, as with previous hematopoietic SCGT clinical trials, an integrating vector is used, where an internal ubiquitous promoter drives transgene expression. Taking this into consideration, the hypothesis was made that the expression of gp91phox could occur in nontargeted cell populations resulting in nonphysiological consequences. By adopting the same multicolor analysis as described previously, cells were first assessed for gp91phox expression. In mature cells, gp91phox expression was found in all except for the XCGD group (Figure 4a, top row). As an additional control, the alpharetroviral vector was modified by using the SFFV promoter (Supplementary Figure S5a), which is known to have stronger activity21 in general than the EFS promoter in driving transgene expression. Surprisingly, restoration of gp91phox expression in the SFFV group was lower compared with the EFS group on a per cell basis (EFS versus SFFV). This may in part be attributed to the susceptibility of this vector to silencing in pluripotent stem cells.22 In the developing fraction (Figure 4a, bottom row), no gp91phox expression was detected in either one of the control groups including those differentiated from a healthy genetic background (XCGD and Healthy). However, in neutrophils differentiated from transduced XCGD iPSCs, gp91phox expression can now be observed in the developing fraction (EFS and SFFV). In the context of this study, the expression of gp91phox in this cell fraction is herein defined as “ectopic.” Thereafter, cells were assessed for the generation of ROS firstly in the mature fraction (Figure 4b, top row). As expected in the transduced groups (EFS and SFFV), ROS production was incomplete on a per-cell basis compared with the healthy control. In the developing fraction (Figure 4b, bottom row), only marginal oxidase activity was detected in the untransduced controls (XCGD and Healthy). However, ROS production could now be detected in the developing fraction of the transduced groups, which may be described as “nonphysiological” (EFS and SFFV). Therefore, a more refined hypothesis could be made that ectopic gp91phox expression in the developing fraction of neutrophils allows these cells to acquire the potential of producing nonphysiological ROS. This could result in the occurrence of detrimental effects that could impede further differentiation such that in cells those are able to transit into a mature hierarchy, cellular recovery is incomplete.
Figure 4.

Identification of neutrophil populations expressing ectopic gp91phox with ROS producing capacity. EFS and SFFV represents neutrophils differentiated from iPSCs where gp91phox expression is driven by these two promoters specifically. Control cells were those differentiated from untransduced healthy or XGCD-iPSCs. (a) Expression of gp91phox. Top row indicates expression in mature (CD64highCD15high) cells. Bottom row indicates expression in developing (CD64dullCD15dull) neutrophils. In this population, gp91phox expression could only be detected in the transduced groups. Gray indicates the isotype control and black line indicates gp91phox expression. (b) Generation of ROS in mature (top row) and developing (bottom row) neutrophils. Nonphysiological ROS production was only detected in the transduced groups where cells were ectopically expressing gp91phox. Gray indicates no stimulation and black line indicates stimulation with phorbol myristate acetate. All numbers indicate the MFI.
Hierarchical transition during neutrophil differentiation
To support the hypothesis that gp91phox may impede neutrophil differentiation, it is important to confirm that within this culture system, differentiating neutrophils could make the transition from developing to a mature hierarchy. To demonstrate this, after sac harvest, HPCs were differentiated for 3 days to obtain a heterogeneous mixture of both developing and mature neutrophils (Figure 5, pre-sorting). At this point, the developing fraction (CD64dullCD15dull) was sorted and checked for its purity in composition (Figure 5, purity). This sorted population was then differentiated for an additional 2 days by which point, both developing (CD64dullCD15dull) and mature (CD64highCD15high) populations could be identified regardless of whether cells were differentiated from XCGD or healthy iPSCs (Figure 5, differentiated). These data confirm that in this culture system, cells follow this pathway of differentiation.
Figure 5.

Developing neutrophils become mature neutrophils with an extended differentiation period. After 3 days of differentiation, a combination of both developing (CD64dullCD15dull) and mature (CD64highCD15high) neutrophilss could be seen in the culture at this point (first column; Pre-sorting). The developing fraction was sorted by FACS and the purity of this sorted fraction was checked (second column; Purity). After two further days of differentiation, the appearance of mature cells that originated from the sorted developing fraction could be identified (third column; Differentiated).
Ectopic expression of gp91phox impedes neutrophil maturation
In determining the effect of ectopic gp91phox expression acting as an impediment to neutrophil maturation, it is important to carry out a broad assessment of whether this correlates with all stages of differentiation within the conditions of this culture system. To exclude puromycin as an additional factor that could compromise neutrophil viability, selection pressure was removed after HPC harvest from iPSC sacs. In order to detect transgene-positive cells and analyze them by multicolor FACS, the design of alpharetroviral vectors was further modified to include a truncated nonsignaling version of the neural growth factor receptor (NGFR) (Supplementary Figure S5b). This is to allow for the detection of gp91phox-expressing neutrophils transduced with the therapeutic (gp91phox-containing) vector but not the control, which contains only puromycin resistance gene and NGFR.
On day 3, the expression of gp91phox in mature neutrophils differentiated from the healthy control (Figure 6a, Healthy) or therapeutic vector-transduced (Figure 6a, Therapeutic) XCGD iPSCs were comparable. The level of gp91phox expression remained comparable between the same groups of cells on day 5 (Figure 6b, Healthy and Therapeutic). Ectopic gp91phox expression could only be detected in developing neutrophils from the therapeutic group. On day 7, mature neutrophils differentiated from healthy iPSCs (Figure 6c, Healthy) showed a considerable increase in gp91phox expression over earlier time points, indicating that further cell maturation had occurred despite the same characterization by immunophenotype. In stark contrast, gp91phox expression has nearly disappeared in the therapeutic vector-transduced group (Figure 6c, Therapeutic) in both the mature and developing fractions. Interestingly, the presence of ROS high producing neutrophils at the same time points of differentiation reflects the expression of gp91phox. Similar to the healthy control group, mature ROS high producing neutrophils from the therapeutic vector transduced group could be detected up to day 5, but these cells were abruptly lost by day 7 (Supplementary Figure S6).
