

Abstract
The mitochondrial permeability transition pore (mtPTP) is a Ca2+-requiring mega-channel which, under pathological conditions, leads to the deregulated release of Ca2+ and mitochondrial dysfunction, ultimately resulting in cell death. Although the mtPTP is a potential therapeutic target for many human pathologies, its potential as a drug target is currently unrealized. Herein we describe an optimization effort initiated around hit 1, 5-(3-hydroxyphenyl)-N-(3,4,5-trimethoxyphenyl)isoxazole-3-carboxamide, which was found to possess promising inhibitory activity against mitochondrial swelling (EC50 < 0.39 µm) and showed no interference on the inner mitochondrial membrane potential (rhodamine 123 uptake EC50 > 100 µm). This enabled the construction of a series of picomolar mtPTP inhibitors that also potently increase the calcium retention capacity of the mitochondria. Finally, the therapeutic potential and in vivo efficacy of one of the most potent analogues, N-(3-chloro-2-methylphenyl)-5-(4-fluoro-3-hydroxyphenyl)isoxazole-3-carboxamide (60), was validated in a biologically relevant zebrafish model of collagen VI congenital muscular dystrophies.
Keywords: calcium retention capacity, mitochondria, muscular dystrophy, permeability transition, zebrafish
Introduction
The vital roles of mitochondria involve ATP synthesis, redox signaling, and regulation of cytoplasmic and mitochondrial matrix calcium (Ca2+) levels.[1] The latter function is reflected by the mitochondrion’s high capacity to store Ca2+ in response to signals arising from elevated cytoplasmic Ca2+ concentrations.[2] The mitochondrial permeability transition pore (mtPTP) is a high-conductance channel of the inner mitochondrial membrane (IMM) sensitive to cyclosporin (Cs) A that opens transiently under normal physiological conditions, possibly acting as a mitochondrial Ca2+-release channel to help maintain cellular Ca2+ homeostasis.[3] However, under pathological conditions, the mtPTP opens persistently and results in fatal consequences such as IMM depolarization, deregulated release of matrix Ca2+, termination of oxidative phosphorylation, swelling, and eventually rupture of the outer mitochondrial membrane (OMM) with the release of apoptotic proteins, resulting in cell death.[3c, 4] Persistent opening of the mtPTP may contribute to a host of chronic and therapeutically challenging diseases, such as multiple sclerosis,[5] amyotrophic lateral sclerosis,[6] Alzheimer’s disease,[7] muscular dystrophies,[8] myocardial infarction,[9] stroke,[10] and diabetes.[11] A key pathophysiological hallmark in these diseases is likely based in mitochondrial dysfunction triggered by Ca2+ and potentiated by oxidative stress.[12]
The molecular composition of the mtPTP is still incompletely understood.[13] The previous long-standing belief that the mtPTP forms at the adjoining sites of the inner and outer membranes through association of the voltage-dependent anion channel (VDAC)[14] of the OMM and the adenine nucleotide translocator (ANT)[15] of the IMM has not been supported by rigorous genetic tests, as VDAC- and ANT-null mitochondria still display a CsA-sensitive mtPTP.[16] Over the past decade, similar genetic analyses have discounted the participation of many other components of the earlier models of the mtPTP.[17] Other studies suggest that the mtPTP forms from dimers of the FOF1 ATP synthase (F-ATP synthase), the IMM nanomachine responsible for the synthesis of ATP.[18] In this context, the mtPTP represents a unique, Ca2+-dependent channel originating from F-ATP synthase that is highly favored by formation of disulfide bonds during oxidative stress.[19] How dimers of F-ATP synthase switch from a Mg2+-dependent catalytic system that makes or hydrolyses ATP into a Ca2+-dependent pore that eliminates the IMM transmembrane potential, which is required for ATP synthesis, remains to be established. While many mtPTP inhibitors have been reported (a few are shown in Figure 1), most of them are not selective and likely inhibit the mtPTP through indirect effects.[20] The most studied inhibitor of the mtPTP is CsA. CsA binds to cyclophilin (CyP) D, a matrix peptidylprolyl cis–trans isomerase (PPIase) and inhibits its activity. However, CyPD inhibitors only desensitize the mtPTP without blocking its activity, as demonstrated by the fact that the mtPTP can still open after elimination of the nuclear gene encoding CyPD.[21] CsA binds to and inhibits the action of all members of the CyP family (16 different CyP proteins are encoded by mammalian genomes).[22] As a result, through interaction with another member of this family, CyPA, CsA also mediates inhibition of calcineurin, resulting in immunosuppression, which is a major side effect of therapy for mtPTP-dependent diseases.[23] Consequently, non-immunosuppressive CyPD inhibitors (e.g., NIM811, Debio025, and antamanide)[24] derived from CsA have been used as therapeutic agents in mtPTP-dependent disease models.[25] However, all CsA derivatives inhibit all CyPs to some extent—not just the mitochondrial isoform—and, like CsA, desensitize, but do not block, the mtPTP. Furthermore, CsA and its derivatives cannot be used to treat mtPTP-based neurological diseases because they do not cross the blood–brain barrier.[23, 26]
Figure 1.

Structures of prominent mtPTP inhibitors.
Despite the above advances, more potent and selective mtPTP inhibitors are needed for use as tool compounds and for possible drug discovery activities. Herein we report the results of a screening/chemical optimization approach that has yielded small-molecule inhibitors of the mtPTP that are ~1000-fold more potent than GNX-865 (which is one of the most relevant prior-art compounds, as reported by Fancelli et al.[27]) in the mitochondrial swelling assay and also exhibit high in vivo efficacy in a zebrafish model of collagen VI (ColVI) congenital muscular dystrophies, a biologically accurate model of a mtPTP based disease.[28]
Results and Discussion
Identification of small-molecule mtPTP inhibitors
As a part of the Molecular Libraries Program of the US National Institutes of Health (NIH), a high-throughput screen was performed within the Molecular Libraries Probe Production Centers Network (MLPCN) to seek new structural hits for the development of potent, small-molecule inhibitors of mtPTP (PubChem Summary AID: 602491).[29] The NIH Molecular Libraries Small Molecule Repository (MLSMR) collection of 363 827 compounds was screened at a concentration of 10 µm in isolated mouse liver mitochondria using the mtPTP swelling assay. Compounds were selected as hits if they were found to inhibit mitochondrial swelling by at least 50% relative to controls. Using this selection criterion, 5040 compounds were identified, corresponding to a hit rate of 1.4% (PubChem AID: 602449). These compounds were subsequently tested in the mitochondrial rhodamine (Rh) 123 uptake assay to identify and exclude probes that interfered with maintenance of the IMM potential (thus preventing Ca2+ uptake) rather than inhibiting the mtPTP. Compounds exhibiting >20% inhibition at 10 µm in this counter-screen assay were excluded from further consideration, which resulted in 1097 compounds of interest. This list was condensed further to 819 compounds based on cheminformatic identification of biologically promiscuous compounds (PubChem Promiscuity) and PAINS (Pan Assay Interference Compounds).[30] The concentration–response effect of these compounds was measured in the mitochondrial swelling (PubChem AID: 651561) and Rh123 uptake assays (PubChem AID: 651564) and yielded 248 compounds with EC50 < 20 µm in the swelling assay, and a Rh123-uptake-to-swelling EC50 ratio of >5-fold, benchmark criteria that were established at the outset of the project. A final prioritization of compounds was based on lack of reactive functionalities and synthetic tractability, followed by validation of activity on high-quality compound samples from the solid physical state. A total of 27 hits, along with CsA as a positive control, were assayed by the calcium retention capacity (CRC) test, a good alternative to the mitochondrial swelling assay used as the primary screen. Amongst all of the validated hits, compounds from the isoxazole chemotype exhibited activity similar to that of CsA in isolated mouse liver mitochondria and also increased the CRC of permeabilized mouse (murine embryonic fibroblasts) and human (HeLa) cells, thereby demonstrating that their effects were not mouse-specific. Based on biological activity and physicochemical properties, the isoxazole scaffold, represented by hit compound 1 (Figure 2), was selected as a starting point for further structure–activity relationship (SAR) studies.
Figure 2.

Regions of hit 1 earmarked for optimization.
Isoxazole compounds are novel structural entities that target the mtPTP
Isoxazole compounds are known for their potential use in a broad array of diseases across the infectious and parasitic diseases and oncology therapeutic areas, etc.[31] However, the potential use of isoxazole compounds to probe mtPTP-related diseases has not been reported. Although not directly comparable with activity against the mtPTP, piperazinylalkylisoxazole compounds have been reported as inhibitors of T-type Ca2+ channel blockers.[32] Regardless, the isoxazole compounds reported herein contain a phenyl ring at the 5-position of the isoxazole unit and an anilide functionality off the 3-position, and these features make the isoxazole compounds reported herein structurally unique to the above-mentioned piperazinylalkylisoxazole-containing compounds.
Isoxazole compound analogue synthesis
Generally, analogues of 1 were assembled by coupling the isoxazole-core-containing carboxylic acids 2 and anilines 3 using thionyl chloride and triethylamine in tetrahydrofuran (THF) (Scheme 1A) to afford the corresponding isoxazole amides 4 in 20–80% yield. For some of these isoxazole amides, the corresponding carboxylic acid partners, as well as the aniline coupling partners, were commercially available. For the assembly of non-isoxazole-core-containing amides, a similar coupling strategy was used (Scheme 1B).
Scheme 1.

Synthetic route for the assembly of isoxazole analogues. Reagents and conditions: a) SOCl2, Et3N, 65°C→RT, THF, 20–80%.
For the isoxazole-containing or different heterocycle-containing carboxylic acids that were not commercially available, a three-step procedure was required for their assembly (Scheme 2). The acetophenone derivatives 8 were treated with dimethyl oxalate and sodium methoxide in diethyl ether for 24 h at room temperature to give the corresponding 1,3-diketones[33] 9 in 60–80% yield, which are present predominantly in the enol form, 9a. At this point, depending on the desired heterocyclic motif, the enols 9a were treated with hydroxylamine hydrochloride in methanol under reflux conditions to form the isoxazole[34] core 10a, or, upon treatment with hydrazine hydrate in acetic acid, afforded the pyrazole core as a 1:1 mixture of regioisomers 10b/10b′, whereas using methylhydrazine in methanol under reflux conditions afforded a 1:1 mixture of regioisomers of the methylpyrazoles 10c/10c′ in 40–90% overall yield. Subsequent saponification of the methyl esters in these compounds with sodium hydroxide in ethanol/THF (2:1) for 2 h at reflux provided the corresponding carboxylic acid intermediates 11a, 11b, 11b′, 11 c, and 11 c′ in 70–98% yields.
