

Abstract
Antibiotic resistance represents one of the greatest threats to public health. The adenylation inhibitor 5′-O-[N-(salicyl)sulfamoyl]adenosine (SAL-AMS) is the archetype for a new class of nucleoside antibiotics that target iron acquisition in pathogenic microorganisms and is especially effective against Mycobacterium tuberculosis, the causative agent of tuberculosis. Strategic incorporation of fluorine at the 2′ and 3′ positions of the nucleoside was performed by direct fluorination to enhance activity and improve drug disposition properties. The resulting SAL-AMS analogs were comprehensively assessed for biochemical potency, whole-cell antitubercular activity, and in vivo pharmacokinetic parameters. Conformational analysis suggested a strong preference of fluorinated sugar rings for either a 2'-endo, 3'-exo (South) or a 3'-endo, 2'-exo (North) conformation. The structure–activity relationships revealed a strong conformational bias for the C3′-endo conformation to maintain potent biochemical and whole-cell activity whereas improved pharmacokinetic properties were associated with the C2′-endo conformation.
Graphical Abstract

INTRODUCTION
Iron is the most abundant element in the universe and essential for all domains of life where it serves as a redox cofactor for enzymes involved in diverse metabolic pathways. However, in many environments, iron is severely depleted, which is particularly challenging for microorganisms that can only assimilate nutrients from their immediate surroundings. To prevent bacterial colonization and growth, mammals use iron binding proteins such as transferrin to withhold iron. The concentration of unbound Fe3+ in human serum and body fluids is ~10−24 M, an astonishingly low concentration close to the reciprocal of Avogadro's number.1 To survive under these iron-restricted conditions, many pathogenic bacteria synthesize, secrete, and reimport small-molecule iron-chelators known generically as siderophores.2 Mycobacterium tuberculosis, the leading cause of bacterial infectious disease mortality, and the focus of the present report, relies on mycobactins 1 (Figure 1), which are aryl-capped siderophores, for iron acquisition in vivo.3 Given the vital role that siderophores play in bacterial iron acquisition, disruption of siderophore biosynthesis or trafficking could provide a new strategy to combat drug resistant bacteria. This strategy is particularly effective for M. tuberculosis since it relies exclusively on the mycobactins for siderophore iron mobilization, whereas other bacteria such as Escherichia coli and Acinetobacter baumanii synthesize multiple siderophores.
Figure 1.

Structures of representative aryl-capped siderophores. The siderophore name is given below each structure along with the producing organism(s). The aryl caps are highlighted in blue (salicylic acid or 2,3-dihydrobenzoic acid). The aryl acid adenylating enzymes (AAAEs) responsible for incorporation of the respective aryl moieites have high homology at the overall protein level and nearly identical active-site residues (MbtA, EntE, BasE, YbtE, PchD, and VibE).
The nucleoside antibiotic 5′-O-[N-(salicyl)sulfamoyl]adenosine (SAL-AMS, 10, Figure 2B) is a prototype for a new class of antibiotics that targets iron acquisition through inhibition of aryl acid adenylating enzymes (AAAEs) in several pathogenic bacteria including MbtA from M. tuberculosis, EntE from Escherichia coli and Klebsiella pneumoniae, BasE from Acinetobacter baumannii, YbtE from Yersinia pestis, VibE from Vibrio cholerae, and PchA from Pseudomonas aeruginosa.4 AAAEs catalyze the first committed step in the synthesis of the respective aryl-capped siderophores 2–6 produced by each bacteria (Figure 1). Adenylation, the ligation of a carboxylic acid substrate with AMP to form an acyl-AMP intermediate, is a ubiquitous process and adenylation inhibitors like SAL-AMS (10) mimic the acyl-AMP intermediate, through replacement of the labile acyl-phosphate moiety with an acyl-sulfamate bioisostere (Figure 2).5 The specificity of SAL-AMS is derived from the salicyl moiety, which allows it to bind to adenylating enzymes that utilize salicylic acid or related carboxyl acid substrates. Affinity-based protein profiling of SAL-AMS in M. tuberculosis showed it binds only to MbtA from more than 60 functionally related adenylating enzymes in mycobacteria.6 Isosteric substitution of the phenol of SAL-AMS with an amino group, completly obliterated biological activity, due to a H-bond repulsion in the enzyme active site, illustrating the high level of selectivity that one can obtain with such adenylation inhibitors.7 SAL-AMS has demonstrated proof-of-concept in vitro4a,c and in vivo,8 but like other adenylation inhibitors that mimic their native acyl-adenylates, it is marred by poor physicochemical properties. To further advance this new class of antibiotics, key improvements in the molecular properties of SAL-AMS will be required.
Figure 2.

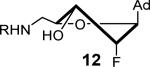
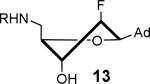
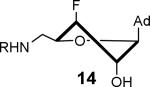
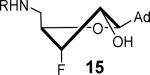
Mycobactin biosynthesis and inhibition. (A) The biosynthesis of mycobactins in M. tuberculosis is initiated by MbtA that catalyzes two partial reactions at the same active site. In the first half-reaction MbtA condenses salicylic acid (7) with ATP to form the reactive mixed anhydride SAL-AMP (8). In the second half-reaction MbtA transfers the salicyl moiety onto MbtB, another protein in the biochemical pathway, to afford 9 that is ultimately elaborated to the mycobactins. The initial biosynthetic step of other aryl-capped siderophores is performed by homologous aryl acid adenylation enzymes (AAAEs). (B) The inhibitors Sal-AMS 10 and its sulfamide isostere 11 mimic the acyl-adenylate intermediate 8, thereby blocking siderophore biosynthesis. The sugar can adopt the Northern (C3′-endo pucker) or Southern (C2′-endo pucker) conformation as depicted. (C) Fluorinated sugar analogs described in this study.
One of the most common means to modulate chemical properties of small molecules is to introduce fluorine, often considered an isostere of hydrogen; although fluorine's van der Waal radius (1.47 Å) is closer to oxygen (1.52 Å) than hydrogen (1.20 Å). Fluorination also has a long tradition in nucleoside chemistry and the synthetic provenance can be traced to a report by Fox and co-workers.9 The replacement of the 2′ or 3′ hydroxyl groups of a nucleoside with a fluorine atom causes only a minor change in the overall structure, but profoundly affects the stereoelectronic properties of the sugar moiety.10 Such dominating effects can control the conformational equilibria and lock the sugar ring into either a North (C3′-endo pucker) or a South (C2′-endo pucker) conformation,11 can stabilize the glycosidic bond towards hydrolysis,12 and can also enhance the lipophilicity.13
Based on the beneficial effects of fluorination, we postulated that strategic introduction of fluorine at the 2′ and/or 3′ position in both α and β- configuration of SAL-AMS may enhance the biological activity and pharmacokinetic behavior. We predicted compounds that mimic the C3′-endo pucker of the native acyl-adenylate intermediate14 would exhibit greater biological activity than compounds that adopt the C2′-endo pucker. However, we could not anticipate the impact of ring pucker or extent of fluorination on in vivo pharmacokinetic parameters. Herein we describe synthesis and conformational analysis of a systematic series of fluorinated nucleoside derivatives of SAL-AMS 12–17 (Figure 2C), along with their enzyme inhibition, antibacterial activity, and complete in vivo pharamacokinetic characterization.
RESULTS AND DISCUSSION
Synthesis
The synthesis of 12–17 was accomplished in two stages: synthesis of the requisite fluorinated nucleoside followed by installation of the N-(salicyl)sulfamide moiety at C-5′. For the second stage, we developed a new methodology while for the first stage, we used an amalgamation of methods for the direct introduction of fluorine into the targeted adenine nucleosides.15 The main synthetic challenge was to install fluorine at the desired positon of the sugar moiety and with the desired stereochemistry. There are two general synthetic approaches towards fluorinated nucleosides: (1) transformation of natural nucleosides using various fluorinating reagents and (2) glycosylation of fluorinated sugar with an appropriate heterocyclic base. With the first approach, one can start with a pre-defined α or β configuration at C-1′, but the main limitation is the sluggish reactivity of certain pentofuranose hydroxyls towards fluorinating reagents. In the second approach, a wide variety of nucleosides can be prepared, but the formation of regioisomers and anomers (α and β isomers) are the major drawbacks. We selected the first approach since it usually involves fewer synthetic steps when suitable protecting groups are available.
The direct introduction of fluorine into each nucleoside building block is illustrated in Schemes 1–4. For the preparation of 3′-deoxy-3′-F-xylo 23 and 2′-deoxy-2′-F-arabino 24, a unified synthetic approach was employed (Scheme 1). Regioselective PMB protection16 of the secondary alcohols of adenosine 18 provided a 3:1 mixture of 2′-OPMB adenosine 19 and 3′-OPMB adenosine 20 in 75% yield. The ratio of 19 and 20 in the mixture was determined by 1H NMR integrations of the anomeric protons. Without further separation of regioisomers, subsequent tritylation of the 5′-OH and 6-NH2 afforded a 3:1 mixture of 21 and 22 that was fluorinated with attendant inversion of configuration using DAST17 to obtain 23 (55% from 21) and 24 (42% from 22), which were easily separated by chromatography. The assignment of fluorine configuration was based on proton and fluorine spectra. In the case of 3′-deoxy-3′-F-xylo 23, the vicinal coupling of fluorine with both 2′-H and 4′-H (3J(2′,F3′) = 13.9, 3J(4′,F3′) = 31.4 Hz), and germinal coupling with 3′-H (2J(3′,F3′) = 50.6 Hz) confirmed the position of fluorine at 3′. The coupling constants also suggest 3′-F has an anti relationship with 4′-H and a syn relationship with 2′-H, which indicates a xylo configuration. The absence of detectable 3J(1′,2′) is consistent with a 3′-endo conformation.18 The configuration at C2′ of 2′-deoxy-2′-F-arabino 24 was unequivocally assigned by the presence of a five bond coupling between 2′-F and C8-H of the adenine base (5J(8,F2′) = 3.1 Hz), which is only observed in arabino-2′-F compounds.17
Scheme 1.

Synthesis of 3′-deoxy-3′-F-xylo and 2′-deoxy-2′-F-arabino nucleosides
Scheme 4.