Figure 6.
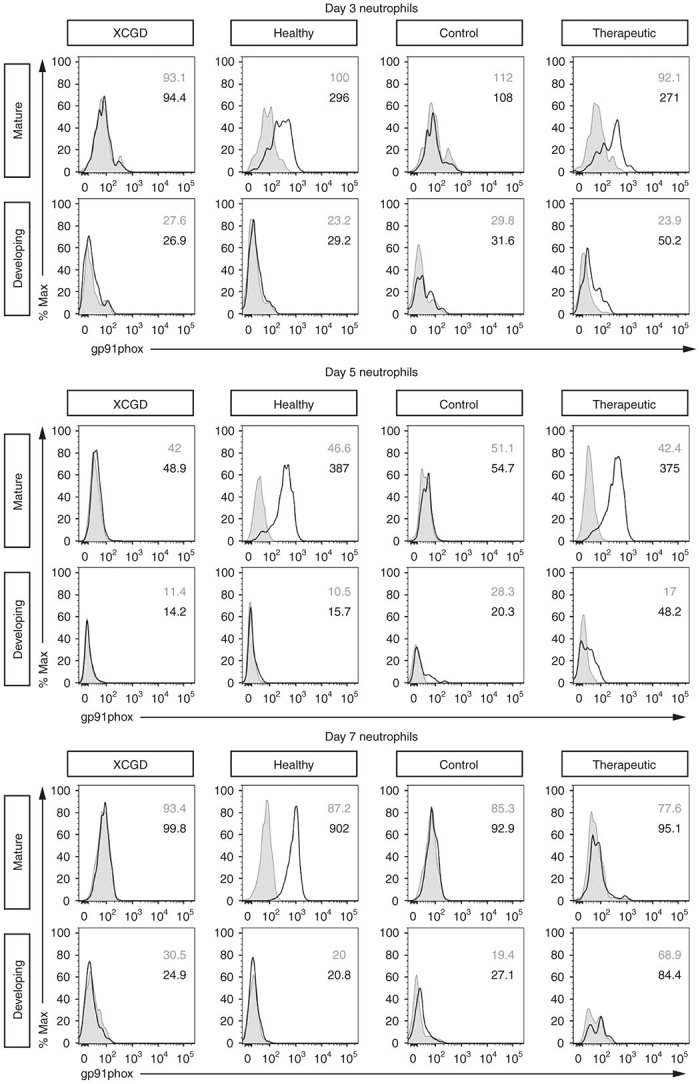
Time-course analysis of gp91phox expression in differentiating neutrophils. XCGD iPSCs were transduced with either the control vector (Control) or gp91phox-containing (Therapeutic) vectors. Control cells were those differentiated from untransduced healthy (Healthy) or XGCD-iPSCs (XCGD). On days 3, 5, and 7 after initiating differentiation, the expression of gp91phox was assessed in both developing and mature neutrophils. Gray indicates the isotype control and black line indicates gp91phox expression. All numbers indicate the MFI.
Disappearance of XCGD-developing neutrophils expressing ectopic gp91phox
In addition to potential detrimental effects occurring secondary to ectopic gp91phox expression, it is possible that the loss in cellular recovery is also due to abrupt silencing of the vector. Since the differentiation impediment is thought to occur at the developing stage of differentiation, at the same time points, NGFR expression was determined in this fraction of neutrophils. No expression was observed for the untransduced controls (Figure 7, XCGD and Healthy) at any time point. On days 3 and 5, NGFR expression can clearly be detected in developing neutrophils differentiated from XCGD iPSCs transduced with either the control (Figure 7, Control) or gp91phox-containing (Figure 7, Therapeutic) vectors. However, by day 7, only the control vector group showed significant NGFR expression. Considering that both vectors share similar designs and were used to transduce XCGD-iPSCs under the same experimental conditions, neither would be considered as being more susceptible to silencing than the other. Therefore, gene silencing alone is unlikely to be a significant factor leading to the loss of NGFR and also gp91phox expression in the therapeutic vector-transduced cells.
Figure 7.

NGFR expression in developing neutrophils. Following the same differentiating conditions for the time-course analysis of gp91phox, at the same time points, the expression of NGFR in developing neutrophils was assessed to determine whether significant vector silencing has occurred. Gray numbers indicate the percentage of NGFR –ve population and black numbers indicate the percentage of NGFR +ve population.
Developing neutrophils ectopically expressing gp91phox are susceptible to increased cell death
Cells were subjected to the 7-AAD/Annexin V apoptosis assay to determine viability. The percentage of cell death was calculated by combining the total percentage of 7-AAD-positive only and 7-AAD/AnnexinV double-positive cells (Supplementary Figure S7) in both the NGFR-negative and NGFR-positive fractions. Despite ectopic gp91phox being detectable on day 3 in developing neutrophils differentiated from therapeutic vector transduced XCGD iPSCs, no significant cell death can be detected at this time point (Figure 8a). However, by day 5, it was found that NGFR-positive developing neutrophils differentiated from iPSCs transduced with the gp91phox-containing vector exhibited significant levels of cell death compared with the NGFR-negative fraction (Figure 8b). At this time point, the same profile of cell death could be observed regardless of whether gp91phox was ectopically expressed in cells differentiated from either an XCGD or healthy (Supplementary Figure S8) genetic background. These findings suggest that ectopic gp91phox expression could exert detrimental effects by compromising the viability of developing neutrophils, which may act as an impediment to further differentiation. By day 7, where previously gp91phox expression was no longer detectable in mature therapeutic vector-transduced cells, there were no longer significant levels of cell death (Figure 8c).