Scheme 2.

Synthetic route for isoxazole carboxylic acid intermediates. Reagents and conditions: a) dimethyl oxalate, NaOMe, Et2O, RT, 24 h, 60–80 %; b) NH2OH·HCl/NH2-NH2·H2O/MeNH-NH2, MeOH/AcOH, reflux, 1/18 h, 40–90%; c) NaOH, EtOH/THF (2:1), reflux, 2 h, 70–98%.
Structure–activity relationship optimization
Using CsA and GNX-865 as positive controls, SAR optimization was driven using three assays, namely: 1) mitochondrial swelling, which is an absorbance-based assay used to assess the concentration–response activity of mtPTP inhibitors; 2) Rh123 uptake, which is a fluorescence-based counter-screen assay performed to identify compounds that prevent mitochondrial swelling by interfering with IMM potential rather than blocking or binding to the mtPTP; and 3) the CRC test, which measures the amount of Ca2+ accumulated and retained by the mitochondria before the precipitous Ca2+ release marking opening of the mtPTP. The ratio between the amounts of Ca2+ required to trigger pore opening in the presence (CRC) versus absence of test compound (CRC0), is a direct measure of the inhibitory effect on the mtPTP and is known as the CRC ratio (CRC/CRC0). Guided by these three key assays, we explored structural modifications primarily around the western and the eastern aryl rings and the isoxazole central core (highlighted regions in Figure 2) to discover efficacious inhibitors.
With respect to SAR for the western aryl ring, it became evident that introduction of the 3-chlorophenyl moiety improved activity (Table 1, entry 14) and migration of the chlorine atom or its replacement around the ring (entry 15 versus 16) caused activity to decrease. Introducing 3,5-dichloro or 3-chloro-5-methyl substitution on the phenyl ring (entries 23 and 30), as well as the 5-chloro-2-methyl, 5-chloro-2-cyano, 5-chloro-2-methoxy pattern on the phenyl ring (entries 29, 25, and 27), which all have the 3-chlorophenyl moiety intact, resulted in considerable enhancement in activity. Interestingly, introducing the 2,6-dimethylphenyl moiety enhanced activity (entry 33), which might suggest a role for sterics in perturbing the coplanarity of the 2,6-dimethylphenyl ring and the amide bond. Introducing a heteroatom in the phenyl ring (entry 31) was tolerated with a modest loss of activity.
Table 1.
Structure–activity relationships around the western aryl ring.
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Ar | CRC/CRC0[a,b] | EC50 [µm][a] | Entry | Ar | CRC/CRC0[a,b] | EC50 [µm][a] | ||
| Mito[c] | Rh123[d] | Mito[c] | Rh123[d] | ||||||
| 12 |  |
1.06±0.04 | >50 | >100 | 23 |  |
5.72±0.25 | 0.190±0.017 | >100 |
| 13 |  |
2.36±0.14 | 0.189±0.028 | >100 | 24 |  |
7.69±0.61 | 0.0296±0.0026 | >100 |
| 14 |  |
4.13±0.17 | 0.0788±0.0058 | >100 | 25 |  |
1.00±0.00 | 17.6±2.2 | >100 |
| 15 | 2.41±0.05 | 0.887±0.125 | >100 | 26 |  |
4.96±0.31 | 0.0300±0.0022 | >100 | |
| 16 |  |
1.55±0.11 | 12.7±1.9 | >100 | 27 |  |
6.95±1.15 | 0.0603±0.0076 | >100 |
| 17 |  |
3.77±0.10 | 0.172±0.014 | >100 | 28 |  |
1.57±0.14 | 1.61±0.87 | >100 |
| 18 |  |
1.75±0.13 | 7.06±0.65 | >100 | 29 |  |
9.63±0.29 | 0.0035±0.0007 | >100 |
| 19 |  |
4.30±0.26 | 0.0075±0.0012 | >100 | 30 |  |
4.55±0.45 | 0.0781±0.0050 | >100 |
| 20 |  |
1.84±0.07 | 12.5±0.7 | >100 | 31 |  |
6.62±0.69 | 0.0266±0.0028 | >100 |
| 21 |  |
2.51±0.30 | 1.52±0.05 | 38.5±4.0 | 32 |  |
3.53±0.37 | 0.100±0.010 | >100 |
| 22 | 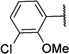 |
1.27±0.11 | 42.7±2.6 | >100 | 33 |  |
8.12±0.59 | 0.0121±0.0014 | >100 |
| CsA | 4.46±0.25 | 0.0953±0.0029 | >100 | GNX-865 | 4.53±0.12 | 0.105±0.007 | >100 | ||
Data are the average ±SEM of n≥3 experiments.
At 1.56 µm.
Mitochondrial swelling.
Rhodamine 123 uptake.
With respect to the eastern aryl ring, varying the position of the phenolic hydroxy group (Table 2, entries 38–43), bioisosteric replacement of the hydroxy group with N-methylsulfonamide (NHSO2Me; entries 44 and 45), protection of the hydroxy group as methoxy (entry 37), and deletion of the hydroxy group (entry 35), resulted in diminished or no activity, indicating the importance of the 3-hydroxyphenyl moiety.
Table 2.
Structure–activity relationships around the eastern aryl ring.
| Entry | Structure | CRC/CRC0[a,b] | EC50 [µm][a] | |
|---|---|---|---|---|
| Mito[c] | Rh123[d] | |||
| 34 |  |
1.00±0.00 | >50 | >100 |
| 35 |  |
1.00±0.00 | >50 | >100 |
| 36 |  |
1.00±0.00 | >50 | >100 |
| 37 |  |
1.03±0.08 | 44.6±3.0 | >100 |
| 38 | 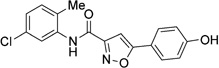 |
1.00±0.00 | >50 | >100 |
| 39 |  |
2.10±0.20 | 2.21±0.17 | >100 |
| 40 |  |
1.46±0.15 | 2.45±0.15 | >100 |
| 41 |  |
1.24±0.09 | >50 | >100 |
| 42 |  |
1.00±0.00 | >50 | >100 |
| 43 |  |
1.25±0.09 | 6.05±0.75 | >100 |
| 44 |  |
0.98±0.02 | >50 | >100 |
| 45 |  |
1.02±0.02 | >50 | >100 |
Data are the average ±SEM of n≥3 experiments.
At 1.56 µm.
Mitochondrial swelling.
Rhodamine 123 uptake.
Having determined desirable substitution patterns on the eastern and western aryl rings, we explored the effect of additional substituents in the eastern aryl ring. To that end, we explored the effect of electron-withdrawing and electron-donating groups on the adjacent phenol group. Analogues containing the 3-hydroxy-4-methoxyphenyl moiety resulted in very-good-to-excellent activity (Table 3, entries 46–55), particularly the 5-chloro-2-methylphenyl amalgamation (entry 46), which resulted in single-digit picomolar activity in the mitochondrial swelling assay and a very high CRC ratio of 10 at 1.56 µm, whereas the 3-chloro-2-methylphenyl combination (entry 52) showed similar potency of 28 pm activity in the mitochondrial swelling assay and a CRC ratio of 9.6 at 1.56 µm. Furthermore, the 5-chloro-2-methylpyridin-3-yl counterpart displayed 10 nm activity in the mitochondrial swelling assay and a CRC ratio of 10 (entry 54). However, complete loss of activity was observed with the incorporation of 3,4-dimethoxyphenyl substitution in the eastern ring, reiterating the importance of the 3-hydroxyphenyl moiety for activity (entries 56 and 57).
Table 3.
Structure–activity relationships around the eastern aryl ring: effect of methoxy, fluoro, and chloro group substitution.
| Entry | Structure | CRC/CRC0[a,b] | EC50 [µm][a] | Entry | Structure | CRC/CRC0[a,b] | EC50 [µm][a] | ||
|---|---|---|---|---|---|---|---|---|---|
| Mito[c] | Rh123[d] | Mito[c] | Rh123[d] | ||||||
| 46 |  |
10.02±0.34 | 0.00000756±0.00000122 | >100 | 58 |  |
6.50±0.77 | 0.0661±0.0032 | >100 |
| 47 |  |
3.83±0.12 | 0.0348±0.0042 | >100 | 59 |  |
6.41±1.19 | 0.0756±0.0092 | >100 |
| 48 |  |
1.71±0.11 | 0.277±0.001 | >100 | 60 |  |
12.14±0.55 | 0.000890±0.000141 | >100 |
| 49 |  |
5.21±0.50 | 0.108±0.010 | >100 | 61 |  |
6.02±0.78 | 0.0554±0.0041 | >100 |
| 50 |  |
6.62±0.84 | 0.0971±0.0034 | >100 | 62 |  |
6.36±0.66 | 0.0133±0.0020 | >100 |
| 51 |  |
4.34±0.53 | 0.146±0.011 | >100 | 63 |  |
15.21±1.26 | 0.000134±0.000016 | >100 |
| 52 |  |
9.59±0.85 | 0.0000283±0.0000048 | >100 | 64 |  |
7.63±0.64 | 0.00905±0.00141 | >100 |
| 53 |  |
2.80±0.18 | 0.365±0.048 | >100 | 65 |  |
3.35±0.28 | 0.819±0.059 | >100 |
| 54 |  |
10.10±1.14 | 0.0105±0.0011 | >100 | 66 |  |
9.12±0.88 | 0.0155±0.0018 | >100 |
| 55 |  |
2.90±0.15 | 0.217±0.052 | >100 | 67 |  |
2.01±0.23 | 2.19±0.15 | >100 |
| 56 |  |
1.00±0.00 | >50 | >100 | 68 |  |
8.22±0.58 | 0.000180±0.000042 | >100 |
| 57 |  |
1.00±0.00 | >50 | >100 | 69 |  |
6.91±0.47 | 0.00134±0.00026 | >100 |
| 70 |  |
6.21±0.66 | 0.00216±0.00045 | >100 | |||||
Data are the average ±SEM of n≥3 experiments.