Synthesis of 2′,3′-dideoxy-2′,3′-FF-ribo nucleoside
The key intermediate 21 also served as an entry point for synthesis of 3′-deoxy-3′-F-ribo 27 through a two-step oxidation–reduction sequence19 that inter-converted the ribo sugar to a xylo sugar (Scheme 2). This was accomplished by Dess-Martin periodinane (DMP) mediated oxidation of 21 to 3'-ketoadenosine derivative 25, which was stereoselectively reduced with sodium triacetoxyborohydride to furnish alcohol 26. The bulky reducing agent NaBH(OAc)3 delivers hydride from the more accessible α-face of the nucleoside. Fluorination of 26 with DAST gave 27 in 61% yield. Once again, the coupling constants of fluorine with 2′-H and 4′-H (3J(2′,F3′) = 21.6, 3J(4′,F3′) = 26.4 Hz) compared to those of 3′-deoxy-3′-F-xylo 23 indicate a ribo-configuration. A significantly large coupling between 1′-H and 2′-H (3J(1′,2′) = 7.5) and the absence of 3J(3′,4′) further confirms the 2′-endo conformation of 3′-deoxy-3′-F-ribo compounds.
Scheme 2.

Synthesis of 3′-deoxy-3′-F-ribo nucleoside
The novel difluorinated nucleoside 2′,3′-dideoxy-2′,3′-FF-xylo 31 was synthesized from 2′-fluoroadenosine 2820 by trityl protection to provide 29 and subsequent treatment with DAST furnished 31 in 62% overall yield (Scheme 3). The 2′-deoxy-2′-F-ribo 30 building block was prepared by PMB protection of 29. As the configuration of fluorine at C2′ is known, a strong vicinal trans coupling between two the fluorines (3J(F2′,F3′) = 15.0) in 31 indicate the 2′,3′-xylo difluoro configuration. The absence of 3J(1′,2′) is consistent with the 3′-endo conformation.
Scheme 3.

Synthesis of 2′,3′-dideoxy-2′,3′-FF-xylo and 2′-deoxy-2′-F-ribo nucleosides
The difluorinated 2′,3′-dideoxy-2′,3′-FF-ribo 35 was prepared from the known lyxo-epoxide21 32 (Scheme 4) following the Watanabe two-step process initiated by epoxide opening with a fluoride ion exclusively from the α-face of the sugar to afford a 3:1 ratio of regioisomers 3′-fluoro-arabino 33 and 2′-fluoro-xylo 34 in a 65% combined yield. Treatment of this mixture with DAST furnished 35 in a 62% yield.22 The regioisomeric ratio was determined by integration of the relevant peaks in 19F NMR spectrum of the crude products. There was no need for the separation of 33 and 34 because both provide the same difluoro product 35 after treatment with DAST. The presence of rather weak cis vicinal coupling between two fluorines (3J(F2′,F3′) = 3.8) and a strong coupling between 1′-H and 2′-H (3J(1′,2′) = 5.0) indicates the 2′,3′-ribo difluoro configuration and C2′-endo conformation.
Each of the fully protected fluorinated nucleosides was globally deprotected with 80% TFA in chloroform to obtain fluorinated nucleosides 36–40 (Scheme 5). This allowed unequivocal structural assignment for 36,23 37,24 38,17, 25 and 4026 by comparison to the literature data of these nucleosides synthesized through alternate synthetic routes, but not 39 since this has not been previously reported. Illustrating the efficiency of direct fluorination, our synthesis of 40 proceeded in 6 steps and 8.5% overall yield from adenosine, whereas, the only described synthesis that utilized glycosylation of adenine with a fluorinated carbohydrate required 13 steps in 0.4% overall yield.26
Scheme 5.

Synthesis of fully unprotected fluorinated nucleoside
The fluorinated nucleoside building blocks were elaborated to the corresponding SAL-AMS derivatives 12–17 (Scheme 6). The primary motivation for using the trityl protecting group among the plethora of common nucleoside protecting groups was based upon prior reports that had shown O-Tr groups could be chemoselectively removed in the presence of a N-Tr group27 as well as the improved lipophilicity engendered by this nonpolar group that facilitated chromatographic purification. De-O-tritylation of 30, 24, 23, 27, 31, and 35 was successively achieved with 0.4 M HCl in 1,4-dioxane to afford 41a–f. Mono-deprotection was complete within 1 h and longer reaction times led to undesired PMB deprotection. The resulting C5' alcohols in 41a–f were converted to the C5' azides using the methodology of Liu and Austin28 to provide 42a–f. This two-step one-pot method converts the 5′-OH to a phosphate leaving group in the first-step, which is displaced by azide in the second step. We observed that the presence of fluorine in the β (‘up’) configuration found in 41b, 41c, and 41e significantly suppressed the reactivity of 5′ phosphates towards nucleophilic azides. For comparision, 42c was obtained in 57% yield and conversion was still incomplete after 24 h of reflux, whereas 42d was obtained in 81% yield and complete conversion was observed within 3 h. This can be explained in terms of electrostatic repulsion between electron rich fluorine and the incoming azide anion. The azides 42a–f were reduced to amino derivatives in quantitative yields by catalytic hydrogenation or with zinc in AcOH. The crude 5′-aminonucleosides were then transformed to the 5′-sulfamides 43a–f in 84–94% yield by refluxing with sulfamide (NH2SO2NH2) in 1,4-dioxane for 2 h. This new method of sulfamide synthesis is superior to previous examples in terms of yield and experimental simplicity.4c, 29 The synthesis of the target molecules was completed by salicylation of sulfamides 43a–f with NHS-ester 44 to obtain fully protected coupled products 45a–f and global deprotection furnished 7–12 in 39–50% yield.30
Scheme 6.

Completion of synthesis of 7–12
Conformational Analysis
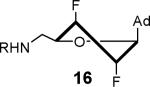
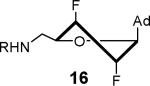
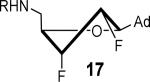
The solution phase conformations adopted by the sugar rings of nucleosides 12–17 were studied by 1H NMR 3J(H,H) scalar coupling constants of ring protons and electronegativities of ring substituents through the use of pseudorotation analysis (program: Matlab Pseudorotation GUI).31 1H NMR were obtained in DMSO-d6 at 23 °C and the multiplets were analyzed (Table 1). Detailed conformation analysis including coupling constants and optimized pseudorotation parameters are provided in Supporting Information (Table S2). The percent North or C3′-endo conformation (%N) adopted by 12–17 agree closely with the reported values for the free nucleosides (Table 1). Most compounds displayed a substantial conformational preference for either the C2′-endo or C3′-endo pucker except for 2′-deoxy-2′-F-ribo 12 and 2′-deoxy-2′-F-arabino 13. The 3′-deoxy-3′-F-xylo 14 primarily adopted the C3′-endo conformation (~90% N) whereas, 3′-deoxy-3′-F-ribo 15 was almost exclusively present in the C2′-endo conformation. Similar conformational rigidity was also observed with 2′,3′-dideoxy-2′,3′-FF-xylo 16 and 2′,3′-dideoxy-2′,3′-FF-ribo 17; the former nearly exclusively in the C3′-endo (96% N) whereas the latter was largely in the C2′-endo (20% N) conformation. Marquez and co-workers previously observed similar extreme inclination of 2′,3′-dideoxy-2′,3′-FF-xylo uridine nucleosides towards C3′-endo conformation (~100% N).32
Table 1.
Relationship between bioactivity and sugar ring conformation of inhibitors 12–17
| Compound | 1H NMR resonances of sugar protonsa | %N (lit.)b | Kiapp (nM) | MIC (μM) |
|---|---|---|---|---|
|
|
65 (67) 33a,b | 1.7 ± 0.1 | 0.19 |
|
|
39 (36)33b | 1.8 ± 0.1 | 0.15 |
|
|
88 (93)10 | 0.6 ± 0.3 | 0.60 |
|
|
0 (2)26a | 6.4 ± 0.3 | 2.3 |
|
|
96 | 1.4 ± 0.3 | 0.78 |

|
|
20 (20)26a | 139 ± 10 | 37 |
NMR was obtained in DMSO-D6, peaks are in expansion view and are ordered according to their chemical shifts
literature values for free nucleosides are reported in Parenthesis.
Biochemical and Antitubercular Evaluation
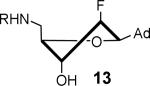
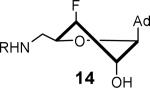
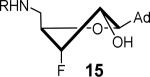
The nucleoside derivatives were evaluated for enzyme inhibition against MbtA, the molecular target of SAL-AMS in M. tuberculosis.4a,c The effect of configuration at C2′ on enzyme inhibition was inconsequential since both 2′-α-fluoro 12 and 2′-β-fluoro 13 were equipotent with appKi's of 1.7–1.8 nM (Table 1). By contrast, the enzyme inhibition was more sensitive to the stereochemical configuration at C3′ and 3′-β-fluoro 14 was approximately 10-fold more potent than 3′-α-fluoro 15. The impact of configuration at C3′ was amplified in difluorinated analogs 16 and 17 (both contain a 2'-α fluoro group), 3′-β-fluoro 16 (appKi = 1.4 nM) was nearly 100-fold more potent than 3'-α-fluoro 17. All compounds were then evaluated for antibacterial activity against virulent M. tuberculosis strain H37Rv and the concentration of inhibitor that resulted in complete inhibition of observable growth was defined as the minimum inhibitory concentration (MIC). The relative trend in antibacterial activities mirrored the biochemical results (Table 1). Thus, the C2′-fluoro diastereomers 12 and 13 had nearly identical MICs while the C3′-fluoro analog 14 was fourfold more potent than 15. The activities of the difluorinated analogs also significantly diverged and 16 (MIC = 0.78 μM) was ca. 50-fold more potent than 17. Comparison of the conformational disposition of each inhibitor with the observed biochemical and antibacterial activities shows an excellent qualitative positive correlation between the C3′-endo conformation and biological activity.
In Vivo Pharmacokinetics
All compounds were then subjected to single dose pharmacokinetic (PK) studies in a crossover experiment using cannulated rats and administered orally (p.o.) at 25 mg/kg then intravenously (i.v.) at 2.5 mg/kg after a 3-day washout period. The serum concentration versus time curves (Figure 2) were employed for the calculation of pharmacokinetic parameters by non-compartmental analysis. Each of the fluorinated analogs exhibited improved oral bioavailability (F%), half-life (t1/2), maximum serum concentration (Cmax), and area under the concentration-time curve (AUC) relative to SAL-AMS 10 as well as decreased clearance (CL) (Table 2). Difluorinated analog 17 achieved the most improved pharmacokinetic profile compared to SAL-AMS (10) with about a fourfold increase in F%, 15-fold higher Cmax and ca. 75-fold increased oral exposure as defined by its oral AUC, which is largely attributed to its remarkable ca. 25-fold increase in half-life to 267 min. This is also clearly noticeable on concentration versus time plots. Pairwise comparison (i.e. 12 vs. 13, 14 vs. 15, and 16 vs. 17) of the oral exposure levels indicated a strong correlation between sugar conformation and relative oral AUC levels. In all cases, analogs favoring the C2′-endo conformation possessed ca. twofold enhanced %F and 3–13-fold greater oral AUCs relative to the diastereomer favoring the C3′-endo conformation.