Figure 8.

Developing neutrophils ectopically expressing gp91phox are susceptible to cell death. On days 3, 5, and 7 of differentiation, the 7-AAD/Annexin-V cell death/apoptosis assay was carried out to determine the total percentage of cell death in the developing neutrophil population of each experimental group. NGFR expression indicates transgene-expressing cells transduced with the control (Control) or gp91phox-transgene containing (Therapeutic) vectors, respectively. Depicted are mean values + SD (n = 3, representative of three independent experiments). *P < 0.05. Statistically significant levels of cell death were only detected in cells ectopically expressing gp91phox. Control cells were those differentiated from untransduced healthy or XGCD-iPSCs.
Discussion
This study demonstrated the important use of patient autologous iPSCs as an in vitro neutrophil differentiation modeling system for XCGD. These cells acted as a platform for developing a multicolored flow cytometry analysis, which allowed for the concomitant identification, within the same culture, of cells according to their differentiation status (developing or mature) followed by other subsequent analyses. Following a time-course assessment of cellular recovery in the context of an iPSC-based modeling system, it could be observed that gp91phox expression in the mature fraction of transduced neutrophils was comparable with that of healthy controls before cells became fully differentiated. However, while endogenous gp91phox expression in healthy control cells continued to increase following extended culture time, transgene-derived gp91phox expression in transduced XCGD cells was abruptly lost. Most critically, ectopic gp91phox expression and nonphysiological production of ROS was detectable in developing neutrophils only in those differentiated from gp91phox-transduced XCGD iPSCs. This appeared to correlate with statistically significant levels of cell death and coincided with a loss in mature gp91phox-expressing cells. Taken together, the data suggest that ectopic gp91phox expression may act as an impediment to further differentiation from XCGD iPSCs by reducing the viability of developing neutrophils.
The key finding from this study was that cellular recovery in neutrophils differentiated from gp91phox-transduced XCGD-iPSCs could not reach levels that were comparable to healthy counterparts. Since this finding was not further verified through applying the same exact methods as described herein using another XCGD modeling system, i.e., CD34+ cells from a patient, there remains the possibility that the detrimental effects of ectopic gp91phox expression is a phenomenon that is unique to the nature of iPSC-based modeling. In fact, Kaufmann et al.23 demonstrated functional correction in myeloid cells differentiated form XCGD CD34+ cells using a similar alpharetroviral vector design. In that report, gp91phox-transduced XCGD CD34+ cells from a patient were induced to undergo myeloid (CD11bhigh+) differentiation by granulocyte colony-stimulating factor. Rather promisingly, the recovery of ROS production in differentiated cells reached to 67% of wild-type levels on a per-cell basis. Future plans to repeat experiments using XCGD CD34+ cells would determine whether similar detrimental effects secondary to ectopic gp91phox expression could also be observed in these cells. However, preliminary experiments aimed at optimizing the in vitro neutrophil differentiation from CD34+ cells from a healthy individual showed that the kinetics of differentiation appeared to be different compared with iPSCs in that a longer period of time was required to obtain mature neutrophils and that clear separation by flow cytometry of developing from mature neutrophils were less distinct (data not shown). Therefore, the focus was placed solely on the use of the iPSC-based system.
Considering that cellular recovery was incomplete following transduction, it is possible that silencing of transgene expression could have occurred. This may be true especially when the SFFV promoter was used, which did not restore cellular recovery closer to wild-type levels on a per-cell basis (Figure 4). Previously, it has been reported that extensive methylation of the SFFV promoter occurred following differentiation in both lentiviral and gammaretroviral vector constructs,24 but it may also apply to the EFS promoter as well.16 As part of the future plans, bisulfite sequencing of the promoter regions for example would indicate the extent of methylation that has occurred in the alpharetroviral vectors used in this study.
However, the data presented here suggests that gene silencing alone would not fully explain the incomplete levels of cell recovery observed. To account for the possible susceptibility of promoter silencing in this system, puromycin selection pressure was sustained throughout from the maintenance of transduced iPSCs to the end of the differentiation process to ensure consistent selection of gp91phox-expressing cells. In addition, it could be confirmed that in some cases, the estimated VCN was as high as 9.25 (EFS) and 14.15 (SFFV), respectively (Table 1). For certain assays, it was necessary to lift puromycin selection pressure at the start of neutrophil differentiation, which may otherwise compromise cell viability in addition to potential ectopic gp91phox involvement. In these situations, an additional selection marker, ΔLNGFR (truncated NGFR), was incorporated into the vector design with the use of the EFS promoter to identify transgene-positive cells throughout the differentiation process. 2A peptides were used for vector construction, which was previously demonstrated to be effective at ensuring efficient and robust multicistronic expression of multiple transgenes in embryonic stem cells.25 Both the control and therapeutic vectors shared similar designs differing only in the gp91phox transgene (Supplementary Figure 5b). Therefore, neither vector construct would be considered to be more susceptible to silencing than the other. However, NGFR abruptly disappeared in therapeutic vector-transduced cells by day 7 of differentiation but expression was retained in control vector–transduced cells (Figure 7). Therefore, these data support the idea that ectopic gp91phox could lead to detrimental effects in this iPSC-based system. Yet it remains unclear in neutrophils whether all transgene-derived gp91phox becomes the stably assembled or whether any excess is subsequently degraded. This information would be necessary for establishing a threshold of tolerance in cells to nonphysiological ROS production. In addition, from the results of this study, it is not possible to rule out the possibility that cell death may be caused by direct toxicity to the gp91phox protein itself. Future work may be necessary to use a mutant form of gp91phox in which the protein is translated but ROS-generating capacity remains abolished.26