At 1.56 µm.
Mitochondrial swelling.
Rhodamine 123 uptake.
With respect to exploring the effect of electron-withdrawing groups by introducing a fluoro functionality adjacent to the phenolic hydroxy group, we obtained inhibitory activity analogous to that observed for the methoxy series, and, in some cases, even better activity (entries 58–67). The 5-chloro-2-methylphenylanilino and 3-chloro-2-methylphenylanilino analogues demonstrated sub-nanomolar activity in the mitochondrial swelling assay and very high CRC ratios of 15 and 12, respectively, at 1.56 µm (entries 63 and 60). The chloro-2-methylpyridin-3-yl aniline analogue displayed 16 nm activity in the mitochondrial swelling assay along with a CRC ratio of 9 (entry 66). Similarly, insertion of a chloro functionality adjacent to the phenolic hydroxy group afforded analogues with good activity (entries 68–70).
Finally, we studied modifications to the central core of the scaffold. Replacing the isoxazole core with phenyl (Table 4, entry 71), 6-methylquinoline (entries 77 and 84), and 5-oxopyrrolidine (entries 78 and 79) rings attenuated the activity. Conversely, replacement with 1H-pyrazole or 1-methyl-1H-pyrazole conserved activity (entries 72–74), whereas their regioisomers (entries 75 and 76) were inactive. More surprising was the effect of introducing thiophene and furan rings (entries 80, 81, and 82), as the resulting compounds showed activity in the Rh123 uptake assay, indicative of preventing mitochondrial swelling by interference with IMM potential rather than blocking or binding to the mtPTP.
Table 4.
Replacement of the isoxazole ring.
| Entry | Structure | CRC/CRC0[a,b] | EC50 [µm][a] | Entry | Structure | CRC/CRC0[a,b] | EC50 [µm][a] | ||
|---|---|---|---|---|---|---|---|---|---|
| Mito[c] | Rh123[d] | Mito[c] | Rh123[d] | ||||||
| 71 |  |
1.47±0.12 | 4.71±0.35 | >100 | 78 |  |
1.09±0.03 | 13.9±0.8 | >100 |
| 72 |  |
10.92±0.74 | 0.0014±0.0002 | 30.4±5.5 | 79 |  |
1.02±0.02 | >50 | >100 |
| 73 |  |
7.51±0.47 | 0.0024±0.0004 | >100 | 80 |  |
1.21±0.07 | 6.43±1.11 | 5.47±0.29 |
| 74 |  |
2.26±0.15 | 0.0211±0.0020 | >100 | 81 |  |
1.04±0.02 | 34.4±3.17 | 21.0±2.2 |
| 75 |  |
1.00±0.00 | >50 | >100 | 82 |  |
0.98±0.02 | 35.2±2.3 | 39.6±3.0 |
| 76 |  |
1.00±0.00 | >50 | >100 | 83 |  |
0.93±0.06 | >50 | >100 |
| 77 |  |
1.00±0.00 | >50 | >100 | 84 |  |
1.00±0.00 | >50 | >100 |
Data are the average ±SEM of n≥3 experiments.
At 1.56 µm.
Mitochondrial swelling.
Rhodamine 123 uptake.
Compound 60 is a key compound based on potency, selectivity, physicochemical properties, and efficiency indices
Overall, the SAR studies revealed a set of seven very potent analogues (Figure 3), with 46 having the lowest mitochondrial swelling activity (EC50 = 7.6 pm) and 63 having the highest CRC ratio of 15 at 1.56 µm. We identified four key compound analogues based on excellent inhibitory activities, CRC ratios, and physicochemical properties. These four analogues shared very promising physicochemical properties (Table 5) and desirable values for binding efficiency[35] metrics, such as ligand efficiency (LE) and lipophilic ligand efficiency (LLE). With respect to in vitro potency and efficacy, all four analogues were comparably matched; however, 60 and 63 were favored in terms of slightly decreased topological polar surface area and the number of hydrogen bond acceptors. In addition, the measured aqueous solubility for compound 60 was 16 µm in PBS at pH 7.4, which was the best of the four key analogues. In light of its promising in vitro potency, selectivity, and physicochemical properties, compound 60 was prioritized for more extensive biological characterization.
Figure 3.

SAR overview. CRC ratios at 1.56 µm compound concentration (bars), swelling (circles), and Rh123 uptake (squares). EC50 values are for isolated mouse liver mitochondria. Data are the average ± SEM of n ≥ 3 experiments. Values equal to those on the dashed line represent EC50 > 50 µm and > 100 µm for the swelling and Rh123 uptake assays, respectively.
Table 5.
Potency, selectivity, physicochemical properties, and efficiency indices for key compounds.
| Parameter | HTS hit 1 | Compd 46 | Compd 54 | Compd 60 | Compd 63 |
|---|---|---|---|---|---|
| CRC/CRC0 [a] | 1.18 −0.06 | 10.0−0.3 | 10.1 −1.1 | 12.1 −0.6 | 15.2 −1.3 |
| Mito EC50 [µm][b] | < 0.39 | 0.00000756 −0.00000122 | 0.0105 −0.0010 | 0.000890−0.000141 | 0.000134 −0.000016 |
| Rh123EC50 [µm][c] | > 100 | > 100 | > 100 | > 100 | > 100 |
| HeLa CC50 [µm][d] | ND[m] | > 50 | > 50 | 39.5−4.65 | 44.5−1.67 |
| Mr [Da][e] | 370.3 | 358.7 | 359.7 | 346.7 | 346.7 |
| tPSA [Å2][e,f] | 98.6 | 80.1 | 92.5 | 70.9 | 70.9 |
| clog P[e] | 2.2 | 3.4 | 2.3 | 3.7 | 3.7 |
| HBA[g,h] | 7 | 5 | 6 | 4 | 4 |
| HBD[g,i] | 2 | 2 | 3 | 2 | 2 |
| Heavy atoms[j] | 27 | 25 | 25 | 24 | 24 |
| LE[k] | 0.32 | 0.60 | 0.43 | 0.51 | 0.56 |
| LLE[l] | 4.2 | 7.6 | 5.5 | 5.2 | 6.1 |
At 1.56 µm.
Mitochondrial swelling.
Rhodamine 123 uptake.
HeLa cell cytotoxicity.
Data were generated using CambridgeSoft ChemBioDraw version 12.
Topological polar surface area.
Data were calculated using SYBYL 8.0, 2010, Tripos Associates, St. Louis, MO (USA).
Hydrogen bond acceptors.
Hydrogen bond donors.
Data were calculated using Marvin 15.3.23.0, 2015, ChemAxon.
Ligand efficiency.
Lipophilic ligand efficiency.
Not determined.
Profiling of compound 60 in Eurofins LeadProfilingScreen and for in vitro drug metabolism and pharmacokinetic properties
Compound 60 was profiled against a panel of 68 G-protein-coupled receptors (GPCRs), ion channels, and transporters. At a concentration of 10µm, 79% inhibition of human norepinephrine transporter (NET) and 51% inhibition of adenosine A3 were observed, whereas inhibition of the other 66 targets in the panel was ≤ 43 %.[36] Moreover, the desired activity of compound 60 in the swelling assay is achieved at >1000-fold lower concentration than the counter-screen activity against the NET and adenosine A3 targets.
Compound 60 was submitted for in vitro drug metabolism and pharmacokinetic profiling to establish a baseline against which future analogues could be compared (Table 6). Compound 60 demonstrated acceptable human plasma protein binding and very high binding for mouse plasma proteins. Assessment of in vitro metabolism revealed a very good half-life in human plasma, whereas a poor half-life was observed in mouse plasma. Similarly, greater compound stability was observed in human liver microsomes, relative to the stability observed in mouse liver microsomes. Clearly, there is room for improvement for an optimized lead compound compared with compound 60.
Table 6.
Summary of in vitro ADME data for compound 60.
| Parameter[a] | Compd 60 |
|---|---|
| Aqueous solubility [µm] (PBS, pH 7.4) | 16.1 |
| Protein binding {recovery} [%] (human; mouse) | 89 {76}; > 99 {0} |
| Plasma stability t1/2 [min] (human; mouse)[b] | > 120; < 30 |
| CLint HLM [µL min−1 mg−1][b,c]; t1/2 [min][b] | <115.5; >60 |
| CLint MLM [µL min−1 mg−1][b,d]; t1/2 [min][b] | > 462; < 15 |
Data collected at Cerep/Eurofins Panlabs.
The remaining compound percentage at multiple time points is reported in the Supporting Information.
Intrinsic clearance, human liver microsomes.
Intrinsic clearance, mouse liver microsomes.
Inhibitory effect of compound 60 on Ca2+-induced mtPTP opening
Elevated levels of matrix Ca2+ concentration and oxidative stress sensitize the mtPTP to open, resulting in an increased permeability of the otherwise impermeant IMM to ions and solutes up to ~1500 Da in size. In vitro mitochondrial volume changes can be followed spectrophotometrically as a decrease in absorbance at λ 540 nm. Consequently, we tested mtPTP opening in isolated mouse liver mitochondria, induced by a 50 µm Ca2+ load (Figure 4A, trace a) which was abolished in the presence of compound 60 at 1.56 µm, (Figure 4A, trace b). A full concentration–response assessment, ranging from 18.6 pm to 1.56 µm, revealed a mitochondrial swelling EC50 value of 0.89 ± 1.42 nm (Figure 4B, trace a), which is two orders of magnitude more potent than that of CsA or GNX-865 (Table 1). Moreover, we confirmed that the lack of mitochondrial swelling in the presence of the inhibitors was due to mtPTP desensitization, and does not decrease the IMM potential (Figure 4B, trace b).
Figure 4.