Table 2.
In Vivo pharmakokinetic parameters of inhibitors in female Sprague-Dawley rats (n = 3, mean ± SD) following a single intravenous (i.v.) and oral (p.o.) administration
| Compound |
i.v. PK parametersa |
p.o. PK parametersb |
||||
|---|---|---|---|---|---|---|
| AUC0-inf (μg.min.mL–1) | CL (mL.min–1.kg–1) | t1/2 (min) | Cmax (μg.mL–1) | AUC0-inf (μg.min.mL–1) | F (%) | |
| 10c | 519 ± 115 | 4.9 ± 0.9 | 11.2 ± 2.2 | 0.40 ± 0.20 | 66 ± 41 | 1.3 ± 0.7 |
| 11c | 304 ± 60 | 8.4 ± 1.9 | 10.6 ± 0.7 | 0.70 ± 0.07 | 100 ± 14 | 3.5 ± 0.2 |

|
1040 ± 330 | 2.5 ± 0.9 | 32.5 ± 7.4 | 0.46 ± 0.30 | 187 ± 11 | 1.8 ± 0.9 |
|
913 ± 65 | 2.7 ± 0.2 | 31.7 ± 1.9 | 1.09 ± 0.03 | 422 ± 70 | 4.7 ± 1.1 |
|
366 ± 33 | 6.9 ± 0.3 | 17.4 ± 1.4 | 0.64 ± 0.22 | 84 ± 12 | 2.3 ± 0.4 |
|
807 ± 78 | 3.1 ± 0.3 | 37.0 ± 1.7 | 0.92 ± 0.04 | 266 ± 2 | 3.5 ± 0.2 |
|
312 ± 54 | 8.5 ± 1.6 | 17.2 ± 1.3 | 0.97 ± 0.42 | 362 ± 85 | 11.4 ± 0.3 |
|
10100 ± 2200 | 0.24 ± 0.06 | 267 ± 36 | 5.99 ± 0.56 | 4730 ± 540 | 5.3 ± 1.3 |
i.v. dose (Di.v.) = 2.5 mg/kg
p.o. dose (Dp.o.) = 25 mg/kg
reference 34.
AUC0-inf: area under the plasma concentration-time curve from time 0 to infinity, CL: clearance, t1/2: terminal elimination half-life, Cmax: maximum plasma concentration, F: relative oral bioavailability calculated as follows F = 100 × [(AUCp.o. × Di.v.)/(AUCi.v. × Dp.o.)].
CONCLUSION
We have reported the efficient synthesis and conformational analysis of a systematic series of fluorinated analogues of SAL-AMS featuring direct fluorination of the nucleoside. The structure–activity relationships (SAR) revealed a strong conformational bias for the C3′-endo conformation to maintain potent biochemical and whole-cell activity. Fluorination of SAL-AMS was also shown to have a dramatic impact on pharmacokinetic properties increasing half-life up to ca. 25-fold, oral exposure 75-fold, and oral bioavailability 10-fold. Further investigations of these fluorinated SAL-AMS derivatives and the application of this strategy to other adenylation inhibitors are ongoing. The results of these studies may have more widespread utility since adenylate-forming enzymes are involved in a myriad of biochemical pathways in DNA, RNA, protein, amino acid, and cofactor biosynthesis as well as post-translational protein modifications.5, 35
EXPERIMENTAL SECTION
General Chemistry Methods
All commercial reagents were used as provided unless otherwise indicated. An anhydrous solvent dispensing system using packed columns of neutral alumina was used for drying THF and CH2Cl2, while packed columns of 4 Å molecular sieves were used to dry DMF, and the solvents were dispensed under nitrogen. All reactions were performed under an inert atmosphere of argon in oven dried (130-150 °C) glassware. Thin-layer chromatography was performed on a pre-coated silica gel 60 F254 plates. The detection of compounds was carried out with UV light. Purification by flash chromatography was performed using a medium-pressure flash chromatography system and flash column silica cartridges with the indicated solvent system. HPLC purifications were performed on instrument equipped with reverse-phase Phenomenex Gemini 10 μm C18 110Å (250 × 21.2 mm) column. All NMR spectra were recorded on a 400 MHz spectrometer at 400 MHz for 1H, 100 MHz for 13C, and 376 MHz for 19F. 1H NMR spectra were referenced to residual CDCl3 (7.27 ppm), DMSO-d6 (2.50 ppm), or CD3OD (3.31 ppm); 13C NMR spectra were referenced to CDCl3 (77.23 ppm) DMSO-D6 (39.51 ppm), or CD3OD (49.15 ppm); and 19F NMR spectra were referenced to hexafluorobenzene (−162.9 ppm)36 or trifluoroacetic acid (−76.5 ppm). NMR chemical shift data are reported as follows: chemical shift, multiplicity (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, ABq = AB quartet, dm = doublet of multiplets), coupling constant, integration. Coupling constants are given in Hertz (Hz). 1H and 13C NMR peak assignments were based on gCOSY and gHMQC NMR spectra, respectively. 19F NMR peaks were assigned using proton-fluorine coupling constants. High-resolution mass spectra (HRMS) were obtained on a TOF instrument.
General Procedure 1: 5′-O,N6-Ditritylation
The nucleoside of choice was dried prior use by coevaporation with anhydrous pyridine. To a stirred solution of the nucleoside (1 mmol, 1 equiv) in pyridine (13 mL) was added DMAP (0.8 equiv) and trityl chloride (2.3 equiv) and the reaction mixture was heated at 80 °C for 4 h. The reaction mixture was cooled down to rt, quenched with EtOH (7 mL), concentrated under reduced pressure and coevaporated with toluene (2 × 10 mL) to obtain a crude residue. Purification by flash chromatography (dry loading, SiO2, EtOAc/hexanes gradient) afforded the 5′-O,N6-ditritylated product.
N6-Trityl-9-[2-O-(4-methoxybenzyl)-5-O-trityl-β-d-ribofuranosyl]adenine (21) and N6-Trityl-9-[3-O-(4-methoxybenzyl)-5-O-trityl-β-d-ribofuranosyl]adenine (22)
An isomeric mixture of 2′-OPMB adenosine 19 and 3′-OPMB adenosine 20 was prepared from adenosine 18 employing a published procedure.16 A mixture of 21 and 22 was prepared from a mixture of 19 and 20 (5.3 g, 13.7 mmol) using the general procedure 1. Flash chromatography (SiO2, gradient: hexanes to 50% EtOAc in hexanes) afforded a mixture of 21 and 22 (5.0 g, 42%) as a white solid which was used in the next step without further separation. The ratio of 21 and 22 in the mixture was determined to be ~3:1 by 1H NMR integrations of anomeric protons.
Major isomer 21: Rf = 0.53 (1:1 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 7.99, 7.85 (2s, 2H; H-2, H-8), 7.42–7.21 (m, 30H; 6×C6H5), 7.14 (d, J = 8.6 Hz, 2H; −PMB), 6.93 (s, 1H; N6-H), 6.77 (d, J = 8.6 Hz, 2H; −PMB), 6.09 (d, 3J(1′,2′) = 4.4 Hz, 1H; H-1′), 4.68–4.66 (m, 1 H; H-2′), 4.64 (ABq, Δδ = 42.8 Hz, J = 11.6 Hz, 2H; −PMB), 4.31–4.27 (m, 1H; H-3′), 4.19 (td, 3J(5′,4′) = 3.3, 3J(3′,4′) ≈ 3J(5″,4′) ≈ 4.6 Hz, 1H; H-4′), 3.77 (s, 3H; −PMB), 3.47 (dd, 3J(5′,4′) = 3.3, 2J(5″,5′) = 10.6 Hz, 1H; H-5′), 3.34 (dd, 3J(5″,4′) = 4.5, 2J(5″,5′) = 10.6 Hz, 1H; H-5″), 2.66 (d, 3J(OH,3′) = 5.6 Hz, 1H; 3′-OH); 13C NMR (100 MHz, CDCl3): δ 160.0, 154.3, 152.5, 148.8, 145.2, 143.8, 138.7, 130.0, 129.2, 128.9, 128.8, 128.1, 128.0, 127.4, 127.1, 121.6, 114.2, 87.3, 87.2, 84.2, 80.5, 73.0, 71.6, 70.3, 63.6, 55.5; HRMS (ESI+): m/z calcd for C56H49N5O5Na [(M + Na)+] 894.3626, found 894.3658 (Δ 3.5 ppm, acquired as a mixture of 21 and 22).
N6-trityl-9-[2-O-(4-methoxybenzyl)-3-deoxy-3-fluoro-5-O-trityl-β-d-xylofuranosyl]adenine (23) and N6-Trityl-9-[3-O-(4-methoxybenzyl)-2-deoxy-2-fluoro-5-O-trityl-β-d-arabinofuranosyl]adenine (24)
To a solution of a mixture of 21 and 22 (4.70 g, 5.39 mmol, 1.0 equiv, 73% 21) in CH2Cl2 (45 mL) and pyridine (4.3 mL, 43.27 mmol, 10 equiv) in a polypropylene tube was added DAST (3.5 mL, 26.49 mmol, 5.0 equiv) dropwise and the mixture was stirred at rt for 24 h. CH2Cl2 (50 mL) was added to the reaction mixture and the organic layer was washed with sat. NaHCO3 and water. The organic layer was dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by flash chromatography (SiO2, gradient: hexanes to 30% EtOAc in hexanes) afforded 23 (1.86 g, 55% from 21) and 24 (0.55 g, 42% from 22) as white solids.