Overall, this study demonstrated the usefulness of multicolor-flow cytometry analysis in the context of an iPSC-based neutrophil differentiation model to test how ectopic gp91phox expression would affect specific cell populations, i.e., developing and mature neutrophils. With optimized procedures, this technique allowed for the concomitant analysis of surface markers alongside other NADPH oxidase components, ER stress markers, and the quantification of cell viability. In a previous study comparing global gene expression in polymorphonuclear neutrophils, it was found that compared with healthy individuals, there was a 3.1-fold downregulation in CYBB (gp91phox) expression in XCGD patients, which correlated with an opposite 2.7-fold increase in NCF1 (p47phox) expression.27 Here, a negative correlation could be identified at the protein level between gp91phox and p47/67phox expression. In addition, previously it had been reported that neutrophils could be induced to undergo apoptosis following sustained ER stress.28 Also, NADPH oxidase-derived ROS has been shown to be capable of inducing ER stress.14 For these reasons, the expression of the ER stress markers GRP78, pEIF2α, pJNK, and XBP-1 was assessed (Supplementary Figures S9 and S10). Multicolor flow cytometry analysis enabled the relative expression of each marker to be calculated by subtracting the mean fluorescence intensity in the NGFR-negative fraction from the NGFR-positive fraction (Supplementary Figure S9). It was found in the XCGD group that there was a significant increase in GRP78 expression in developing neutrophils ectopically expressing gp91phox (Supplementary Figure S10a, XCGD, Ectopic gp91 versus Control). Interestingly, the presence of ectopic gp91phox protein corresponded with a significant increase in pEIF2α expression in healthy cells (Supplementary Figure S10b, Healthy, Ectopic gp91 versus Control). No statistically significant differences could be detected for pJNK and XBP-1 (Supplementary Figure S10c,d). Although it cannot be concluded that induced ER stress is the main underlying mechanism leading to induced apoptosis, the data do suggest that these cells may have the propensity for undergoing such a fate. Therefore, cell viability was determined in cells ectopically expressing gp91phox but given the wide range of physiological functions of NADPH oxidase, it is possible that perturbations in ROS production may lead to other negative effects.
Although the clinical relevance of our iPSC-based neutrophil differentiation system remains to be further addressed, it will be a useful platform for determining how faithfully an artificial promoter would restore gp91phox expression relative to healthy endogenous levels. In fact, it was possible to determine the precise point during hematopoietic differentiation that gp91phox expression appeared and its level of expression at given time points. During the time-course assessment of gp91phox expression in healthy control cells, it was not detected in the developing fractions (CD64dullCD15dull) at any time point, indicating the robust and stringent regulation of this NADPH subunit during development. Expression of gp91phox became detectable only in mature fractions (CD64highCD15high), but the levels varied significantly during the time-course analysis, showing highest levels of expression at day 7 (Figure 6c) compared with those at day 5 (Figure 6b). Consistent with these observations, earlier during hematopoietic differentiation, it is known that gp91phox expression is very low in stem or progenitor hematopoietic cells but becomes much higher in mature phagocytes.12 To date, it remains to be seen whether constitutive overexpression of gp91phox can compromise the functionality or engraftment potential of HSCs.29 Nonetheless, there seems to be a consensus that transgene expression should be avoided in this population unless absolutely necessary in order to minimize any genotoxic risks.30 Recently, a dual-regulatory strategy combining transcriptional and posttranscriptional regulation in which the use of a lentiviral vector with a myeloid-specific promoter and two micro-RNA 126 “sponge” sequences were shown to effectively restrict gp91phox expression to the myeloid compartment with minimal leakage.31 It would be interesting to see whether by using the latest generation of clinical vectors, the levels gp91phox expression achieved could reach closer to healthy wild-type levels using our iPSC-based system. By combining with the site-specific integration techniques, such vector testing could become feasible in the near future.
In summary, by using XCGD iPSCs as a modeling platform to assess the effects of constitutive transgene expression, an alternative prospective of this disease has been presented. There is promise in that ROS antagonists such as NAC is already in clinical use to treat a range of existing diseases including pulmonary and neurological disorders,32 but it remains to be seen how this may apply to XCGD treatment. The evidence here also supports the current trend in hematopoietic SCGT of lineage restricted transgene expression as this strategy may both minimize the genotoxic risk and improve neutrophil viability at the same time. It is hoped that further study using this iPSC modeling system may yet yield additional therapeutic targets the resolution of which could lead to improvements in CGD clinical outcomes.
Materials and Methods
Sendai virus vectors
Development of nonintegrating Sendai virus vectors harboring human OCT3/4, SOX2, KLF4, and c-MYC is as previously described.33 The removal of Sendai virus vectors from iPSCs was carried out using Lipofectamine RNAi Max (Invitrogen, Waltham, MA) to transfect established iPSC lines with small interfering RNA L527.
Alpharetroviral vectors
Alpharetroviral vectors34 were kindly provided by Dr Axel Schambach (Hannover Medical School, Germany). The production of viral supernatant is as previously described.35 To construct the pAlpha.SIN.SFFV.IRES.PURO transfer plasmid, IRES-PURO (SalI-XbaI) from LV.EF.L3T4.IRES.PURO (provided by Dr T. Yamaguchi, University of Tokyo, Japan) was cloned into pAlpha.SIN.SFFV.gp91s.oPRE. For pAlpha.EFS.IRES.PURO, the EF1α short promoter (NotI-BamHI) from pAlpha.EFS.gp91s.oPRE was cloned into pAlpha.SFFV.IRES.PURO. For the construction of pAlpha.SIN.EFS.gp91s.F2A.PURO.T2A.ΔLNGFR, there was initial PCR amplification of gp91s (BamHI-ClaI) from pAS.SFFV.gp91s.oPRE, Puromycin resistance gene (XbaI-BspEI) from CS.CDF.CG.PRE,36 and ΔLNGFR (HpaI-AccI) from SFCMM-2 (ref. 37). These three fragments were then cloned into T7 mOKS38 to replace Oct4, Klf4, and Sox2, respectively, to create pT7.gp91s.PURO.ΔLNGFR. From this, the gp91s.F2A.PURO.T2A.ΔLNGFR cassette (BamHI-AccI) was then cloned into pAlpha.EFS.gp91s.oPRE. For the control vector pAlpha.EFS.PURO.T2A.ΔLNGFR.oPRE, the PURO.ΔLNGFR cassette (BamHI-AccI) from T7.gp91s.PURO.ΔLNGFR was inserted instead.