Effect of 60 on Ca2+-induced mtPTP. A) Mitochondrial swelling was induced with 50 µm Ca2+, which was prevented with 60. B) Concentration–response of experiment described in panel A (■) and interference with Rh123 uptake (□). C) Representative CRC of DMSO-, CsA-, 60-, and CsA + 60-treated mitochondria; D) Concentration–response of 60 (■) and CsA (2 µm) + 60 (□)-to-solvent CRC ratios; assays were performed on isolated mouse liver mitochondria (0.25 mg mL−1). E) Representative CRCs of HeLa cells treated with DMSO (trace a) or 60 (1.56 µm; trace c) before cell permeabilization with digitonin; trace b: cells were treated as in trace a and CRC was performed in the presence of 60 (1.56 µm). F) 60-to-solvent CRC ratios of permeabilized HeLa cells (0.8×106 per condition). Data are the average ± SEM of n ≥ 5 experiments.
Compound 60 and CsA likely act on different biological targets
In the following assays, we used isolated mouse liver mitochondria as the test material because they are easily prepared and give consistent and reproducible results. Ample literature indicates that these mitochondria respond to pore agonists and inhibitors as human mitochondria respond.[3a] A suspension of mouse liver mitochondria supplemented with the membrane-impermeant fluorescent dye Calcium Green-5N at 0.5 µm was loaded with a train of 20 µm Ca2+ pulses until the threshold was reached and precipitous Ca2+ release was registered, marking opening of the mtPTP (e.g., Figure 4C). The presence of 1.56 µm 60 raised the Ca2+ load required for mtPTP opening from 80 to ~960 nmol (mg protein)−1 (Figure 4C traces a and c, respectively), which was further increased by augmenting the concentration of 60, eventually culminating in the compound-to-solvent CRC ratio of 15 (Figure 4D, trace a). Based on our findings that the maximum CRC ratios of isolated mouse liver mitochondria treated with 60 are ~3-fold higher than those treated with CsA (Figure 4C, compare traces c and a, and traces b and a), we suspected that our inhibitors and CsA act on different biological targets. To address this, we treated mitochondria with 2 µm CsA together with various concentrations of 60 and performed the CRC test. We found that the two compounds’ effects are synergistic at all tested concentrations of 60 (Figure 4C,D), indicating the target of diarylisoxazole-3-carboxamides is likely not CyPD.
Compound 60 inhibits human mtPTP
Given that our mtPTP inhibitors were identified and optimized using murine mitochondria, and that species-specific regulation of the mtPTP has been demonstrated,[18b, 37] we tested whether human mtPTP is also affected by 60. The inhibitory effect of 60 on human mitochondria was initially confirmed by the increased CRC ratios of permeabilized HeLa cells upon treatment with increasing concentrations of 60 (Figure 4F). Moreover, we treated intact HeLa cells with 1.56 µm 60 or DMSO for 30 min and washed them to eliminate excess 60 by centrifugation. Cells were then permeabilized with digitonin, and the CRC was assessed. As shown in Figure 4E, we observed that 60 is effective following treatment of intact human cells (compare traces a and c), indicating that it was able to permeate the plasma membrane and reach its mitochondrial target, thus desensitizing mtPTP activation to the same extent as DMSO-treated cells (trace b).
Compound 60 protects against chemical activation of mtPTP
We next investigated whether 60 is protective against known chemical activators of the mtPTP that result in oxidative stress and trigger pore opening. We pretreated isolated mouse liver mitochondria with 10 µm Ca2+, an amount insufficient to induce mtPTP opening per se (Figure 5 traces a), and challenged them with reagents that react with two distinct classes of redox-sensitive thiols and increase the mtPTP sensitivity to Ca2+: 1) PhAsO (Figure 5A) and diamide (Figure 5B), which react with matrix thiols;[38] and 2) Cu(OP)2 and NEM (Figure 5C,D, respectively), which react with intermembrane space exposed thiol groups.[39] In all cases, the mtPTP transition, from closed to open conformation, was delayed by CsA at 1.56 µm and was prevented by compound 60 at the same concentration, as measured by the lack of mitochondrial swelling (Figure 5 traces c and d, respectively). Therefore, the isoxazole inhibitors prevented mtPTP opening irrespective of changes in calcium flux and stimuli that would induce pore opening.
Figure 5.

Effect of 60 on chemical activation of the mtPTP. Suspension of isolated mouse liver mitochondria (0.25 mg mL−1) were supplemented with 10 µm Ca2+ only (traces a); 10 µm Ca2+ and A) 7 µm PhAsO, B) 2 mm diamide, C) 7 µm Cu(OP)2, or D) 2 mm NEM (traces b–d). In traces c and d, 1.56 µm CsA or 60, respectively, were also present. Traces are representative of four separate experiments.
Compound 60 shows no effect on ATP synthesis and HeLa cell proliferation
Because it was recently suggested that the mtPTP is formed by a unique conformation of F-ATP synthase dimers,[18, 37b] we investigated whether apart from preventing mtPTP formation, compound 60 also affects ATP synthesis, a potentially untoward side effect. Thus, we measured mitochondrial respiration both in isolated mouse liver mitochondria and in intact HeLa cells in the presence or absence of the inhibitor. No statistically significant differences in respiratory control ratios (Figure 6B), or FCCP-stimulated and oligomycin-insensitive respiration were observed in both isolated mouse liver mitochondria (Figure 6A) and HeLa cells (Figure 6C), demonstrating that the diarylisoxazole-3-carboxamides do not affect the respiratory chain complexes or inherent function of F-ATP synthase. Indeed, compound 60 was not cytotoxic up to a concentration of 12.50 µm (CC50 = 39.5 ± 4.65 µm; Figure 6D) as confirmed by the similar number of viable HeLa cells after treatment for 24 h. Note that as an additional precaution, the plasma membrane multidrug resistance pump inhibitor CsH (which does not inhibit the PTP)[40] (Figure 6D) or verapamil[41] (data not shown) were added in these experiments to ensure that 60 is not extruded from the cells. Similar results were observed following treatment of HEK293 cells and murine embryonic fibroblasts with 60 (data not shown).
Figure 6.

Effect of 60 on oxygen consumption rate (OCR) and HeLa cell proliferation. A) OCR of ADP + Pi-stimulated isolated mouse liver mitochondria. B) Ratios of OCR before-to-after addition of oligomycin. C) OCR of HeLa cells. D) Interference with HeLa cell proliferation after 24 h treatment with the indicated concentration of compound 60. In panels C and D, the multidrug resistance inhibitor CsH (2 µg mL−1) was also present. Similar results were obtained with 20 µm verapamil. A), C) Data are the average ± SD of a representative experiment performed four separate times. B), D) Data are the average ± SEM of four and five experiments, respectively; ***p < 0.001 as assessed by t-test.
Compound 60 shows therapeutic potential in a validated vertebrate model of mtPTP-based disease
As noted above in the Introduction, activation of the mtPTP has been implicated in some of the most challenging human disorders. Indeed, our true appreciation of the extent of mtPTP activation in human diseases was only fully developed through the creation of mice lacking CyPD and performing tests in murine models of these disorders. Among these are muscular dystrophies due to the absence of the extracellular matrix protein ColVI. ColVI is an essential component of myofiber extracellular matrix; mutations in ColVI result in two major human diseases: Ullrich congenital muscular dystrophy (UCMD) and Bethlem myopathy (BM).[42] Mouse models of UCMD have been created through knock-out of the murine Col6a1 gene, but, in contrast to UCMD patients, homozygous recessive animals exhibit very mild clinical myopathy.[43] However, in both humans and mice, assessment of mitochondrial characteristics amply documented that inappropriate mtPTP activity plays a key role in disease pathogenesis.[43b, 44] Consistent with this idea, modification of the mtPTP, either pharmacologically in humans and in mice (with CsA and non-immunosuppressive derivatives such as Debio025 or NIM811),[8b, 25b, 28b, 45] or elimination of the mouse gene encoding CyPD,[46] was found to improve mitochondrial changes and to decrease myofiber cell death. A more robust model of UCMD has been generated in zebrafish by the injection of antisense morpholino oligonucleotides directed to the exon 9 splicing region of the orthologous ColVIa gene.[28] This treatment results in an in-frame deletion paralleling common mutations in UCMD.[47] Severe myopathy, motor deficits, and dramatic ultrastructural defects are present in morpholino-injected animals that successfully recapitulate the clinical severity of human UCMD.[28] Furthermore, treatment with NIM811[28b] reversed many of these defects. Consequently, we used the zebrafish ColVI myopathic model as a convenient, powerful, and easily assayed in vivo system to validate the therapeutic potential of the most potent diarylisoxazole-3-carboxamide mtPTP inhibitors outlined here.
Zebrafish embryos were injected at the 1–2-cell stage with morpholinos designed to mimic ColVIa exon 9 splicing defects common in UCMD.[47] Control morpholinos represent sequences not found in the zebrafish genome. Exon 9 morphant-treated embryos were then assayed using validated motor assays for defective motor function and muscle defects at 24 and 48 h post-fertilization (hpf). Motor function was assessed by spontaneous coiling events, reflecting twisted myofibrils, and “touch-evoked escape response”.[45a] This assay measures the ability of embryos to “escape” after touching them with a small tip. The responses of embryos in this assay were subdivided into four groups according to their ability to escape: normal embryos with unaltered ability to swim, embryos with minor motility disruptions, embryos showing only spontaneous coiling events around the body axis without the ability to escape, and paralyzed embryos with no motility. As reflected in Figure 7A,B, 87% of embryos injected with exon 9 morpholino showed severe motor impairments relative to control embryos. In contrast, exon 9 morphants treated with 60, simply added to the fish water, showed a dramatic improvement in motor function as demonstrated with spontaneous coiling events (Figure 7A) or touch-evoked response (Figure 7B).
Figure 7.

Effect of 60 on defects present in exon 9 col6a1 morphant zebrafish. A) Coiling events were recorded at 24 hpf in control (CRL_MO) and exon 9 morphant (Ex9_MO) embryos in the presence of the indicated concentrations of 60 added at 21 hpf. Data are the mean ± SEM of at least five independent experiments (n = 52 for each condition); **p < 0.01, ***p < 0.001 as determined by one-way ANOVA test with Bonferroni correction. B) In the presence of the indicated concentrations of 60 added at 21 hpf, response evoked by touching embryos at 24 hpf with a pipette tip were recorded. Embryos (n = 35 for each condition) were assigned to four groups based on their escape response: normal, low motility defects, spontaneous coiling events only, and complete paralysis. Results are reported as the percentage of total embryos analyzed for each group. Comparison between groups at different conditions was made using χ2 test and one-way ANOVA with Bonferroni correction; **p < 0.01, ***p < 0.001. C) Bar graphs report the percentage of birefringence classes at 48 hpf after treatment with compound 60 added at 21 hpf, showing normal birefringence, mild myopathic phenotype, and strong myopathic phenotype, as indicated. The total number of embryos used was n = 35 for each condition; *p < 0.05, ***p < 0.001, as determined by χ2 test and one-way ANOVA with Bonferroni correction.