23: Rf = 0.35 (1:3 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 8.07, 7.77 (2s, 2H; H-2, H-8), 7.49–7.24 (m, 32H; 6×C6H5 and −PMB), 6.96 (s, 1H; N6-H), 6.86 (d, J = 8.8 Hz, 2H; −PMB), 6.31 (br s, 1H; H-1′), 5.07 (dd, 3J(3′,4′) = 2.5, 2J(3′,F3′) = 50.6 Hz, 1H; H-3′), 4.76 (ABq, Δδ = 62.8 Hz, J = 11.6 Hz, 2H; −PMB), 4.56 (dtd, 3J(3′,4′) = 2.3, 3J(5′,4′) = 6.1, 3J(5″,4′) = 6.2, 3J(4′,F3′) = 31.3 Hz, 1H; H-4′), 4.38 (d, 3J(2′,F3′) = 13.9 Hz, 1H; H-2′), 3.79 (s, 3H; −PMB), 3.62 (dd, 3J(5″,4′) = 6.8, 2J(5′,5″) = 9.4 Hz, 1H; H-5″), 3.48 (dd, 3J(5′,4′) = 5.9, 2J(5′,5″) = 9.7 Hz, 1H; H-5′); 13C NMR (100 MHz, CDCl3): δ 159.9, 154.2, 152.5, 148.5, 145.2, 143.7, 137.8 (d, 5J(C8,F3′) = 5.8 Hz), 129.9, 129.2, 128.8, 128.8, 128.1, 128.1, 127.4, 127.1, 121.2, 114.2, 94.1 (d, 1J(C3′,F3′) = 184.9 Hz), 88.4, 87.3, 85.3 (d, 2J(C2′,F3′) = 27.3 Hz), 82.1 (d, 2J(C4′,F3′) = 19.0 Hz), 72.5, 71.6, 60.8 (d, 3J(C5′,F3′) = 9.4 Hz), 55.5; 19F NMR (376 MHz, CDCl3): δ −202.3 (ddd, 3J(2′,F3′) = 14.0, 3J(4′,F3′) = 31.4, 2J(3′,F3′) = 50.6 Hz); HRMS (ESI+): m/z calcd for C56H48FN5O4Na [(M + Na)+] 896.3583, found 896.3604 (Δ 2.3 ppm).
24: Rf = 0.27 (1:3 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 8.04 (s, 1H; H-2), 7.97 (d, 5J(8,F2′) = 3.1 Hz, 1H; H-8), 7.45–7.19 (m, 30H; 6×C6H6), 7.18 (d, J = 8.4 Hz, 2H; −PMB), 6.95 (s, 1H; N6-H), 6.82 (d, J = 8.4 Hz, 2H; −PMB), 6.42 (dd, 3J(1′,2′) = 3.0, 3J(1′,F2′) = 21.6 Hz, 1H; H-1′), 5.05 (dd, 3J(1′,2′) = 3.2, 2J(2′,F2′) = 51.2 Hz, 1H; H-2′), 4.53 (ABq, Δδ = 24.8 Hz, J = 11.5 Hz, 2H; −PMB), 4.26 (dd, 3J(3′,4′) = 3.7, 3J(3′,F2′) = 17.9 Hz, 1H; H-3′), 4.17–4.16 (m, 1H; H-4′), 3.77 (s, 3H; −PMB), 3.40 (dd, 3J(4′,5′) = 5.4, 2J(5′,5″) = 10.4 Hz, 1H; H-5′), 3.33 (dd, 3J(4′,5″) = 5.0, 2J(5′,5″) = 10.2 Hz, 1H; H-5″); 13C NMR (100 MHz, CDCl3): δ 159.8, 154.3, 152.6, 148.8, 145.2, 143.8, 139.6 (d, 4J(C8,F2′) = 6.3 Hz), 129.8, 129.2, 128.8, 128.7, 128.1, 128.0, 127.4, 127.1, 120.4, 114.2, 93.3 (d, 1J(C2′,F2′) = 192.7 Hz), 87.1, 83.2 (d, 2J(C1′,F2′) = 16.8 Hz), 81.8, 81.6 (d, 2J(C3′,F2′) = 25.7 Hz), 72.3, 71.6, 63.3, 55.5; 19F NMR (376 MHz, CDCl3): δ −197.3 (dddd, 5J(8,F2′) = 3.0, 3J(3′,F2′) = 17.9, 3J(1′,F2′) = 21.3, 2J(2′,F2′) = 51.3 Hz); HRMS (ESI+): m/z calcd for C56H48FN5O4Na [(M + Na)+] 896.3583, found 896.3624 (Δ 4.5 ppm).
N6-Trityl-3′-deoxy-2′-O-(4-methoxybenzyl)-3′-oxo-5′-O-trityladenosine (25)
To a solution of Dess-Martin periodinane (2.74 g, 6.468 mmol 1.2 equiv) in CH2Cl2 (50 mL) and t-BuOH (0.67 mL) was added a solution of alcohol 21 (4.7 g, 5.39 mmol, 1 equiv) in CH2Cl2 (50 mL). Anhydrous Na2CO3 powder (0.1 g) was added and the reaction mixture was stirred at rt for 3 h. The reaction was quenched by adding 1 M Na2S2O3 (5 mL), sat. NaHCO3 (5 mL), brine (5 mL), and EtOAc (15 mL). The layers were separated and the aqueous layer was extracted with EtOAc (2 × 15 mL). The combined organic layers were dried (MgSO4), filtered, and concentrated under reduced pressure. Purification by flash chromatography (SiO2, 40:60 EtOAc/hexanes) afforded the product (3.9 g, 83%) as a white solid. Rf = 0.45 (1:1 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 7.92, 7.75 (2s, 2H; H-2, H-8), 7.39–7.17 (m, 30H; 6×C6H5), 7.02 (d, J = 8.5 Hz, 2H; −PMB), 6.96 (s, 1H; N6-H), 6.69 (d, J = 8.5 Hz, 2H; −PMB), 6.10 (d, 3J(1′,2′) = 7.8 Hz, 1H; H-1′), 5.21 (d, 3J(1′,2′) = 7.8 Hz, 1H; H-2′), 4.73 (ABq, Δδ = 34.4 Hz, J = 11.7 Hz, 2H; −PMB), 4.32–4.31 (m, 1H; H-4′), 3.72 (s, 3H; −PMB), 3.49 (dd, 3J(5″,4′) = 4.0, 2J(5′,5″) = 10.5 Hz, 1H; H-5′), 3.40 (dd, 3J(5′,4′) = 2.4, 2J(5′,5″) = 10.5 Hz, 1H; H-5″); 13C NMR (100 MHz, CDCl3): δ 209.1, 159.9, 154.3, 152.7, 149.2, 145.1, 143.4, 138.7, 130.0, 129.2, 128.8, 128.2, 128.1, 128.0, 127.4, 127.2, 121.4, 114.1, 87.4, 84.6, 81.3, 78.3, 72.8, 71.7, 63.3, 55.4; HRMS (ESI+): m/z calcd for C56H47N5O5Na [(M + Na)+] 892.3469, found 892.3425 (Δ – 4.9 ppm).
N6-Trityl-9-[2-O-(4-methoxybenzyl)-5-O-trityl-β-d-xylofuranosyl]adenine (26)
To AcOH (57 mL) cooled to 4 °C was added NaBH4 (1.30 g, 34.37 mmol, 6.5 mmol) and the mixture was stirred for 10 min. Ketone 25 (4.60 g, 5.29 mmol, 1 equiv) was added as a dry powder and the reaction mixture was stirred for 2 h at 4 °C during which time the reaction solidified. The resulting frozen reaction mixture was allowed to melt by warming to rt, immediately evaporated, and coevaporated with EtOH (2 × 50 mL) under reduced pressure. The residue was partitioned between CHCl3 (100 mL) and H2O (100 mL), and the organic layer was washed with sat. NaHCO3, brine, dried (Na2SO4), and evaporated under reduced pressure. Purification by flash chromatography (SiO2, 50:50 EtOAc/hexanes) afforded the product (3.37 g, 73%) as a white solid. Rf = 0.58 (1:1 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 7.89, 7.72 (2s, 2H; H-2, H-8), 7.44–7.16 (m, 32H; 6×C6H5 and −PMB), 7.10 (d, 3J(OH,3′) = 11.0 Hz, 1H; 3′-OH), 7.04 (s, 1H; N6-H), 6.88 (d, J = 8.5 Hz, 2H; −PMB), 5.77 (d, 3J(1′,2′) = 1.7 Hz, 1H; H-1′), 4.57 (ABq, Δδ = 73.2 Hz, J = 11.3 Hz, 2H; −PMB), 4.35 (d, 3J(1′,2′) = 1.7 Hz, 1H; H-2′), 4.28 (ddd, 3J(3′,4′) = 3.2, 3J(5′,4′) = 5.7, 3J(5″,4′) = 6.4, 1H; H-4′), 4.23 (dd, 3J(3′,4′) = 3.2, 3J(OH,3′) = 11.0 Hz, 1H; H-3′), 3.81 (s, 3H; −PMB), 3.58–3.51 (m, 2H; H-5′/5″); 13C NMR (100 MHz, CDCl3): δ 159.8, 154.6, 151.6, 146.9, 144.9, 144.0, 140.3, 129.7, 129.3, 129.1, 128.9, 128.2, 127.9, 127.2, 127.1, 122.1, 114.2, 91.3, 90.1, 87.2, 82.8, 74.6, 72.6, 71.7, 62.3, 55.5; HRMS (ESI+): m/z calcd for C56H49N5O5Na [(M + Na)+] 894.3626, found 894.3673 (Δ 5.2 ppm).
N6-Trityl-9-[3-deoxy-3-fluoro-2-O-(4-methoxybenzyl)-5-O-trityl-β-d-ribofuranosyl]adenine (27)
To a solution of alcohol 26 (2.3 g, 2.64 mmol, 1.0 equiv) in CH2Cl2 (50 mL) and pyridine (2.1 mL, 26.4 mmol, 10 equiv) was added DAST (2.1 ml, 15.84 mmol, 6.0 equiv) dropwise and the mixture was stirred at rt for 24 h. CH2Cl2 (50 mL) was added to the reaction mixture and the organic layer was washed with sat. NaHCO3, water, and brine. The organic layer was dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by flash chromatography (SiO2, 40:60 EtOAc/hexanes) afforded the product (1.4 g, 61%) as a white solid. Rf = 0.35 (1:2 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 7.91, 7.79 (2s, 2H; H-2, H-8), 7.38–7.22 (m, 30H; 6×C6H5), 7.09 (d, J = 8.5 Hz, 2H; −PMB), 6.93 (s, 1H; N6-H), 6.74 (d, J = 8.6 Hz, 2H; −PMB), 6.06 (d, 3J(1′,2′) = 7.5 Hz, 1H; H-1′), 5.11 (ddd, 3J(2′,3′) = 4.3, 3J(1′,2′) = 7.6, 3J(2′,F3′) = 21.6 Hz, 1H; H-2′), 4.99 (ddd, 3J(3′,4′) = 1.4, 3J(2′,3′) = 4.4, 3J(3′,F3′) = 54.4 Hz, 1H; H-3′), 4.54 (ABq, Δδ = 23.2 Hz, J = 11.7 Hz, 2H; −PMB), 4.42 (dm, 3J(4′,F3′) = 26.4 Hz, 1H; H-4′), 3.75 (s, 3H; −PMB), 3.47 (dd, 3J(5′,4′) = 4.8, 2J(5′,5″) = 10.5 Hz, 1H; H-5′), 3.30 (dd, 3J(5″,4′) = 4.1, 2J(5′,5″) = 10.5 Hz, 1H; H-5″); 13C NMR (100 MHz, CDCl3): δ 159.8, 154.3, 152.5, 149.1, 145.2, 143.6, 139.5, 129.8, 129.2, 129.0, 128.8, 128.1 (×2), 127.4, 127.1, 121.8, 114.1, 90.4 (d, 1J(C3′,F3′) = 186.6 Hz), 87.5, 86.5, 82.7 (d, 2J(C2′,F3′) = 23.5 Hz), 77.6 (d, 2J(C4′,F3′) = 15.8 Hz), 72.7, 71.6, 63.0 (d, 3J(C5′,F3′) = 9.9 Hz), 55.5; 19F NMR (376 MHz, CDCl3): δ −199.3 (ddd, 3J(2′,F3′) = 21.6, 3J(4′,F3′) = 26.4, 2J(3′,F3′) = 54.3 Hz); HRMS (ESI+): m/z calcd for C56H48FN5O4Na [(M + Na)+] 896.3583, found 896.3546 (Δ −4.1 ppm).