Generation of patient autologous iPSCs and transduction
PB samples were obtained from either a healthy volunteer or a XCGD patient on informed consent following the Declaration of Helsinki standard ethics procedure with approval from the Institutional Ethical Committees. PB CD34+ cells were isolated using the CD34 MicroBead Kit according to the manufacturer’s protocol (Miltenyi Biotec, Bergisch Gladbach, Germany). These were then transduced with the earlier described Sendai virus vectors to generate iPSCs. Reprogrammed iPSCs were maintained under conditions as previously described.39 For the transduction of iPSCs, healthy or XCGD iPSCs were first maintained under feeder free conditions on matrigel (BD Biosciences, San Jose, CA). Transduced cells were subsequently maintained on puromycin-resistant mouse embryonic fibroblasts under constant puromycin (A11138-03; Life Technologies, Waltham, MA) selection pressure.
Determination of VCN
VCN was determined using the QX200 Droplet Digital PCR System (BIO-RAD, Hercules, CA) as per manufacturer’s instructions (manuscript in preparation for detailed methods). Genomic DNA was extracted from transduced XCGD or healthy iPSCs using the NucleoSpin Tissue XS kit (MACHEREY-NAGEL, Duren, Germany). Target primer-probes were designed to recognize sequences within the puromycin resistance gene.
Immunocytochemical staining of iPSCs
To begin with, iPSC colonies were fixed using 5% paraformaldehyde and permeabilized with 0.1% Triton X-100. Primary phycoerythrin (PE)-conjugated anti-SSEA-4 antibody (FAB1435P; R&D Systems, Minneapolis, MN) was used for SSEA-4 staining. For TRA-1–60 and TRA-1–81, pretreated colonies were incubated with primary mouse antihuman TRA-1–60 (MAB4360; Merck Millipore, Darmstadt, Germany) and mouse antihuman TRA-1–81 (MAB4381; Merck Millipore). This is followed by incubation with secondary Alexa Fluor 488-conjugated goat antimouse antibody (A11029; Molecular Probes-Invitrogen, Waltham, MA). 4′,6-diamidino-2-phenylindol (Roche Diagnostics, Basel, Switzerland) were used to counterstain cell nuclei.
ALP activity in iPSCs
10% formalin neutral buffer solution (Wako, Tokyo, Japan) was used for the fixation of iPSC colonies. ALP activity was then detected using the 1-Step NBT/BCIP (34042; Thermo Fischer Scientific, Waltham, MA) solution.
Teratoma formation
Dissociated iPSCs were injected at a density of 1.0 × 106 cells into the medulla of a single testis of a NOD/ShiJic-scid mouse.40 After 12 weeks, the resulting teratomas were resected and then fixed in 5% paraformaldehyde before being embedded in paraffin. Microscopic sections were stained with hematoxylin and eosinstain to determine evidence of tri-lineage germ layer contribution. Approvals for conducting these experiments were granted by the institutional regulation board for human ethics at the Institute of Medical Science, University of Tokyo.
Karyotyping and point mutation sequencing
Chromosome karyotyping (Q-banding) was performed by Chromosome Science Labo (CSL, Sapporo, Japan). Fifty total metaphases were examined.
Neutrophil differentiation of human iPSCs
Human iPSCs were first differentiated into HPCs as previously described41 with slight modifications. In brief, irradiated puromycin-resistant C3H10T1/2 were co-cultured with detached iPSC colonies in hematopoietic cell differentiation medium (Iscove’s modified Dulbecco’s medium supplemented with 15% fetal bovine serum [FBS] and a cocktail of 10 mg/ml human insulin, 5.5 mg/ml human transferrin, 5 ng/ml sodium selenite, 2 mmol/l L-glutamine, 0.45 mmol/l a-monothioglycerol, and 50 mg/ml ascorbic acid in the presence of VEGF) under sustained puromycin selection pressure. On day14, HPCs were harvested from “sac-like” structures. For neutrophil differentiation, HPCs were continuously co-cultured with irradiated C3H10T1/2 cells in differentiation medium (αMEM + 10% fetal bovine serum [(Biological Industries, Kibbutz Beit Haemek, Israel) + 1% penicillin streptomycin glutamine) including 50 ng/ml G-CSF (R&D Systems) with a half medium change every 3 days.
Cell morphology and expression of cytoplasmic proteins
After methanol fixation, cell morphology of differentiated neutrophils were determined by Wright-Giemsa (WG) staining using Hemacolor solution 2 (Red, 111956) and solution 3 (Blue, 111957) (Merck Millipore). MPO and ALP were stained using the New PO-K and ALP staining kits, respectively (Muto Pure Chemicals, Tokyo, Japan).
NBT-coated yeast phagocytosis assay
Autoclaved Baker yeast was suspended at a density of 1 × 108/ml in 0.5% NBT solution (Sigma-Aldrich, St Louis, MO) dissolved in phosphate-buffered saline. A 5 μl suspension was incubated with 2.5 × 105 differentiated neutrophils suspended in 50 μl of fetal bovine serum. After 1 hour of incubation at 37 °C with periodic tapping, cytospin preparations were made of each sample, which was then stained with 1% safranin-O (Muto Pure Chemcals).