To assess structural muscle organization, muscle birefringence was evaluated at 48 hpf. This technique evaluates muscle structural defects in zebrafish models of muscular dystrophy.[48] Muscle birefringence was analyzed by taking advantage of muscle fiber anisotropy. As can be seen in Figure 7C, exon 9 morphants exhibited severe muscle defects relative to controls, and these defects were largely ameliorated following treatment with 60. Indeed, total birefringence scores demonstrated that treatment with 60 generated significant recovery of muscle defects (ANOVA, p < 0.05).
Conclusions
In summary, a high-throughput screening and SAR optimization effort to identify mtPTP inhibitors has resulted in isoxazole compounds whose structure, activity, and properties were optimized to yield highly valuable probes with picomolar inhibitory activity and potential for further development as treatments for a variety mtPTP-related diseases. We confirmed that the isoxazole inhibitors do not interfere with the IMM potential and are protective against known chemical activators of the mtPTP that induce oxidative stress and trigger pore opening. Moreover, the key analogues demonstrated activity synergistic with CsA, suggesting that the target is not CyPD. We noted that the isoxazole inhibitors do not affect the inherent function of F-ATP synthase or impact the cell viability at effective concentrations. Detailed studies to identify the biological mechanism of action for these compounds are underway. Finally, we validated the therapeutic potential and in vivo efficacy of the most efficacious analogue, 60, in the zebrafish ColVI myopathic model. Future lead optimization work will focus on improving the in vitro and in vivo pharmacokinetic properties for the compound series and on evaluating the resulting analogues for activity in a variety of mtPTP-related disease models.
Experimental Section
Chemistry
General procedures
All solvents and reagents were used as received from commercial suppliers, unless noted otherwise. 1H and 13C NMR spectra were recorded on a Bruker AM 400 spectrometer (operating at 400 and 101 MHz, respectively) or a Bruker AVIII spectrometer (operating at 500 and 126 MHz, respectively) in CDCl3 with 0.03% TMS as an internal standard. Chemical shifts (δ) are reported in parts per million (ppm), and coupling constants (J) are in Hertz (Hz). Spin multiplicities are reported as s=singlet, d=doublet, t=triplet, q=quartet, dd=doublet of doublet, ddd=doublet of doublet of doublet, dt=doublet of triplet, td=triplet of doublet, and m=multiplet. Microwave reactions were carried out using a Biotage Initiator Classic. Column chromatography separations were performed using the Teledyne Isco CombiFlash Rf using RediSep Rf silica gel columns. Analytical RPLC involved an Agilent 1200 RRLC system with UV detection (Agilent 1200 DADSL) and mass detection (Agilent 6224 TOF). Analytical method conditions included a Waters Aquity BEH C18 column (2.1×50 mm, 1.7 µm) and elution with a linear gradient of 5% CH3CN in buffered aqueous ammonium formate (pH 9.8) to 100% CH3CN at a flow rate of 0.4 mL min−1. Automated preparative RP HPLC purification was performed using an Agilent 1200 Mass-Directed Fractionation system (Prep Pump G1361 with gradient extension, make-up pump G1311A, pH modification pump G1311A, HTS PAL autosampler, UVDAD detection G1315D, fraction collector G1364B, and Agilent 6120 quadrupole spectrometer G6120A). Preparative chromatography conditions included a Waters X-Bridge C18 column (19×150 mm, 5 µm, with 19×10 mm guard column), elution with a H2O/CH3CN gradient, which increased to 20% CH3CN content over 4 min at a flow rate of 20 mL min−1 (modified to pH 9.8 by the addition of NH4OH by auxiliary pump), and sample dilution in DMSO. The preparative gradient, triggering thresholds, and UV wavelength were selected according to the analytical RP HPLC analysis of each crude sample. Compound purity was measured on the basis of peak integration (area under the curve) from UV/Vis absorbance at λ 214 nm, and compound identity was determined on the basis of mass spectral and NMR analyses. All compounds had >95% purity as determined by the HPLC methods described above.
General synthesis procedure
To a solution of isoxazole carboxylic acid (0.390 mmol, 1 equiv) in dry THF (1.5 mL) in a 4-dram vial was added SOCl2 (0.558 mmol, 1.43 equiv), and the resulting solution was stirred at reflux for 0.5 h. After cooling the reaction mixture to 35°C, a solution of 5-chloro-2-methylaniline (0.390 mmol, 1 equiv) and Et3N (1.560 mmol, 4 equiv) in dry THF (1 mL) was added dropwise. After stirring at RT for 2 h the reaction mixture was quenched by the addition of 1 n HCl and extracted with EtOAc (3×2 mL). The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated. The residue was analyzed and purified according to the analytical- and preparative-scale RP HPLC methods.
N-(5-Chloro-2-methylphenyl)-5-(3-hydroxy-4-methoxyphenyl)isoxazole-3-carboxamide (46)
This compound was prepared following the general procedure using 5-(3-hydroxy-4-methoxyphenyl)isoxazole-3-carboxylic acid (11 mg, 0.047 mmol) and 5-chloro-2-methylaniline (7 mg, 0.047 mmol). Isolated compound 46 as an off-white powder (8 mg, 47 %): mp: 186–192°C (dec.); 1H NMR (400 MHz, [D6]DMSO): δ=10.28 (s, 1H), 9.49 (s, 1H), 7.52 (d, J=2.2 Hz, 1H), 7.41 (dd, J=8.4, 2.2 Hz, 1H), 7.36–7.29 (m, 2H), 7.30–7.22 (m, 2H), 7.09 (d, J=8.5 Hz, 1H), 3.85 (s, 3H), 2.24 ppm (s, 3H); 13C NMR (101 MHz, [D6]DMSO): δ=170.9, 159.2, 157.4, 150.0, 146.9, 136.5, 132.1, 131.8, 129.8, 126.0, 125.5, 117.8, 112.5, 98.4, 55.7, 17.2 ppm; HRMS (ESI TOF) m/z: [M+H]+ calcd for C18H16ClN2O4: 359.0793, found: 359.0789.
N-(5-Chloro-2-methylpyridin-3-yl)-5-(3-hydroxy-4-methoxyphenyl)isoxazole-3-carboxamide (54)
This compound was prepared following the general procedure using 5-(3-hydroxy-4-methoxyphenyl)isoxazole-3-carboxylic acid (25 mg, 0.106 mmol) and 5-chloro-2-methylpyridin-3-amine (15 mg, 0.106 mmol). Isolated compound 54 as an off-white powder (9 mg, 23%): mp: 216–222 °C (dec.); 1H NMR (500 MHz, [D6]DMSO): δ=10.56 (s, 1H), 9.50 (bs, 1H), 8.44 (d, J=2.3 Hz, 1H), 8.00 (d, J=2.3 Hz, 1H), 7.42 (dd, J=8.4, 2.2 Hz, 1H), 7.34 (d, J=2.2 Hz, 1H), 7.29 (s, 1H), 7.10 (d, J=8.5 Hz, 1H), 3.85 (s, 3H), 2.45 ppm (s, 3H); 13C NMR (126 MHz, [D6]DMSO): δ=171.1, 159.1, 157.8, 152.4, 150.1, 146.9, 144.8, 133.1, 132.2, 127.8, 118.9, 117.9, 112.5, 98.5, 55.7, 20.6 ppm; HRMS (ESI TOF) m/z: [M+H]+ calcd for C17H15ClN3O4: 360.0746, found: 360.0816.
N-(3-Chloro-2-methylphenyl)-5-(4-fluoro-3-hydroxyphenyl)isoxazole-3-carboxamide (60)
This compound was prepared following the general procedure using 5-(4-fluoro-3-hydroxyphenyl)isoxazole-3-carboxylic acid (29 mg, 0.130 mmol) and 3-chloro-2-methylaniline (18 mg, 0.130 mmol). Isolated compound 60 as an off-white powder (14 mg, 30%): mp: 242–246°C (dec.); 1H NMR (500 MHz, [D6]DMSO): δ=10.56 (s, 1H), 10.41 (s, 1H), 7.50 (dd, J=8.3, 2.2 Hz, 1H), 7.47–7.38 (m, 3H), 7.39–7.31 (m, 2H), 7.28 (t, J=8.0 Hz, 1H), 2.26 ppm (s, 3H); 13C NMR (126 MHz, [D6]DMSO): δ=169.9, 158.5 (d, J=247.1 Hz), 153.5, 151.6, 145.6 (d, J=12.8 Hz), 136.7, 133.8, 132.1, 127.3, 127.0, 125.9, 123.0 (d, J=3.4 Hz), 117.8 (d, J=7.2 Hz), 117.3 (d, J=19.2 Hz), 114.9 (d, J=3.6 Hz), 99.9, 15.3 ppm; HRMS (ESI TOF) m/z: [M+H]+ calcd for C17H13ClFN2O3: 347.0593, found: 347.0597.
N-(5-Chloro-2-methylphenyl)-5-(4-fluoro-3-hydroxyphenyl)isoxazole-3-carboxamide (63)
This compound was prepared following the general procedure (isoxazole amide) 1 using 5-(4-fluoro-3-hydroxyphenyl)isoxazole-3-carboxylic acid (25 mg, 0.112 mmol) and 5-chloro-2-methylaniline (16 mg, 0.112 mmol). Isolated compound 63 as an off-white powder (7 mg, 18%): mp: 214–216 °C (dec.); 1H NMR (500 MHz, [D6]DMSO): δ=10.36 (s, 1H), 7.51 (dd, J=7.0, 2.1 Hz, 2H), 7.43 (ddd, J=8.4, 4.3, 2.2 Hz, 1H), 7.40 (s, 1H), 7.39–7.30 (m, 2H), 7.27 (dd, J=8.2, 2.3 Hz, 1H), 2.24 ppm (s, 3H); 13C NMR (126 MHz, [D6]DMSO): δ=169.9, 159.4, 157.3, 152.6 (d, J=246.5 Hz), 145.8 (d, J=12.8 Hz), 136.4, 132.3, 131.9, 129.8, 126.1, 125.6, 117.5 (d, J=7.0 Hz), 117.2 (d, J=19.3 Hz), 114.9 (d, J=3.8 Hz), 99.8, 17.2 ppm; HRMS (ESI TOF) m/z: [M+H]+ calcd for C17H13ClFN2O3: 347.0593, found: 347.0599.