N6-Trityl-9-(2-deoxy-2-fluoro-5-O-trityl-β-D-ribofuranosyl)adenine (29)
This was prepared from 2′-deoxy-2′-fluoroadenosine 28 (0.50 g, 1.86 mmol) using the general procedure 1. Purification by flash chromatography (SiO2, 50:50 EtOAc/hexanes) afforded the product (1.10 g, 79%) as a white solid. Rf = 0.40 (1:1 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 7.98, 7.92 (2s, 2H; H-2, H-8), 7.42–7.18 (m, 30 H; 6×C6H5), 7.10 (s, 1H; N6-H), 6.17 (dd, 3J(1′,2′) = 2.4, 3J(1′,F2′) = 17.6 Hz, 1H; H-1′), 5.54 (ddd, 3J(2′,1′) = 2.4, 3J(2′,3′) = 4.5, 2J(2′,F2′) = 53.1 Hz, 1H; H-2′), 4.73 (ddd, 3J(2′,3′) = 4.5, 3J(3′,4′) = 6.9, 3J(3′,F2′) = 17.4 Hz, 1H; H-3′), 3.55–3.52 (m, 1H; H-4′), 3.53 (dd, 3J(4′,5′) = 3.1, 2J(5′,5″) = 10.7 Hz, 1H; H-5′), 3.41 (dd, 3J(4′,5″) = 4.6, 2J(5′,5″) = 10.7 Hz, 1H; H-5″); 13C NMR (CDCl3): δ 154.3, 152.6, 148.4, 145.0, 143.6, 138.8, 129.2, 128.8, 128.1 (×2), 127.4, 127.1, 121.4, 93.4 (d, 1J(C2′,F2′) = 186.8 Hz), 87.3, 87.0 (d, 2J(C1′,F2′) = 33.1 Hz), 82.6, 71.6, 70.3 (d, 2J(C3′,F2′) = 16.5 Hz), 63.0; 19F NMR (CDCl3): δ −205.7 (dt, 3J(3′,F2′) = 17.4, 3J(1′,F2′) = 17.4, 2J(2′,F2′) = 53.1 Hz); HRMS (ESI+): m/z calcd for C48H41FN5O3 [(M + H)+] 754.3188, found 754.3195 (Δ 0.9 ppm).
N6-Trityl-9-[2-deoxy-2-fluoro-3-O-(4-methoxybenzyl)-5-O-trityl-β-d-ribofuranosyl]adenine (30)
To a solution of 29 (0.35 g, 0.464 mmol, 1 equiv) in THF (7 mL) at 0 °C was added 60% NaH (23 mg, 0.575 mmol, 1.2 equiv) and the mixture was stirred for 10 min. TBAI (23 mg, 0.062 mmol, 0.12 equiv) and PMBCl (77 μL, 0.568 mmol, 1.2 equiv) were sequentially added and the mixture was stirred at that temperature for further 30 min. The reaction was slowly warmed to rt and stirred for 15 h. The reaction was quenched with MeOH (7 mL), evaporated, partitioned between CH2Cl2 and H2O. The aqueous layer was extracted with CH2Cl2 and the combined organics were dried (Na2SO4) and evaporated. Purification by flash chromatography (SiO2, gradient: hexanes to 50:50 EtOAc/hexanes) afforded the product (0.32 g, 78%) as a white solid. Rf = 0.21 (1:2 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 7.98, 7.91 (2s, 2H; H-2, H-8), 7.34–7.17 (m, 32H; 6×C6H6, −PMB), 7.01 (s, 1H; N6-H), 6.88 (d, J = 8.6 Hz, 2H; −PMB), 6.15 (d, 3J(1′,F2′) = 18.0 Hz, 1H; H-1′), 5.64 (dd, 3J(3′,2′) = 4.0, 2J(2′,F2′) = 53.1 Hz, 1H; H-2′), 4.68–4.66 (m, 1H; H-3′), 4.53 (ABq, Δδ = 48.0 Hz, J = 11.4 Hz, 2H; −PMB), 4.27 (dt, 3J(4′,5′) = 3.5, 3J(4′,5″) = 3.5, 3J(3′,4′) = 7.4 Hz, 1H; H-4′), 3.80 (s, 3H; −PMB), 3.48 (dd, 3J(4′,5′) = 3.1, 2J(5′,5″) = 10.9 Hz, 1H; H-5′), 3.24 (dd, 3J(4′, 5″) = 4.2, 2J(5′,5″) = 10.3 Hz, 1H; H-5″); 19F NMR (376 MHz, CDCl3): δ −204.6 (dt, 3J(1′,F2′) = 18.5, 3J(3′,F2′) = 18.5, 2J(2′,F2′) = 52.8 Hz); HRMS (ESI+): m/z calcd for C56H49FN5O4 [(M + H)+] 874.3763, found 874.3756 (Δ −0.8 ppm).
N6-Trityl-9-(2,3-dideoxy-2,3-difluoro-5-O-trityl-β-d-xylofuranosyl)adenine (31)
To a solution of alcohol 29 (2.30 g, 3.05 mmol, 1.0 equiv) in CH2Cl2 (50 mL) and pyridine (2.40 mL, 29.8 mmol, 10 equiv) was added DAST (2.0 mL, 15.14 mmol, 5.0 equiv) dropwise and the mixture was stirred at rt for 25 h. CH2Cl2 (50 mL) was added to the reaction mixture and the organic layer was washed with sat. NaHCO3 and water. The organic layer was dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by flash chromatography (SiO2, 30:70 EtOAc/hexanes) afforded the product (1.79 g, 78%) as a white solid. Rf = 0.45 (1:2 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 8.08, 7.77 (2s, 2H; H-2, H-8), 7.51–7.24 (m, 30H; 6×C6H5), 6.97 (s, 1H; N6-H), 6.40 (d, 3J(1′,F2′) = 20.2 Hz, 1H; H-1′), 5.42 (dd, 3J(2′,F3′) = 9.8, 2J(2′,F2′) = 48.0 Hz, 1H; H-2′), 5.27 (ddd, 3J(3′,4′) = 3.0, 3J(3′,F2′) = 7.7, 2J(3′,F3′) = 50.0 Hz, 1H; H-3′), 4.56 (dm, 3J(4′,F3′) = 31.2 Hz, 1H; H-4′), 3.68 (dd, 3J(5′,4′) = 6.5, 2J(5′,5″) = 9.8 Hz, 1H; H-5′), 3.54 (dd, 3J(5″,4′) = 6.0, 2J(5′,5″) = 9.8 Hz, 1H; H-5″); 13C NMR (100 MHz, CDCl3): δ 154.2, 152.8, 148.6, 145.1, 143.5, 137.5 (d, J = 6.2 Hz), 129.2, 128.8, 128.2, 128.1, 127.5, 127.1, 121.1, 96.1 (dd, 2J(C2′,F3′) = 31.7, 1J(C2′,F2′) = 186.1 Hz), 92.4 (dd, 2J(C3′,F2′) = 30.1, 1J(C3′,F3′) = 184.9 Hz), 87.7 (d, 2J(C1′,F2′) = 36.6 Hz), 87.5, 81.8 (d, 2J(C4′,F3′) = 19.4 Hz), 71.6, 60.4 (d, 3J(C5′,F3′) = 9.0 Hz); 19F NMR (376 MHz, CDCl3): δ −191.9 (ddddd, 4J(4′,F2′) = 3.4, 3J(3′,F2′) = 7.9, 3J(F2′,F3′) = 15.0, 3J(1′,F2′) = 20.3, 2J(2′,F2′) = 47.7 Hz; F-2′), −208.4 (dddd, 3J(2′,F3′) = 9.6, 3J(F2′,F3′) = 14.9, 3J(4′,F3′) = 31.1, 2J(3′,F3′) = 49.5 Hz; F-3′); HRMS (ESI+): m/z calcd for C48H40F2N5O2 [(M + H)+] 756.3145, found 756.3138 (Δ −0.9 ppm).
N6-Trityl-9-(3-deoxy-3-fluoro-5-O-trityl-β-d-arabinofuranosyl)adenine (33) and N6-Trityl-9-(2-deoxy-2-fluoro-5-O-trityl-β-d-xylofuranosyl)adenine (34)
To a solution of epoxide 3221 3.20 g, 4.36 mmol, 1.0 equiv) in 2-ethoxyethanol (40 mL) was added KHF2 (1.50 g, 19.2 mmol, 4.4 equiv) and NaF (2.20 g, 52.39 mmol, 12.0 equiv) and the mixture was heated under reflux at 145 °C for 15 h. The reaction mixture was allowed to cool to rt, solvent was removed under reduced pressure and the residue was diluted with CHCl3 (40 ml) and washed with water. The organic layer was dried (MgSO4), filtered, and concentrated under reduced pressure. The ratio of 33 and 34 was determined to be 3.1:1 by 1H NMR of the crude product. Purification by flash chromatography (SiO2, 35:65 EtOAc/hexanes) afforded the product as a mixture of 33 and 34 (2.12 g, 65% combined) as a white solid. A few pure fractions of each regioisomer were separately collected and characterized.