Formation of neutrophil extracellular traps
Differentiated neutrophils were seeded onto poly-l-lysine (Sigma-Aldrich) coated glass coverslips (Matsunami, Osaka, Japan). The method used for generating neutrophil extracellular traps were adapted from a previously described protocol.42 After permeabilization and fixation, cells were stained with primary purified polyclonal anti-MPO antibody (Abcam, Cambridge, UK) and then secondary Alexa Fluor 546-conjugated goat antirabbit antibody (A11029, Molecular Probes-Invitrogen) along with SYTO13 (Life Technologies, Thermo Fischer, MA) to stain nucleic acids.
Flow cytometry determination of surface marker and NGFR expression
For surface staining, cells were first stained with primary monoclonal rabbit anti-human FCGR1A antibody (EPR4623, Abcam). This was followed by secondary Alexa Fluor 488-conjugated goat anti-rabbit antibody (A11034, Molecular Probes-Invitrogen) and BV605-conjugated anti-human CD15 (W6D3, BioLegend, San Diego, CA). Where the detection of NGFR expression is subsequently desired, cells are then treated with FcR blocking reagent (130-059-901; Miltenyi Biotec). This was followed by staining with primary anti-CD271 (NGFR) (HE20.4, BioLegend) and then secondary APC/Cy7-conjugated goat anti-mouse IgGI (Southern Biotech, Birmingham, AL) or BV421-conjugated antimouse IgG (RMG1-1, BioLegend). Data were acquired on FACSAria I or II sorter (BD Biosciences) and analyzed using FlowJo X software (Tree Star, Ashland, OR).
Intracellular detection of gp91/p47/p67 phox
Following surface staining of CD64 and CD15, cells were fixed and permeabilized using the Fixation/Permeabilization solution (Becton Dickinson Franklin Lakes, NJ). For the detection of gp91phox, cells were then stained with PE-conjugated monoclonal anti-human gp91phox antibody (7D5, MBL, Nagoya, Japan) or control PE-conjugated anti-mouse IgG1 (BioLegend). Alternatively, cells were instead incubated with anti-p47phox (clone 1, Becton Dickinson), anti-p67phox mAb (clone D-6, Santa Cruz Biotechnology, CA) or purified mouse IgG1 (Becton Dickinson). This was followed by reaction with PE-conjugated anti-mouse IgG1 (Southern Biotech).
Detection of ROS
The production of ROS by test cells including PB, and differentiated developing and mature neutrophils was detected using the dihydrorhodamine123 flow cytometry assay as previously described.43 In brief, cell samples were suspended in ~400 μl of reaction buffer (HBSS + 0.5% BSA). Catalase (Sigma-Aldrich) was added at a final concentration of 1400 U/μl alongside 1.8 μl of a 29 mmol/l stock solution of dihydrorhodamine123 (Sigma-Aldrich). Phorbol myristate acetate (Sigma-Aldrich) was added to stimulate samples at a final concentration of 0.2 μmol/l. After 15-minute reaction at 37 °C, the samples were washed and analyzed by flow cytometry. When indicated, cell surface antigen was stained as previously described.
Reverse transcription polymerase chain reaction
The RNeasy Micro kit (Qiagen, Hilden, Germany) was used to extract RNA from iPSCs, which was then reverse transcribed using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Thermo Fischer, Waltham, MA) with random 6-mer primers. Reverse transcription polymerase chain reaction was performed using ExTaq HS (Takara, Shiga, Japan). Individual PCR reactions were normalized against GAPDH rRNA. Primer sequences can be found in Table 2.
Table 2. Primer sequences for RT-PCR.
| Gene | Primer orientation | Sequence |
|---|---|---|
| OCT3/4 | F | 5′-ATTCAGCCAAACGACCATC-3′ |
| R | 5′-GGAAAGGGACCGAGGAGTA-3′ | |
| SOX2 | F | 5′-CAGCGCATGGACAGTTAC-3′ |
| R | 5′-GGAGTGGGAGGAAGAGGT-3′ | |
| c-MYC | F | 5′-AGTTTCATCTGCGACCCG-3′ |
| R | 5′-CCTCATCTTCTTGTTCCTCCT-3′ | |
| KLF4 | F | 5′-GCGGGAAGGGAGAAGACA-3′ |
| R | 5′-CCGGATCGGATAGGTGAA-3′ | |
| NANOG | F | 5′-CAGCCCTGATTCTTCCACCAGTCCC-3′ |
| R | 5′-TGGAAGGTTCCCAGTCGGGTTCACC-3′ | |
| GAPDH | F | 5′-AACAGCCTCAAGATCATCAGC-3′ |
| R | 5′-TTGGCAGGTTTTTCTAGACGG-3′ |
RT-PCR, reverse transcription polymerase chain reaction.
Intracellular detection of ER stress markers
Cells were first stained for CD64 and CD15 as followed by treatment with FcR blocking reagent (130-059-901, Miltenyi Biotec). Cells were then treated with the fixation/permeabilization solution (Becton Dickinson). Staining of ER-stress markers were done firstly with primary rabbit antihuman EIF2S1 (E90, Abcam), rabbit antihuman BiP (C50B12, Cell Signaling Technology, Danvers, MA), mouse anti-human phosphor SAPK/JNK (G9, Cell Signaling Technology), and mouse anti-human XBP1 (S Isoform, R&D Systems) antibodies. This was followed by secondary staining with either PE-conjugated mouse IgG1 (RMG1-1, Biolegend) or goat antirabbit AF546 antibody (A11010, Molecular Probes-Invitrogen). In the final step, detection of NGFR expression is as previously described.
Cell death apoptosis assay
Following cells surface and NGFR staining, cell viability was assessed using the PE Annexin V Apoptosis Detection Kit I (BD Pharmingen San Jose, CA) as per the manufacturer’s instructions.