Biology
Reagents
Sucrose, 3-(N-morpholino) propanesulfonic acid (MOPS), phosphoric acid, tris(hydroxymethyl)aminomethane (Tris), ethylene glycol tetraacetic acid (EGTA), glutamic acid, malic acid, CsA, phenylarsine oxide (PhAsO), diamide, N-ethylmaleimide (NEM), bovine serum albumin (BSA), CaCl2, carbonylcyanide-p-trifluoromethoxyphenyl hydrazone (FCCP), verapamil, and dimethyl sulfoxide (DMSO) were from Sigma–Aldrich, CsH was from Enzo Life Sciences, digitonin was from Calbiochem, Rh123 and Calcium Green-5N were from Invitrogen, copper-o-phenanthroline (Cu(OP)2) was prepared just before use by mixing CuSO4 with o-phenanthroline at a 1:2 molar ratio in double-distilled water. All chemicals were of the highest purity commercially available; 96-well plates were from Sacco S.r.l., Italy.
Animal studies
All procedures were approved by the CEASA of the University of Padova (72/2012 and 39/214 to P.B.) and authorized by the Italian Ministry of Health.
General methodology
Assays on biological samples, including purified mitochondria, cultured cells, and zebrafish were treated with compounds described in this study at a final DMSO concentration of 1%. The compound concentrations stated in biological experiments refer to the “formal” concentration, that is, moles added/volume, making no assumption as to the influence of biological materials on the concentration of added compound.
Isolation of mitochondria
C57BL6/J mouse liver mitochondria were prepared from mice aged between 3 and 6 months by standard differential centrifugation. Mice fed ad libitum were sacrificed by cervical dislocation, their livers were removed and placed in a glass beaker containing ice-cold isolation buffer (IB: 0.25m sucrose, 10 mm Tris·HCl, 0.1 mm EGTA-Tris, pH 7.4) supplemented with BSA. Livers were then cut into small pieces with scissors, rinsed with ice-cold IB, and passed through a pre-chilled Potter homogenizer with Teflon pestle. The homogenate (~30 mL per liver) was transferred to centrifuge tubes, and unbroken cells and nuclei were removed by centrifugation at 685×g for 10 min at 4 °C. The supernatant containing mitochondria and other organelles was transferred to new tubes and centrifuged at 6010×g for 10 min at 4 °C. The resulting supernatant was discarded, and mitochondrial pellet was carefully suspended in ice-cold IB buffer and spun at 9390×g for 5 min at 4°C. The pellet was suspended in IB to give a protein concentration of ~60–80 mg mL−1 and stored on ice. Experiments were started immediately and completed within 5 h. Protein concentration was determined by the Biuret method.[49]
Cell culture
HeLa cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Invitrogen), 100 U mL−1 penicillin and 100 µg mL−1 streptomycin (Invitrogen) in a humidified atmosphere of 5% CO2/95% air at 37°C.
Assessment of mitochondrial swelling
Changes in mitochondrial volume of isolated mouse liver mitochondria were followed in a 96-well clear assay plate (Falcon 353072) at a final volume of 0.2 mL. First, 0.1 mL of sucrose assay buffer (SAB: 250 mm sucrose, 10 mm MOPS-Tris, 0.01 mm EGTA-Tris, 1.0 mm phosphoric acid-Tris, 10 mm glutamate, and 5 mm malate, pH 7.4) supplemented with twice the Ca2+ concentration required to induce mitochondrial swelling (which was determined for each preparation of mitochondria, typically 50 µm) was dispensed to the assay plate. A set of wells also contained 2.0 mm EGTA (to prevent mitochondrial swelling). The test wells contained a range of concentrations of the compounds of interest or 2% DMSO. Experiments were started by the addition of 0.1 mL of mitochondrial suspension (0.5 mg mL−1 mitochondria in SAB without respiratory substrates) to the assay plates. Absorbance was read for 30 min at λ 540 nm with a Multi-Skan EX (Thermo Scientific) plate reader.
Assessment of mitochondrial membrane potential
Mitochondrial membrane potential of isolated mouse liver mitochondria was assessed based on accumulation of the cationic fluorescent dye Rh123. First, 0.1 mL of SAB was dispensed to a 96-well black assay plate (Falcon 353376). A set of wells also contained 0.8 µm FCCP (to prevent Rh123 uptake). The test wells contained a range of concentrations of the compounds of interest or 2% DMSO. Then, 0.1 mL of 0.5 mg mL−1 mitochondria in SAB devoid of respiratory substrates and supplemented with 0.8 µm Rh123 were added to all wells of the assay plate. Following incubation at room temperature for 5 min, fluorescence intensity (λex 485 nm, λem 538 nm) was read on a Fluoroskan Ascent FL (Thermo Scientific) plate reader.
Assessment of calcium retention capacity
Calcium retention capacity (CRC) of isolated mouse liver mitochondria was assessed as follows: First, 0.1 mL of SAB were dispensed to a 96-well black assay plate (Falcon 353376) in the presence of 2% DMSO (control wells) or varying concentrations of test compounds. Then, 0.1 mL of 0.5 mg mL−1 mitochondria in SAB devoid of respiratory substrates but supplemented with 1 µm Calcium Green-5N were added to all wells of the assay plate. A train of 5, 10, or 20 µm Ca2+ pulses was added at 1.5, 3, or 4.5 min intervals, respectively, and fluorescence intensity (λex 485 nm, λem 538 nm) was read on Fluoroskan Ascent FL (Thermo Scientific) plate reader.
CRC of HeLa cells
The cells were cultured for 48 h to reach 70–80% confluency, then harvested by trypsinization and washed in KCl buffer (KB: 130 mm KCl, 10 MOPS-Tris, 1 mm phosphoric acid-Tris, 0.1 mm EGTA-Tris, pH 7.4). Cells were then suspended in the above buffer to 8×106 cells per mL and treated with 1.56 µm 60 or DMSO on ice for 30 min, followed by excess compound elimination by centrifugation (experiments presented in Figure 4E only; the step was omitted in experiments of Figure 4F). The pellet was resuspended in KB (except that EGTA-Tris was increased to 1.0 mm) to give a concentration of 2×107 cells per mL and treated with 0.1 mm digitonin for 10 min on ice to permeabilize the plasma membrane. Following excess digitonin elimination by washing cells twice in KB, the cells were suspended to 8×106 cells per mL in KB containing 10 µm EGTA-Tris and supplemented with 1 µm Calcium Green-5N and processed as described for isolated mouse liver mitochondria.
Measurement of respiration
Mitochondrial oxygen consumption was assessed with the Seahorse Extracellular Flux Analyzer XF24 (Seahorse Bioscience, Billerica, MA, USA) essentially as described previously.[50] Briefly, mitochondrial assay solution contained 220 mm mannitol, 70 mm sucrose, 25 mm MOPS-Tris, 10 mm Pi-Tris, 5 mm MgCl2, 1 mm EGTA-Tris, 0.2% fatty-acid-free BSA, 5 mm succinate, and 2 µm rotenone, pH 7.4. Mitochondria (5 µg, suspended in 50 µL mitochondrial assay solution) were added to each well of an XF24 cell culture microplate, centrifuged at 2000×g for 20 min at 4 °C, and then supplemented with 450 µL of mitochondrial assay solution containing 4 mm ADP to initiate the experiments. Additions were as indicated in Figure 6A. Cellular oxygen consumption was assessed with the Seahorse Extracellular Flux Analyzer XF24[51] on monolayers of HeLa cells seeded at a density of 3×104 per well in 0.2 mL DMEM and cultured for 24 h. Assays were started by replacing the growth medium with 0.5 mL serum and antibiotic-free unbuffered DMEM (pH 7.4) supplemented with the multidrug resistance inhibitor verapamil (20 µm, data not shown) or CsH (2 µg mL−1), and additions were made as indicated in Figure 6C. At the end of the experiment the protein content per well was quantified with a BCA Protein Assay Kit (Thermo Scientific-Pierce).
Cell viability assay
HeLa cells were seeded at a density of 1×104 per well in 96-well plates and let to adhere for 6 h before treatment with varying concentrations of 60 or vehicle (1% DMSO) in the presence of the multidrug resistance inhibitor verapamil (20 µm) or CsH (2 µg mL−1). After treatment for 24 h the relative viable cell number was determined with a CellTiter 96 Aqueous One Solution Cell Proliferation Assay Kit (Promega).
Zebrafish and embryo maintenance
Adult zebrafish were maintained in the facility of the University of Padova containing aerated, 28.5 °C-conditioned saline water according to standard protocols. Fish were kept under a 14 h light–10 h dark cycle. For mating, males and females were separated in the late afternoon, and were freed to start courtship the next morning, which ended with egg deposition and fecundation. Eggs were collected, washed with fish water (0.5 mm NaH2PO4, 0.5 mm NaHPO4, 0.2 mg L−1 methylene blue, 3 mg L−1 instant ocean) and embryos were maintained at 28.5 °C. All protocols and manipulations with zebrafish were performed as described.[52]
Morpholino injections
To reproduce the dominant negative UCMD or BM phenotype in zebrafish, we used a previous published exon 9 morpholino,[28] which targets exon 9 of the zebrafish col6a1 gene. Exon 9, col6a1: GAG AGC GGA AGA CGA ACC TTC ATTC (GeneTools, Inc.). A control morpholino, with no sequence homology in zebrafish genome was used. Embryos isolated after paired matings of wild-type zebrafish were injected at 1–2 cell stage using a WPI pneumatic PicoPump PV820 injector. Morpholino was injected at a concentration of 0.1 mm, corresponding to ~4 ng per embryo.