33: Rf = 0.44 (1:1 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 8.05, 7.96 (2s, 2H; H-2, H-8), 7.44–7.18 (m, 30H; 6×C6H5), 7.07 (s, 1H; N6-H), 6.18–6.17 (m, 1H; H-1′), 5.52 (d, 3J(2′,OH) = 7.6 Hz, 1H; −OH), 5.04 (dm, 2J(3′,F3′) = 51.6 Hz, 1H; H-3′), 4.53–4.48 (m, 1H; H-4′), 4.30 (dm, 3J(2′,F3′) = 28.8 Hz, 1H; H-2′), 3.57–3.44 (m, 2H; H-5′, H-5″); 13C NMR (CDCl3): δ 154.4, 152.1, 148.3, 144.9, 143.4, 140.5, 129.1, 128.8, 128.2, 128.1, 127.5, 127.1, 120.8, 96.9 (d, 1J(C3′,F3′) = 183.0 Hz), 87.8, 86.0, 81.7 (d, 2J(C2′,F3′) = 25.4 Hz), 74.4 (d, 2J(C4′,F3′) = 25.2 Hz), 71.6, 63.4 (d, 3J(C5′,F3′) = 9.6 Hz); 19F NMR (CDCl3): δ −182.9 ppm (dddd, 4J(5′,F3′) = 2.5, 3J(4′,F3′) = 12.2, 3J(2′,F3′) = 28.9, 2J(3′,F3′) = 51.7 Hz); HRMS (ESI+): m/z calcd for C48H40FN5O3Na [(M + Na)+] 776.3007, found 776.3028 (Δ 2.7 ppm).
34: Rf = 0.51 (1:1 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 7.86, 7.84 (2s, 2H; H-2, H-8), 7.43–7.13 (m, 31H; 6×C6H5 and −OH), 7.12 (s, 1H; N6-H), 5.93 (d, 3J(1′,F2′) = 25.6 Hz, 1H; H-1′), 5.29 (dm, 2J(2′,F2′) = 49.6 Hz, 1H; H-2′), 4.34–4.28 (m, 2H; H-3′, H-4′), 3.59–3.52 (m, 2H; H-5′/5″); 13C NMR (CDCl3): δ 154.7, 151.7, 146.7, 144.8, 143.9, 140.1, 129.1, 128.9, 128.2, 127.9, 127.3, 127.2, 121.9, 101.0 (d, 1J(C2′,F2′) = 186.1 Hz), 90.1 (d, 2J(C1′,F2′) = 39.6 Hz), 87.3, 82.9, 74.6 (d, 2J(C3′,F2′) = 25.8 Hz), 71.8, 62.0; 19F NMR (CDCl3): δ −177.8 (ddd, 3J(3′,F2′) = 15.6, 3J(1′,F2′) = 25.5, 2J(2′,F2′) = 49.5 Hz); HRMS (ESI+): m/z calcd for C48H40FN5O3Na [(M + Na)+] 776.3007, found 776.3021 (Δ 1.8 ppm).
N6-Trityl-9-(2,3-dideoxy-2,3-difluoro-5-O-trityl-β-d-ribofuranosyl)adenine (35)
To a solution of a mixture of 33 and 34 (1.94 g, 2.57 mmol, 1.0 equiv) in toluene (30 mL) and pyridine (0.57 mL, 4.72 mmol, 1.8 equiv) was added DAST (2.04 ml, 15.44 mmol, 6.0 equiv) dropwise and the mixture was stirred at rt for 1 h before heating the mixture at 80 °C for an additional 3 h. EtOAc (100 mL) was added to the reaction mixture and the organic layer was washed with sat. NaHCO3 and water. The organic layer was dried (MgSO4), filtered, and concentrated under reduced pressure. Purification by flash chromatography (SiO2, 20:80 EtOAc/hexanes) afforded the product (1.20 g, 62%) as a white solid. Rf = 0.33 (1:2 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 7.93, 7.88 (2s, 2H; H-2, H-8), 7.39–7.19 (m, 30H; 6×C6H5), 6.79 (s, 1H; N6-H), 6.18 (ddd, 4J(1′,F3′) = 1.7, 3J(1′,2′) = 5.0, 3J(1′,F2′) = 14.2 Hz, 1H; H-1′), 6.02 (ddt, 3J(2′,3′) ≈ 3J(1′,2′) ≈ 4.8, 3J(2′,F3′) = 13.2, 2J(2′,F2′) = 50.9 Hz, 1H; H-2′), 5.45 (ddt, 3J(3′,2′) ≈ 3J(3′,4′) ≈ 4.0, 3J(3′,F2′) = 7.6, 2J(3′,F3′) = 53.0 Hz, 1H; H-3′), 4.46 (dm, 3J(4′,F3′) = 21.8 Hz, 1H; H-4′), 3.54 (dd, 3J(5′,4′) = 4.2, 2J(5′,5″) = 10.8 Hz, 1H; H-5′), 3.39 (dd, 3J(5″,4′) = 4.3, 2J(5′,5″) = 10.8 Hz, 1H; H-5″); 13C NMR (100 MHz, CDCl3): δ 154.4, 152.7, 148.7, 145.1, 143.5, 139.3, 129.2, 128.8, 128.1 (×2), 127.5, 127.2, 121.7, 89.7 (dd, 2J(C2′,F3′) = 14.4, 1J(C2′,F2′) = 196.3 Hz), 88.9 (dd, 2J(C3′,F2′) = 13.9, 1J(C2′,F2′) = 190.4 Hz), 87.5, 86.1 (dd, 3J(C1′,F3′) = 2.5, 2J(C1′,F2′) = 31.1 Hz), 81.3 (dd, 3J(C4′,F2′) = 2.5, 2J(C4′,F3′) = 22.7 Hz), 71.6, 62.5 (d, 3J(C5′,F3′) = 6.4 Hz); 19F NMR (376 MHz, CDCl3): δ −206.9 (dddd, 3J(F2′,F3′) = 3.8, 3J(2′,F3′) = 13.5, 3J(4′,F3′) = 21.8, 2J(3′,F3′) = 53.3 Hz; F-3′), −213.0 (dm, 2J(2′,F2′) = 51.1 Hz; F-2′); HRMS (ESI+): m/z calcd for C48H39F2N5O2Na [(M + Na)+] 778.2964, found 778.2970 (Δ 0.8 ppm).
9-(3-Deoxy-3-fluoro-β-d-xylofuranosyl)adenine (36)
To a flask containing compound 23 (0.075 g, 0.086 mmol) was added 80% aqueous TFA in CHCl3 (5 mL). After 1 h MeOH (5 mL) was added and the reaction mixture was then concentrated under reduced pressure. The residue was dissolved in MeOH (5 mL), residual TFA was neutralized by adding Et3N (10 drops), and the mixture was concentrated under reduced pressure. To the residue was added H2O (5 mL) and Et2O (5 mL) and the mixture was stirred vigorously. The phases were separated and MeCN (2 mL) was added to the aqueous layer. The aqueous layer was filtered through a syringe filter and purification was performed by preparative reverse-phase HPLC using a Phenomenex Gemini 10 μm C18 110Å (250 × 21.2 mm) column at a flow rate of 21 mL/min with a gradient from 5% to 30% MeCN in H2O over 12 min. The appropriate fractions containing the product were pooled and lyophilized to afford the product (0.020 g, 87%) as a white solid. 1H NMR (400 MHz, CD3OD): δ 8.22, 8.15 (2s, 2H; H-2, H-8), 6.09 (d, 3J(1′,2′) = 2.0 Hz, 1H; H-1′), 5.10 (ddd, 3J(2′,3′) = 1.8, 3J(3′,4′) = 3.3, 2J(3′,F3′) = 51.7 Hz, 1H; H-3′), 4.72 (dt, 3J(2′,3′) = 1.8, 3J(1′,2′) = 1.9, 3J(2′,F3′) = 13.9 Hz, 1H; H-2′), 4.47 (dtd, 3J(3′,4′) = 3.2, 3J(5′,4′) ≈ 3J(5″,4′) = 5.8, 3J(4′,F3′) = 27.9 Hz, 1H; H-4′), 3.98 (dd, 3J(5′,4′) = 5.7, 3J(5″,5′) = 11.8 Hz, 1H; H-5′), 3.93 (ddd, 4J(5″,F3′) = 1.6, 3J(5″,4′) = 6.1, 3J(5″,5′) = 11.9 Hz, 1H; H-5″); 13C NMR (100 MHz, CD3OD): δ 157.5, 154.1, 150.4, 140.4 (d, 5J(C8,F3′) = 6.4 Hz), 120.4, 96.9 (d, 1J(C3′,F3′) = 184.0 Hz), 91.6 (d, 3J(C1′,F3′) = 1.5 Hz), 84.1 (d, 2J(C4′,F3′) = 19.3 Hz), 79.9 (d, 2J(C2′,F3′) = 26.6 Hz), 60.2 ppm (d, 3J(C5′,F3′) = 10.2 Hz); 19F NMR (376 MHz, CD3OD): δ −203.2 (ddd, 3J(2′,F3′) = 13.9, 3J(4′,F3′) = 27.9, 2J(3′,F3′) = 51.7 Hz); HRMS (ESI+): m/z calcd for C10H12FN5O3Na [(M + Na)+] 292.0816, found 292.0823 (Δ 2.4 ppm).
9-[2-Deoxy-2-fluoro-β-d-arabinofuranosyl]adenine (37)
To a flask containing compound 24 (0.060 g, 0.069 mmol) was added 80% aqueous TFA in CHCl3 (5 mL) to form a brown solution. After 1 h MeOH (5 mL) was added and the reaction mixture was then concentrated under reduced pressure. The residue was dissolved in MeOH (5 mL), residual TFA was neutralized by adding Et3N (10 drops), and the mixture was concentrated under reduced pressure. To the residue was added H2O (5 mL) and Et2O (5 mL) and the mixture was stirred vigorously. The phases were separated and MeCN (2 mL) was added to the aqueous layer. The aqueous layer was filtered through a syringe filter and purification was performed by preparative reverse-phase HPLC using a Phenomenex Gemini 10 μm C18 110Å (250 × 21.2 mm) column at a flow rate of 21 mL/min with a gradient from 5% to 30% MeCN in H2O over 12 min. The appropriate fractions containing the product were pooled and lyophilized to afford the product (0.017 g, 89%) as a white solid. 1H NMR (400 MHz, CD3OD): δ 8.33 (d, 5J(8,F2′) = 2.1 Hz, 1H; H-8), 8.21 (s, 1H; H-2), 6.48 (dd, 3J(1′,2′) = 4.1, 3J(1′,F2′) = 16.1 Hz, 1H; H-1′), 5.14 (ddd, 3J(2′,3′) = 3.0, 3J(1′,2′) = 4.2, 2J(2′,F2′) = 52.2 Hz, 1H; H-2′), 4.53 (ddd, 3J(2′,3′) = 3.0, 3J(3′,4′) = 4.8, 3J(3′,F2′) = 18.2 Hz, 1H; H-3′), 4.01 (q, 3J(4′,5′) ≈ 3J(4′, 5″) ≈ 3J(4′,3′) = 4.6 Hz, 1H; H-4′), 3.87 (ddd, 5J(F2′,5′) = 1.4, 3J(4′,5′) = 4.0, 2J(5′,5″) = 12.2 Hz, 1H; H-5′), 3.81 (dd, 3J(4′,5″) = 5.2, 2J(5′,5″) = 12.2 Hz, 1H; H-5″); 13C NMR (100 MHz, CDCl3): δ 157.5, 154.2, 150.5, 141.8 (d, 4J(C8,F2′) = 4.3 Hz), 119.8, 97.1 (d, 1J(C2′,F2′) = 192.9 Hz), 85.9 (d, 3J(C4′,F2′) = 4.0 Hz), 84.5 (d, 2J(C1′,F2′) = 17.2 Hz), 74.9 (d, 2J(C3′,F2′) = 24.5 Hz), 62.3 (d, 4J(C3′,F2′) = 1.5 Hz); 19F NMR (376 MHz, CD3OD): δ −199.1 (dt, 3J(1′,F2′) = 17.2, 3J(3′,F2′) = 17.2, 2J(2′,F2′) = 52.2 Hz); HRMS (ESI+): m/z calcd for C10H12FN5O3Na [(M + Na)+] 292.0816, found 292.0803 (Δ −4.4 ppm).