Statistical analysis
Mean grouped values were compared using one-way or two-way ANOVA. All statistical analyses were carried out using Prism 6 software (Graphpad, San Diego, CA).
Acknowledgments
Special thank goes to Y. Ishii (FACS Core Laboratory, University of Tokyo) for her technical support on the flow cytometry analyses. H. Takagi assisted with optimizing ROS assays. Further gratitude extends to A. Schambach for supplying the alpharetroviral SIN vector alongside M. Grez and K. Kaufmann for their technical insight and discussions. Additional thanks also go to T. Wada for providing the XCGD patient’s peripheral blood sample and M. Nakanishi for providing the Sendai virus vector. Mr. Yukinori Yatsuda and his team at Bio-Rad kindly collaborated on the ddPCR work. The authors have no other conflict of interest to declare. This work was supported by funding from the Project for the Realization of Regenerative Medicine and Support for Core Institutes for iPS Cell Research from the Ministry of Education, Culture; Support for the Core Institutes for iPS Cell Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT; to H.N.); and a Grant-in-Aid for the Global COE Program from MEXT to the University of Tokyo. This work was also supported by The Program for Intractable Disease Research utilizing Disease-specific iPS cells, Research Center Network for realization of Regenerative Medicine, Japan Science and Technology Agency (JST). H.-T.L. was supported by the Interchange Association of Japan, the Academic Research Grant for Graduate School of Frontier Sciences Doctor Course Students and the EGS research assistant program, IMSUT, University of Tokyo.
References
- Segal, AW (1996). The NADPH oxidase and chronic granulomatous disease. Mol Med Today 2: 129–135. [DOI] [PubMed] [Google Scholar]
- Segal, BH, Leto, TL, Gallin, JI, Malech, HL and Holland, SM (2000). Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine (Baltimore) 79: 170–200. [DOI] [PubMed] [Google Scholar]
- Segal, BH, Veys, P, Malech, H and Cowan, MJ (2011). Chronic granulomatous disease: lessons from a rare disorder. Biol Blood Marrow Transplant 17(suppl. 1): S123–S131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candotti, F, Shaw, KL, Muul, L, Carbonaro, D, Sokolic, R, Choi, C et al. (2012). Gene therapy for adenosine deaminase-deficient severe combined immune deficiency: clinical comparison of retroviral vectors and treatment plans. Blood 120: 3635–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavazzana-Calvo, M, Hacein-Bey, S, de Saint Basile, G, Gross, F, Yvon, E, Nusbaum, P et al. (2000). Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science 288: 669–672. [DOI] [PubMed] [Google Scholar]
- Scaramuzza, S, Biasco, L, Ripamonti, A, Castiello, MC, Loperfido, M, Draghici, E et al. (2013). Preclinical safety and efficacy of human CD34(+) cells transduced with lentiviral vector for the treatment of Wiskott-Aldrich syndrome. Mol Ther 21: 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malech, HL, Choi, U and Brenner, S (2004). Progress toward effective gene therapy for chronic granulomatous disease. Jpn J Infect Dis 57: S27–S28. [PubMed] [Google Scholar]
- Bianchi, M, Hakkim, A, Brinkmann, V, Siler, U, Seger, RA, Zychlinsky, A et al. (2009). Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood 114: 2619–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, S, Ott, MG, Schultze-Strasser, S, Jauch, A, Burwinkel, B, Kinner, A et al. (2010). Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med 16: 198–204. [DOI] [PubMed] [Google Scholar]
- Chen, C, Liu, Y, Liu, R, Ikenoue, T, Guan, KL, Liu, Y et al. (2008). TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med 205: 2397–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen, L, Yu, L and Dinauer, MC (1998). Probing the role of the carboxyl terminus of the gp91phox subunit of neutrophil flavocytochrome b558 using site-directed mutagenesis. J Biol Chem 273: 6575–6581. [DOI] [PubMed] [Google Scholar]
- Bedard, K and Krause, KH (2007). The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87: 245–313. [DOI] [PubMed] [Google Scholar]
- Kuwabara, WM, Zhang, L, Schuiki, I, Curi, R, Volchuk, A and Alba-Loureiro, TC (2015). NADPH oxidase-dependent production of reactive oxygen species induces endoplasmatic reticulum stress in neutrophil-like HL60 cells. PLoS One 10: e0116410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, G, Scull, C, Ozcan, L and Tabas, I (2010). NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J Cell Biol 191: 1113–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, K, Tanabe, K, Ohnuki, M, Narita, M, Ichisaka, T, Tomoda, K et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131: 861–872. [DOI] [PubMed] [Google Scholar]
- Zou, J, Sweeney, CL, Chou, BK, Choi, U, Pan, J, Wang, H et al. (2011). Oxidase-deficient neutrophils from X-linked chronic granulomatous disease iPS cells: functional correction by zinc finger nuclease-mediated safe harbor targeting. Blood 117: 5561–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y, Cowley, SA, Siler, U, Melguizo, D, Tilgner, K, Browne, C et al. (2012). Derivation and functional analysis of patient-specific induced pluripotent stem cells as an in vitro model of chronic granulomatous disease. Stem Cells 30: 599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama, Y, Suzuki, T, Sakata-Yanagimoto, M, Kumano, K, Higashi, K, Takato, T et al. (2009). Derivation of functional mature neutrophils from human embryonic stem cells. Blood 113: 6584–6592. [DOI] [PubMed] [Google Scholar]
- Brinkmann, V, Reichard, U, Goosmann, C, Fauler, B, Uhlemann, Y, Weiss, DS et al. (2004). Neutrophil extracellular traps kill bacteria. Science 303: 1532–1535. [DOI] [PubMed] [Google Scholar]