Compound treatment
Morphant embryos were dechorionated at 20 hpf and then treated with 60 at 21 hpf. Untreated morphants and wild-type embryos were used as controls. Compound 60 was used at 5 and 10 µm and dissolved in fish water with 1% DMSO. Vehicle control treatment consisted of fish water with 1% DMSO. Analyses of compound effects on embryos were performed as described at 24 and 48 hpf.
Motor activity
Spontaneous coiling rates were recorded by observing the number of coiling events in 15 s for single embryos at 24 hpf using light microscopy. Touch-evoked escape response was measured at 48 hpf by observing the ability of larvae to escape after touching the body with a little tip. Embryos were subdivided into four groups according to their ability to escape: paralyzed with no ability to move, showing coiling events only, embryos with minor motor impairments, or normal embryos swimming in the fish water; these were assigned a score of 0, 1, 2, or 3, respectively. Statistical analysis was performed on mean scores at each experimental condition.
Birefringence assay
Muscle birefringence was measured at 48 hpf on tricaine-anesthetized embryos by taking advantage of muscle fiber anisotropy. It was measured using two polarizing filters on a Leica M165FC stereomicroscope. Briefly, anesthetized embryos were placed on a glass slide, and muscle light refraction was analyzed by using two polarizing filters. The first filter produces the polarized light to illuminate the sample, and the second polarizing filter, called the analyzer, calculates the angle of light refracted from muscle fibers. In particular, the top polarizing filter was twisted at a 90° angle until the light refracting through the muscle was visible through a stereomicroscope. Integrated area of birefringence was calculated by using ImageJ software.[48] Birefringence values ≥2×106 (typical of wild-type individuals) were rated as normal, values between 1.9 and 0.6×106 were considered as an indication of mild disease, and values ≤ 0.6×106 were rated as an indication of severe myopathy. Statistical analysis was performed on the mean birefringence values at each experimental condition.
Statistical analysis
Differences between control and compound-treated samples were determined by one-way ANOVA test with Bonferroni correction using GraphPad Prism (version 5.1 for Windows). Data represent the mean of at least five independent experiments (n=52 for each condition) ± SEM; **p < 0.01, ***p < 0.001 for Figure 7A. For Figure 7B, comparison between groups at different conditions was made using χ2 test and one-way ANOVA with Bonferroni correction; **p < 0.01, ***p < 0.001. For Figure 7B,C, the total number of embryos used is n=35 for each condition; *p < 0.05, ***p < 0.001, as determined by χ2 test and one-way ANOVA with Bonferroni correction.
Supplementary Material
Acknowledgements
The authors gratefully acknowledge funding from the US National Institutes of Health (NIH) and Telethon-Italy. Chemistry efforts at the University of Kansas Specialized Chemistry Center were supported by NIH U54HG005031 awarded to J.A. Support for the University of Kansas NMR instrumentation was provided by NIH Shared Instrumentation Grant number S10RR024664 and National Science Foundation (NSF) Major Research Instrumentation Grant number 0320648. The authors thank Patrick Porubsky (University of Kansas) for compound management. Initial assay validation, high-throughput screening, and hit confirmation efforts at the Conrad Prebys Center for Chemical Genomics were supported by NIH grant U54HG005033 awarded to J.C.R. Funding for the biological assays was provided by NIH R03A033978 awarded to M.F. and P.B., NIH U54HG005031-05S1 awarded to J.A., and by Telethon-Italy GGP14037 to P.B.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cmdc.201500284.
Contributor Information
Michael A. Forte, Email: forte@ohsu.edu.
Paolo Bernardi, Email: bernardi@bio.unipd.it.
Frank J. Schoenen, Email: schoenen@ku.edu.
References
- 1.a) Siemen D, Ziemer M. IUBMB Life. 2013;65:255–262. doi: 10.1002/iub.1130. [DOI] [PubMed] [Google Scholar]; b) Bernardi P, Rasola A, Forte M, Lippe G. Physiol. Rev. 2015;95:1111–1155. doi: 10.1152/physrev.00001.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Szabadkai G, Duchen MR. Physiology. 2008;23:84–94. doi: 10.1152/physiol.00046.2007. [DOI] [PubMed] [Google Scholar]; b) Gunter TE, Sheu S-S. Biochim. Biophys. Acta J Bioenerg. 2009;1787:1291–1308. doi: 10.1016/j.bbabio.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Bernardi P, Krauskopf A, Basso E, Petronilli V, Blachly-Dyson E, De Lisa F, Forte M. FEBS J. 2006;273:2077–2099. doi: 10.1111/j.1742-4658.2006.05213.x. [DOI] [PubMed] [Google Scholar]; b) Barsukova AG, Bourdette D, Forte M. Eur. J. Neurosci. 2011;34:437–447. doi: 10.1111/j.1460-9568.2011.07760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Barsukova A, Komarov A, Hajnoczky G, Bernardi P, Bourdette D, Forte M. Eur. J. Neurosci. 2011;33:831–842. doi: 10.1111/j.1460-9568.2010.07576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Bernardi P, Scorrano L, Colonna R, Petronilli V, Di Lisa F. Eur. J. Biochem. 1999;264:687–701. doi: 10.1046/j.1432-1327.1999.00725.x. [DOI] [PubMed] [Google Scholar]; b) Forte M, Bernardi P. J. Bioenerg. Biomembr. 2005;37:121–128. doi: 10.1007/s10863-005-6565-9. [DOI] [PubMed] [Google Scholar]
- 5.Forte M, Gold BG, Marracci G, Chaudhary P, Basso E, Johnsen D, Yu X, Fowlkes J, Bernardi P, Bourdette D. Proc. Natl. Acad. Sci. USA. 2007;104:7558–7563. doi: 10.1073/pnas.0702228104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin LJ, Gertz B, Pan Y, Price AC, Molkentin JD, Chang Q. Exp. Neurol. 2009;218:333–346. doi: 10.1016/j.expneurol.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Nat. Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Millay DP, Sargent MA, Osinska H, Baines CP, Barton ER, Vuagniaux G, Sweeney HL, Robbins J, Molkentin JD. Nat. Med. 2008;14:442–447. doi: 10.1038/nm1736. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Merlini L, Angelin A, Tiepolo T, Braghetta P, Sabatelli P, Zamparelli A, Ferlini A, Maraldi NM, Bonaldo P, Bernardi P. Proc. Natl. Acad. Sci. USA. 2008;105:5225–5229. doi: 10.1073/pnas.0800962105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 10.Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Proc. Natl. Acad. Sci. USA. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fujimoto K, Chen Y, Polonsky KS, Dorn GW., II Proc. Natl. Acad. Sci. USA. 2010;107:10214–10219. doi: 10.1073/pnas.0914209107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Battigelli A, Russier J, Venturelli E, Fabbro C, Petronilli V, Bernardi P, Da Ros T, Prato M, Bianco A. Nanoscale. 2013;5:9110–9117. doi: 10.1039/c3nr02694a. [DOI] [PubMed] [Google Scholar]
- 13.a) Leung AW, Varanyuwatana P, Halestrap AP. J. Biol. Chem. 2008;283:26312–26323. doi: 10.1074/jbc.M805235200. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Azarashvili T, Grachev D, Krestinina O, Evtodienko Y, Yurkov I, Papadopoulos V, Reiser G. Cell calcium. 2007;42:27–39. doi: 10.1016/j.ceca.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 14.a) Szabu I, De Pinto V, Zoratti M. FEBS lett. 1993;330:201–205. doi: 10.1016/0014-5793(93)80273-w. [DOI] [PubMed] [Google Scholar]; b) Szabu I, De Pinto V, Zoratti M. FEBS lett. 1993;330:206–210. doi: 10.1016/0014-5793(93)80274-x. [DOI] [PubMed] [Google Scholar]
- 15.Halestrap AP, Brennerb C. Curr. Med. Chem. 2003;10:1507–1525. doi: 10.2174/0929867033457278. [DOI] [PubMed] [Google Scholar]
- 16.a) Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Nat. Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Kwong JQ, Davis J, Baines CP, Sargent MA, Karch J, Wang X, Huang T, Molkentin JD. Cell Death Differ. 2014;21:1209–1217. doi: 10.1038/cdd.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gutiérrez-Aguilar M, Douglas DL, Gibson AK, Domeier TL, Molkentin JD, Baines CP. J. Mol. Cell. Cardiol. 2014;72:316–325. doi: 10.1016/j.yjmcc.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Šileikyte J, Blachly-Dyson E, Sewell R, Carpi A, Menabo R, Di Lisa F, Ricchelli F, Bernardi P, Forte M. J. Biol. Chem. 2014;289:13769–13781. doi: 10.1074/jbc.M114.549634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabo I, Lippe G, Bernardi P. Proc. Natl. Acad. Sci. USA. 2013;110:5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Carraro M, Giorgio V, Sileikyte J, Sartori G, Forte M, Lippe G, Zoratti M, Szabo I, Bernardi P. J. Biol. Chem. 2014;289:15980–15985. doi: 10.1074/jbc.C114.559633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bernardi P. Front. Physiol. 2013;4:95. doi: 10.3389/fphys.2013.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.For a detailed discussion on prior art, see reference [29].