9-(3-Deoxy-3-fluoro-β-d-ribofuranosyl)adenine (38)
To a flask containing compound 27 (0.055 g, 0.063 mmol) was added 80% aqueous TFA in CHCl3 (5 mL). After 1 h MeOH (5 mL) was added and the reaction mixture was then concentrated under reduced pressure. The residue was dissolved in MeOH (5 mL), residual TFA was neutralized by adding Et3N (10 drops), and the mixture was concentrated under reduced pressure. To the residue was added H2O (5 mL) and Et2O (5 mL) and the mixture was stirred vigorously. The phases were separated and MeCN (2 mL) was added to the aqueous layer. The aqueous layer was filtered through a syringe filter and purification was performed by preparative reverse-phase HPLC using a Phenomenex Gemini 10 μm C18 110Å (250 × 21.2 mm) column at a flow rate of 21 mL/min with a gradient from 5% to 30% MeCN in H2O over 12 min. The appropriate fractions containing the product were pooled and lyophilized to afford the product (0.017 g, 97%) as a white solid. 1H NMR (400 MHz, CD3OD): δ 8.29, 8.18 (2s, 2H; H-2, H-8), 6.01 (d, 3J(1′,2′) = 8.0 Hz, 1H; H-1′), 5.12 (dd, 3J(2′,3′) = 4.3, 2J(3′,F3′) = 54.5 Hz, 1H; H-3′), 4.98 (ddd, 3J(2′,3′) = 4.3, 3J(1′,2′) = 8.0, 3J(2′,F3′) = 25.2 Hz, 1H; H-2′), 4.44 (dt, 3J(4′,5′) = 2.5, 3J(4′,5″) = 2.5, 3J(4′,F3′) = 27.6 Hz, 1H; H-4′), 3.87 (dt, 4J(4′,F3′) = 2.4, 3J(4′,5′) = 2.4, 2J(5′,5″) = 12.6 Hz, 1H; H-5′), 3.79 (dd, 3J(4′,5″) = 2.4, 2J(5′,5″) = 12.7 Hz, 1H; H-5″); 13C NMR (100 MHz, CD3OD): δ 157.9, 153.7, 150.2, 142.3, 121.3, 94.7 (d, 1J(C3′,F3′) = 182.0 Hz), 90.5, 86.6 (d, 2J(C4′,F3′) = 22.1 Hz), 74.7 (d, 2J(C2′,F3′) = 16.5 Hz), 63.2 (d, 3J(C5′,F3′) = 11.6 Hz); 19F NMR (376 MHz, CD3OD): δ −199.6 (dt, 3J(2′,F3′) ≈ 3J(4′,F3′) = 26.5, 2J(3′,F3′) = 53.8 Hz); HRMS (ESI+): m/z calcd for C10H12FN5O3Na [(M + Na)+] 292.0816, found 292.0816 (Δ <1 ppm).
9-(2,3-Dideoxy-2,3-difluoro-β-d-xylofuranosyl)adenine (39)
To a flask containing compound 31 (0.089 g, 0.118 mmol) was added 80% aqueous TFA in CHCl3 (5 mL). After 1 h MeOH (5 mL) was added and the reaction mixture was then concentrated under reduced pressure. The residue was treated with sat. NaHCO3 (20 ml) and extracted with EtOAc (3 × 20 ml). The organic layer was dried (MgSO4), filtered, and concentrated under reduced pressure. Purification by flash chromatography (SiO2, 98:2 EtOAc/MeOH) afforded the product (0.023 g, 80%) as a white solid. Rf = 0.28 (5:95 MeOH/EtOAc); 1H NMR (400 MHz, CD3OD): δ 8.23, 8.17 (2s, 2H; H-2, H-8), 6.39 (dd, 3J(1′,2′) = 2.4, 3J(1′,F2′) = 18.9 Hz, 1H; H-1′), 5.73 (ddt, 3J(1′,2′) ≈ 3J(2′,3′) ≈ 1.9, 3J(2′,F3′) = 11.6, 2J(2′,F2′) = 48.5 Hz, 1H; H-2′), 5.47 (dddd, 3J(2′,3′) = 1.8, 3J(3′,4′) = 3.6, 3J(3′,F2′) = 10.1, 2J(3′,F3′) = 50.1 Hz, 1H; H-3′), 4.50 (dm, 3J(4′,F3′) = 27.2 Hz, 1H; H-4′), 4.00 (dd, 3J(4′,5′) = 5.5, 2J(5′,5″) = 11.9 Hz, 1H; H-5′) , 3.97–3.92 (m, 1H; H-5″); 13C NMR (100 MHz, CD3OD): δ 157.6, 154.3, 150.5, 140.3 (d, J = 5.7 Hz), 120.3, 97.5 (dd, 2J(C2′,F3′) = 31.1, 1J(C2′,F2′) = 184.9 Hz), 93.9 (dd, 2J(C3′,F2′) = 29.2, 1J(C2′,F2′) = 183.9 Hz), 88.5 (dd, 3J(C1′,F3′) = 1.9, 2J(C1′,F2′) = 36.2 Hz), 84.0 (d, 2J(C4′,F3′) = 19.7 Hz), 59.8 (d, 3J(C5′,F3′) = 9.8 Hz); 19F NMR (376 MHz, CD3OD): δ −192.3 (ddm, 3J(1′,F2′) = 19.0, 2J(2′,F2′) = 48.5 Hz; F-2′), −207.3 (ddt, 3J(F2′,F3′) = 11.8, 3J(2′,F3′) = 11.8, 3J(4′,F3′) = 27.0, 2J(3′,F3′) = 50.5 Hz; F-3′); HRMS (ESI+): m/z calcd for C10H11F2N5O2Na [(M + Na)+] 294.0773, found 294.0768 (Δ −1.7 ppm).
9-(2,3-Dideoxy-2,3-difluoro-β-d-ribofuranosyl)adenine (40)
To a flask containing compound 35 (0.138 g, 0.182 mmol) was added 80% aqueous TFA in CHCl3 (3 mL). After 1 h MeOH (5 mL) was added and the reaction mixture was then concentrated under reduced pressure. The residue was treated with sat. NaHCO3 (7 mL) and extracted with EtOAc (3 × 7 mL). The organic layer was dried (MgSO4), filtered, and concentrated under reduced pressure. Purification by flash chromatography (SiO2, 98:2 EtOAc/MeOH) afforded the product (0.039 g, 80%) as a white solid. Rf = 0.40 (5:95 MeOH/EtOAc); 1H NMR (400 MHz, [D6]DMSO): δ 8.57 (s, 1H), 8.52 (br s, 2H; −NH2), 8.38 (s, 1H), 6.36 (ddd, 4J(1′,F3′) = 1.6, 3J(1′,2′) = 5.8, 3J(1′,F2′) = 13.6 Hz, 1H; H-1′), 5.90 (dddd, 3J(2′,3′) = 4.3, 3J(1′,2′) = 5.8, 3J(2′,F3′) = 16.6, 2J(2′,F2′) = 50.3 Hz, 1H; H-2′), 5.53 (dm, 2J(3′,F3′) = 52.9 Hz, 1H; H-3′), 4.42 (dm, 3J(4′,F3′) = 24.0 Hz, 1H; H-4′), 3.70–3.67 (m, 2H; H-5′/5″); 13C NMR ([D6]DMSO): δ 153.6, 149.4, 148.6, 140.8, 119.1, 90.6 (dd, 2J(C2′,F3′) = 14.3, 1J(C2′,F2′) = 193.8 Hz), 89.8 (dd, 2J(C3′,F2′) = 13.3, 1J(C2′,F2′) = 184.8 Hz), 84.7 (dd, 3J(C1′,F3′) = 2.0, 2J(C1′,F2′) = 31.5 Hz), 83.4 (dd, 3J(C4′,F2′) = 2.4, 2J(C4′,F3′) = 21.4 Hz), 60.2 (d, 3J(C5′,F3′) = 8.4 Hz); 19F NMR (376 MHz, [D6]DMSO): δ −206.2 (dddd, 3J(F2′,F3′) = 4.9, 3J(2′,F3′) = 16.6, 3J(4′,F3′) = 22.6, 2J(3′,F3′) = 53.4 Hz; F-3′), −214.4 (dm, 2J(2′,F2′) = 50.7 Hz; F-2′); HRMS (ESI+): m/z calcd for C10H12F2N5O2 [(M + H)+] 272.0954, found 272.0963 (Δ 3.3 ppm).
General Procedure 2: Selective Deprotection of 5′-O-Trityl
A 0.4 M HCl solution in 1,4-dioxane was prepared by diluting commercially available 4 M HCl in 1,4-dioxane. A solution of 5′-O,N6-ditritylated compound (1 mmol) in 0.4 M HCl in 1,4-dioxane (38 mL) was stirred at rt for 1 h. MeOH (25 mL) was added and the reaction mixture was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (50 mL) and washed with sat. NaHCO3, water, and brine. The organic layer was dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by flash chromatography (SiO2, EtOAc/hexanes gradient) afforded the product.