- Metzler, KD, Fuchs, TA, Nauseef, WM, Reumaux, D, Roesler, J, Schulze, I et al. (2011). Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood 117: 953–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, XB (2013). Cellular reprogramming of human peripheral blood cells. Genomics Proteomics Bioinformatics 11: 264–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaff, N, Lachmann, N, Ackermann, M, Kohlscheen, S, Brendel, C, Maetzig, T et al. (2013). A ubiquitous chromatin opening element prevents transgene silencing in pluripotent stem cells and their differentiated progeny. Stem Cells 31: 488–499. [DOI] [PubMed] [Google Scholar]
- Kaufmann, KB, Brendel, C, Suerth, JD, Mueller-Kuller, U, Chen-Wichmann, L, Schwäble, J et al. (2013). Alpharetroviral vector-mediated gene therapy for X-CGD: functional correction and lack of aberrant splicing. Mol Ther 21: 648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst, F, Ball, CR, Tuorto, F, Nowrouzi, A, Wang, W, Zavidij, O et al. (2012). Extensive methylation of promoter sequences silences lentiviral transgene expression during stem cell differentiation in vivo. Mol Ther 20: 1014–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa, K, Cowan, AB, Nakatsuji, N and Suemori, H (2007). Efficient multicistronic expression of a transgene in human embryonic stem cells. Stem Cells 25: 1707–1712. [DOI] [PubMed] [Google Scholar]
- Yoshida, LS, Saruta, F, Yoshikawa, K, Tatsuzawa, O and Tsunawaki, S (1998). Mutation at histidine 338 of gp91(phox) depletes FAD and affects expression of cytochrome b558 of the human NADPH oxidase. J Biol Chem 273: 27879–27886. [DOI] [PubMed] [Google Scholar]
- Kobayashi, SD, Voyich, JM, Braughton, KR, Whitney, AR, Nauseef, WM, Malech, HL et al. (2004). Gene expression profiling provides insight into the pathophysiology of chronic granulomatous disease. J Immunol 172: 636–643. [DOI] [PubMed] [Google Scholar]
- Kim, SY, Jun, HS, Mead, PA, Mansfield, BC and Chou, JY (2008). Neutrophil stress and apoptosis underlie myeloid dysfunction in glycogen storage disease type Ib. Blood 111: 5704–5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grez, M, Reichenbach, J, Schwäble, J, Seger, R, Dinauer, MC and Thrasher, AJ (2011). Gene therapy of chronic granulomatous disease: the engraftment dilemma. Mol Ther 19: 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, C and Dunbar, CE (2011). Stem cell gene therapy: the risks of insertional mutagenesis and approaches to minimize genotoxicity. Front Med 5: 356–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiriaco, M, Farinelli, G, Capo, V, Zonari, E, Scaramuzza, S, Di Matteo, G et al. (2014). Dual-regulated lentiviral vector for gene therapy of X-linked chronic granulomatosis. Mol Ther 22: 1472–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd, S, Dean, O, Copolov, DL, Malhi, GS and Berk, M (2008). N-acetylcysteine for antioxidant therapy: pharmacology and clinical utility. Expert Opin Biol Ther 8: 1955–1962. [DOI] [PubMed] [Google Scholar]
- Nishimura, K, Sano, M, Ohtaka, M, Furuta, B, Umemura, Y, Nakajima, Y et al. (2011). Development of defective and persistent Sendai virus vector: a unique gene delivery/expression system ideal for cell reprogramming. J Biol Chem 286: 4760–4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suerth, JD, Maetzig, T, Galla, M, Baum, C and Schambach, A (2010). Self-inactivating alpharetroviral vectors with a split-packaging design. J Virol 84: 6626–6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suerth, JD, Maetzig, T, Brugman, MH, Heinz, N, Appelt, JU, Kaufmann, KB et al. (2012). Alpharetroviral self-inactivating vectors: long-term transgene expression in murine hematopoietic cells and low genotoxicity. Mol Ther 20: 1022–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya, K, Shirakawa, J, Kameyama, T, Honda, S, Tahara-Hanaoka, S, Miyamoto, A et al. (2003). CD226 (DNAM-1) is involved in lymphocyte function-associated antigen 1 costimulatory signal for naive T cell differentiation and proliferation. J Exp Med 198: 1829–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonini, C, Ferrari, G, Verzeletti, S, Servida, P, Zappone, E, Ruggieri, L et al. (1997). HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science 276: 1719–1724. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, T, Hamanaka, S, Kamiya, A, Okabe, M, Kawarai, M, Wakiyama, Y et al. (2012). Development of an all-in-one inducible lentiviral vector for gene specific analysis of reprogramming. PLoS One 7: e41007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama, N, Nishimura, S, Nakamura, S, Shimizu, T, Ohnishi, R, Endo, H et al. (2010). Transient activation of c-MYC expression is critical for efficient platelet generation from human induced pluripotent stem cells. J Exp Med 207: 2817–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masaki, H, Ishikawa, T, Takahashi, S, Okumura, M, Sakai, N, Haga, M et al. (2007). Heterogeneity of pluripotent marker gene expression in colonies generated in human iPS cell induction culture. Stem Cell Res 1: 105–115. [DOI] [PubMed] [Google Scholar]
- Takayama, N, Nishikii, H, Usui, J, Tsukui, H, Sawaguchi, A, Hiroyama, T et al. (2008). Generation of functional platelets from human embryonic stem cells in vitro via ES-sacs, VEGF-promoted structures that concentrate hematopoietic progenitors. Blood 111: 5298–5306. [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Laube B, Abu Abed U, Goosmann C, Zychlinsky A (2010). Neutrophil extracellular traps: how to generate and visualize them. J Vis Exp 36: e1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vowells, SJ, Sekhsaria, S, Malech, HL, Shalit, M and Fleisher, TA (1995). Flow cytometric analysis of the granulocyte respiratory burst: a comparison study of fluorescent probes. J Immunol Methods 178: 89–97. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.