- 21.Basso E, Fante L, Fowlkes J, Petronilli V, Forte M, Bernardi P. J. Biol. Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- 22.Wang P, Heitman J. Genome Biol. 2005;6:226. doi: 10.1186/gb-2005-6-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 24.Waldmeier PC, Feldtrauer JJ, Qian T, Lemasters JJ. Mol. Pharmacol. 2002;62:22–29. doi: 10.1124/mol.62.1.22. [DOI] [PubMed] [Google Scholar]
- 25.a) Taddeo EP, Laker RC, Breen DS, Akhtar YN, Kenwood BM, Liao JA, Zhang M, Fazakerley DJ, Tomsig JL, Harris TE, Keller SR, Chow JD, Lynch KR, Chokki M, Molkentin JD, Turner N, James DE, Yan Z, Hoehn KL. Mol. Metab. 2014;3:124–134. doi: 10.1016/j.molmet.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tiepolo T, Angelin A, Palma E, Sabatelli P, Merlini L, Nicolosi L, Finetti F, Braghetta P, Vuagniaux G, Dumont JM, Baldari CT, Bonaldo P, Bernardi P. Br. J. Pharmacol. 2009;157:1045–1052. doi: 10.1111/j.1476-5381.2009.00316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.a) Schinkel AH, Wagenaar E, van Deemter L, Mol CA, Borst P. J. Clin. Invest. 1995;96:1698–1705. doi: 10.1172/JCI118214. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Murasawa S, Iuchi K, Sato S, Noguchi-Yachide T, Sodeoka M, Yokomatsu T, Dodo K, Hashimoto Y, Aoyama H. Bioorg. Med. Chem. 2012;20:6384–6393. doi: 10.1016/j.bmc.2012.08.062. [DOI] [PubMed] [Google Scholar]; c) Yun B, Lee H, Ghosh M, Cravatt BF, Hsu KL, Bonventre JV, Ewing H, Gelb MH, Leslie CC. J. Biol. Chem. 2014;289:1491–1504. doi: 10.1074/jbc.M113.497651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fancelli D, Abate A, Amici R, Bernardi P, Ballarini M, Cappa A, Carenzi G, Colombo A, Contursi C, Di Lisa F, Dondio G, Gagliardi S, Milanesi E, Minucci S, Pain G, Pelicci PG, Saccani A, Storto M, Thaler F, Varasi M, Villa M, Plyte S. J. Med. Chem. 2014;57:5333–5347. doi: 10.1021/jm500547c. [DOI] [PubMed] [Google Scholar]
- 28.a) Telfer WR, Busta AS, Bonnemann CG, Feldman EL, Dowling JJ. Hum. Mol. Genet. 2010;19:2433–2444. doi: 10.1093/hmg/ddq126. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zulian A, Rizzo E, Schiavone M, Palma E, Tagliavini F, Blaauw B, Merlini L, Maraldi NM, Sabatelli P, Braghetta P, Bonaldo P, Argenton F, Bernardi P. Hum. Mol. Genet. 2014;23:5353–5363. doi: 10.1093/hmg/ddu254. [DOI] [PubMed] [Google Scholar]
- 29.Sileikyte J, Roy S, Porubsky P, Neuenswander B, Wang J, Hedrick M, Pinkerton AB, Salaniwal S, Kung P, Mangravita-Novo A, Smith LH, Bourdette DN, Jackson MR, Aubé J, Chung TD, Schoenen FJ, Forte MF, Bernardi P, editors. NIH Molecular Libraries. Probe Reports from the Molecular Libraries Program. Bethesda (MD): National Center for Biotechnology Information (USA); 2010. Small Molecules Targeting the Mitochondrial Permeability Transition. [ http://www.ncbi.nlm.nih.gov/books/NBK47352/: April 15, 2014 (updated January 16, 2015)] [PubMed] [Google Scholar]
- 30.a) Baell JB. Future Med. Chem. 2010;2:1529–1546. doi: 10.4155/fmc.10.237. [DOI] [PubMed] [Google Scholar]; b) Baell JB, Holloway GA. J. Med. Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 31.Kumar KA, Jayaroopa P. Int. J. Pharm. Chem. Biol. Sci. 2013;3:294–304. [Google Scholar]
- 32.Jung HK, Doddareddy MR, Cha JH, Rhim H, Cho YS, Koh HY, Jung BY, Pae AN. Bioorg. Med. Chem. 2004;12:3965–3970. doi: 10.1016/j.bmc.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 33.Bertenshaw SR, Carter JS, Collins PW, Docter SH, Granets MJ, Khanna IK, Malecha JW, Miyashiro JM, Penning TD, Rogers KL, Rogier DJ, Jr, Talley JJ, Yu SS. Pat. No. EP 0924201. B1. G.D. Searle & Co.; 1999. [Google Scholar]
- 34.Bischoff A, Sundaresan K, Bala Koteswara RP, Ainan B, Ayyamperumal H, Grish A. R, Tatiparthy S, Prabhu G, Subramanya H. Int. PCT Pub. No. WO 2009117676. A3. Forest Laboratories Holdings Ltd.; 2010. [Google Scholar]
- 35.Hopkins AL, Keseru GM, Leeson PD, Rees DC, Reynolds CH. Nat. Rev. Drug Discovery. 2014;13:105–121. doi: 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- 36.Full panel inhibition data for compound 60 are provided in the Supporting Information.
- 37.a) Azzolin L, von Stockum S, Basso E, Petronilli V, Forte MA, Bernardi P. FEBS lett. 2010;584:2504–2509. doi: 10.1016/j.febslet.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) von Stockum S, Giorgio V, Trevisan E, Lippe G, Glick GD, Forte MA, Da-Re C, Checchetto V, Mazzotta G, Costa R, Szabo I, Bernardi P. J. Biol. Chem. 2015;290:4537–4544. doi: 10.1074/jbc.C114.629766. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ricchelli F, Dabbeni-Sala F, Petronilli V, Bernardi P, Hopkins B, Bova S. Biochim. Biophys. Acta Bioenerg. 2005;1708:178–186. doi: 10.1016/j.bbabio.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 38.Petronilli V, Costantini P, Scorrano L, Colonna R, Passamonti S, Bernardi P. J. Biol. Chem. 1994;269:16638–16642. [PubMed] [Google Scholar]
- 39.a) Costantini P, Colonna R, Bernardi P. Biochim. Biophys. Acta Bioenerg. 1998;1365:385–392. doi: 10.1016/s0005-2728(98)00090-5. [DOI] [PubMed] [Google Scholar]; b) Sileikyte J, Petronilli V, Zulian A, Dabbeni-Sala F, Tognon G, Nikolov P, Bernardi P, Ricchelli F. J. Biol. Chem. 2011;286:1046–1053. doi: 10.1074/jbc.M110.172486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.a) Ford JM, Hait WN. Pharmacol. Rev. 1990;42:155–199. [PubMed] [Google Scholar]; b) Bernardi P. Ital. J. Neurol. Sci. 1999;20:395–400. doi: 10.1007/s100720050057. [DOI] [PubMed] [Google Scholar]
- 41.a) Tsuruo T, Iida H, Tsukagoshi S, Sakurai Y. Cancer Res. 1981;41:1967–1972. [PubMed] [Google Scholar]; b) Bosch I, Croop J. Biochim. Biophys. Acta Rev. Cancer. 1996;1288:F37–F54. doi: 10.1016/0304-419x(96)00022-4. [DOI] [PubMed] [Google Scholar]
- 42.a) Pepe G, Bertini E, Bonaldo P, Bushby K, Giusti B, de Visser M, Guicheney P, Lattanzi G, Merlini L, Muntoni F, Nishino I, Nonaka I, Yaou RB, Sabatelli P, Sewry C, Topaloglu H, van der Kooi A. Neuromuscular Disord. 2002;12:984–993. doi: 10.1016/s0960-8966(02)00139-6. [DOI] [PubMed] [Google Scholar]; b) Lampe AK, Bushby KMD. J. Med. Genet. 2005;42:673–685. doi: 10.1136/jmg.2002.002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.a) Maraldi NM, Sabatelli P, Columbaro M, Zamparelli A, Manzoli FA, Bernardi P, Bonaldo P, Merlini L. Adv. Enzyme Regul. 2009;49:197–211. doi: 10.1016/j.advenzreg.2008.12.009. [DOI] [PubMed] [Google Scholar]; b) Irwin WA, Bergamin N, Sabatelli P, Reggiani C, Megighian A, Merlini L, Braghetta P, Columbaro M, Volpin D, Bressan GM, Bernardi P, Bonaldo P. Nat. Genet. 2003;35:367–371. doi: 10.1038/ng1270. [DOI] [PubMed] [Google Scholar]
- 44.Angelin A, Tiepolo T, Sabatelli P, Grumati P, Bergamin N, Golfieri C, Mattioli E, Gualandi F, Ferlini A, Merlini L, Maraldi NM, Bonaldo P, Bernardi P. Proc. Natl. Acad. Sci. USA. 2007;104:991–996. doi: 10.1073/pnas.0610270104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.a) Zulian A, Tagliavini F, Rizzo E, Pellegrini C, Sardone F, Zini N, Maraldi NM, Santi S, Faldini C, Merlini L, Petronilli V, Bernardi P, Sabatelli P. Front. Aging Neurosci. 2014;6:324. doi: 10.3389/fnagi.2014.00324. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Merlini L, Sabatelli P, Armaroli A, Gnudi S, Angelin A, Grumati P, Michelini ME, Franchella A, Gualandi F, Bertini E, Maraldi NM, Ferlini A, Bonaldo P, Bernardi P. Oxid. Med. Cell. Longevity. 2011;2011:139194. doi: 10.1155/2011/139194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palma E, Tiepolo T, Angelin A, Sabatelli P, Maraldi NM, Basso E, Forte MA, Bernardi P, Bonaldo P. Hum. Mol. Genet. 2009;18:2024–2031. doi: 10.1093/hmg/ddp126. [DOI] [PubMed] [Google Scholar]
- 47.a) Lucarini L, Giusti B, Zhang R-Z, Pan T-C, Jimenez-Mallebrera C, Mercuri E, Muntoni F, Pepe G, Chu M-L. Hum. Genet. 2005;117:460–466. doi: 10.1007/s00439-005-1318-8. [DOI] [PubMed] [Google Scholar]; b) Lampe AK, Zou Y, Sudano D, O’Brien KK, Hicks D, Laval SH, Charlton R, Jimenez-Mallebrera C, Zhang RZ, Finkel RS, Tennekoon G, Schreiber G, van der Knaap MS, Marks H, Straub V, Flanigan KM, Chu ML, Muntoni F, Bushby KMD, Bönnemann CG. Hum. Mutat. 2008;29:809–822. doi: 10.1002/humu.20704. [DOI] [PubMed] [Google Scholar]
- 48.Berger J, Sztal T, Currie PD. Biochem. Biophys. Res. Commun. 2012;423:785–788. doi: 10.1016/j.bbrc.2012.06.040. [DOI] [PubMed] [Google Scholar]
- 49.Gornall AG, Bardawill CJ, David MM. J. Biol. Chem. 1949;177:751–766. [PubMed] [Google Scholar]
- 50.Rogers GW, Brand MD, Petrosyan S, Ashok D, Elorza AA, Ferrick DA, Murphy AN. PloS one. 2011;6:e21746. doi: 10.1371/journal.pone.0021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, Ferrick DA. Am. J. Physiol. 2006;292:C125–C136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
- 52.Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Dev. Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.