N6-Trityl-9-[2-deoxy-2-fluoro-3-O-(4-methoxybenzyl)-β-d-ribofuranosyl]adenine (41a)
This was prepared from ditritylated compound 30 (0.10 g, 0.114 mmol) using general procedure 2. Purification by flash chromatography (SiO2, 70:30 EtOAc/hexanes) afforded the product (0.06 g, 86%) as a white solid. Rf = 0.22 (1:1 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 7.98, 7.81 (2s, 2H; H-2, H-8), 7.35–7.21 (m, 17H; 3×C6H6, −PMB), 7.10 (s, 1H; N6-H), 6.90 (d, J = 8.6 Hz, 2H; −PMB), 6.47 (br s, 1H; −OH), 6.10 (dd, 3J(1′,2′) = 6.8, 3J(1′,F2′) = 12.0 Hz, 1H; H-1′), 5.79 (ddd, 3J(2′,3′) = 4.8, 3J(1′,2′) = 6.9, 2J(2′,F2′) = 51.6 Hz, 1H; H-2′), 4.69 (ABq, Δδ = 72.8 Hz, J = 11.2 Hz, 2H; −PMB), 4.40 (dt, 3J(3′,F2′) = 1.4, 3J(3′,4′) = 1.4, 3J(2′,3′) = 4.6 Hz, 1H; H-3′), 4.34–4.33 (m, 1H; H-4′), 3.86 (dm, 2J(5′,5″) = 13.1 Hz, 1H; H-5′), 3.80 (s, 3H; −PMB), 3.70 (dd, 3J(4′,5″) = 1.8, 2J(5′,5″) = 13.2 Hz, 1H; H-5″); 13C NMR (100 MHz, CDCl3): δ 159.7, 154.8, 152.0, 147.5, 144.8, 140.1, 129.8, 129.6, 129.1, 128.1, 127.2, 122.6, 114.2, 91.8 (d, 1J(C2′,F2′) = 197.3 Hz), 88.8 (d, 2J(C1′,F2′) = 31.0 Hz), 86.8 (d, 3J(C4′,F2′) = 3.2 Hz), 77.1 (d, 2J(C3′,F2′) = 13.0 Hz), 73.2 (d, 4J(PMB-CH2-,F2′) = 3.2 Hz), 71.7, 63.0, 55.5; 19F NMR (376 MHz, CDCl3): δ −213.9 (dd, 3J(1′,F2′) = 11.9, 2J(2′,F2′) = 51.6 Hz); HRMS (ESI+): m/z calcd for C37H34FN5O4Na [(M + Na)+] 654.2487, found 654.2538 (Δ 7.8 ppm).
N6-Trityl-9-[2-deoxy-2-fluoro-3-O-(4-methoxybenzyl)-β-d-arabinofuranosyl]adenine (41b)
This was prepared from ditritylated compound 24 (0.45 g, 0.515 mmol) using general procedure 2. Purification by flash chromatography (SiO2, 70:30 EtOAc/hexanes) afforded the product (0.25 g, 78%) as a white solid. Rf = 0.29 (1:1 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 8.02 (s, 1H; H-2), 7.92 (d, 5J(8,F2′) = 1.9 Hz, 1H; H-8), 7.36–7.22 (m, 18H; 3×C6H6, −PMB, N6-H), 6.90 (d, J = 8.6 Hz, 2H; −PMB), 6.34 (dd, 3J(1′,2′) = 4.1, 3J(1′,F2′) = 17.2 Hz, 1H; H-1′), 5.18 (ddd, 3J(2′,3′) = 2.3, 3J(1′,2′) = 4.1, 2J(2′,F2′) = 52.1 Hz, 1H; H-2′), 5.01 (br s, 1H; −OH), 4.61 (ABq, Δδ = 24.4 Hz, J = 11.3 Hz, 2H; −PMB), 4.51 (ddd, 3J(2′,3′) = 2.4, 3J(3′,4′) = 4.9, 3J(3′,F2′) = 18.7 Hz, 1H; H-3′), 4.09 (q, 3J(4′,5′) = 3.7, 3J(4′, 5″) = 3.8, 3J(4′,3′) = 3.8 Hz, 1H; H-4′), 3.89 (dd, 3J(4′,5′) = 3.1, 2J(5′,5″) = 12.4 Hz, 1H; H-5′), 3.01 (s, 3H; −PMB), 3.70 (dd, 3J(4′,5″) = 4.0, 2J(5′,5″) = 12.4 Hz, 1H; H-5″); 13C NMR (100 MHz, CDCl3): δ 159.8, 154.3, 152.5, 148.7, 145.0, 139.2 (d, J = 4.3 Hz), 129.8, 129.2, 128.9, 128.1, 127.0, 120.5, 114.2, 94.1 (d, 1J(C2′,F2′) = 195.0 Hz), 84.1 (d, 2J(C1′,F2′) = 17.4 Hz), 83.2 (d, 3J(C4′,F2′) = 3.4 Hz), 80.8 (d, 2J(C3′,F2′) = 25.0 Hz), 72.6, 71.7, 61.5, 55.5; 19F NMR (376 MHz, CDCl3): δ −195.8 (dt, 3J(1′,F2′) = 17.9, 3J(3′,F2′) = 17.9, 2J(2′,F2′) = 51.9 Hz).
N6-Trityl-9-[3-deoxy-3-fluoro-2-O-(4-methoxybenzyl)-β-d-xylofuranosyl]adenine (41c)
This was prepared from ditritylated compound 23 (0.88 g, 1.007 mmol) using general procedure 2. Purification by flash chromatography (SiO2, 80:20 EtOAc/hexanes) afforded the product (0.47 g, 75%) as a white solid. Rf = 0.20 (1:1 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 8.02, 7.80 (2s, 2H; H-2, H-8), 7.36–7.21 (m, 15H; 3×C6H5), 7.12 (d, J = 8.6 Hz, 2H; −PMB), 7.07 (s, 1H; N6-H), 6.77 (d, J = 8.6 Hz, 2H; −PMB), 5.97 (d, 3J(1′,2′) = 4.4 Hz, 1H; H-1′), 5.21 (ddd, 3J(2′,3′) = 4.2, 3J(3′,4′) = 5.6, 2J(3′,F3′) = 53.2 Hz, 1H; H-3′), 4.90 (br s, 1H; −OH), 4.80 (dt, 3J(2′,3′) = 4.3, 3J(1′,2′) = 4.3, 3J(2′,F3′) = 15.7 Hz, 1H; H-2′), 4.61 (ABq, Δδ = 21.6 Hz, J = 11.5 Hz, 2H; −PMB), 4.39 (dq, 3J(3′,4′) = 4.2, 3J(5′,4′) = 4.3, 3J(5″,4′) = 4.3, 3J(4′,F3′) = 17.9 Hz, 1H; H-4′), 3.96–3.87 (m, 2H; H-5′/5″), 3.74 (s, 3H; −PMB); 13C NMR (100 MHz, CDCl3): δ 159.8, 154.4, 152.3, 148.0, 145.0, 138.9 (d, 5J(C8,F3′) = 3.3 Hz), 129.8, 129.2, 128.8, 128.1, 127.1, 121.6, 114.1, 94.5 (d, 1J(C3′,F3′) = 190.1 Hz), 87.8 (d, 3J(C1′,F3′) = 7.1 Hz), 83.2 (d, 2J(C2′,F3′) = 24.0 Hz), 81.1 (d, 2J(C4′,F3′) = 19.9 Hz), 72.7, 71.7, 60.4 (d, 3J(C5′,F3′) = 8.2 Hz), 55.4; 19F NMR (376 MHz, CDCl3): δ −204.6 (dt, 3J(2′,F3′) = 16.7, 3J(4′,F3′) = 16.7, 2J(3′,F3′) = 53.1 Hz); HRMS (ESI+): m/z calcd for C37H35FN5O4 [(M + H)+] 632.2668, found 632.2658 (Δ −1.6 ppm).
N6-Trityl-9-[3-deoxy-3-fluoro-2-O-(4-methoxybenzyl)-β-d-ribofuranosyl]adenine (41d)
This was prepared from ditritylated compound 27 (0.73 g, 0.835 mmol) using general procedure 2. Purification by flash chromatography (SiO2, 80:20 EtOAc/hexanes) afforded the product (0.39 g, 75%) as a white solid. Rf = 0.36 (1:1 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 7.92, 7.74 (2s, 2H; H-2, H-8), 7.36–7.23 (m, 15H; 3×C6H5), 7.28 (d, J = 8.6 Hz, 2H; −PMB), 7.05 (s, 1H; N6-H), 6.86 (dd, 5J(OH,F3′) = 2.3 Hz, 3J(OH,5″) = 12.2 Hz, 1H; 5′-OH), 6.72 (d, J = 8.6 Hz, 2H; −PMB), 5.90 (d, 3J(1′,2′) = 8.1 Hz, 1H; H-1′), 5.19 (dd, 3J(2′,3′) = 4.0, 2J(3′,F3′) = 54.7 Hz, 1H; H-3′), 4.89 (ddd, 3J(2′,3′) = 3.9, 3J(1′,2′) = 8.1, 3J(2′,F3′) = 24.6 Hz, 1H; H-2′), 4.49 (d, 3J(4′,F3′) = 28.4 Hz, 1H; H-4′), 4.47 (ABq, Δδ = 53.6 Hz, J = 11.3 Hz, 2H; −PMB), 3.88 (dm, 2J(5′,5″) = 12.7 Hz, 1H; H-5′), 3.74 (s, 3H; −PMB), 3.69 (t, 3J(OH,5″) ≈ 2J(5′,5″) ≈ 12.7 Hz, 1H; H-5″); 13C NMR (100 MHz, CDCl3): δ 159.8, 154.8, 151.7, 147.3, 144.9, 140.6, 129.8, 129.2, 128.8, 128.1, 127.3, 122.8, 114.0, 91.5 (d, 1J(C3′,F3′) = 184.0 Hz), 89.7, 86.7 (d, 2J(C2′,F3′) = 21.5 Hz), 79.0 (d, 2J(C4′,F3′) = 15.9 Hz), 72.8, 71.8, 62.8 (d, 3J(C5′,F3′) = 11.9 Hz), 55.5; 19F NMR (376 MHz, CDCl3): δ −198.9 (dddd, 5J(OH,F3′) = 2.9, 3J(2′,F3′) = 24.1, 3J(4′,F3′) = 27.8, 2J(3′,F3′) = 54.9 Hz); HRMS (ESI+): m/z calcd for C37H35FN5O4 [(M + H)+] 632.2668, found 632.2615 (Δ −8.4 ppm).
N6-Trityl-9-(2,3-dideoxy-2,3-difluoro-β-d-xylofuranosyl)adenine (41e)
This was prepared from ditritylated compound 31 (1.60 g, 2.12 mmol) using general procedure 2. Purification by flash chromatography (SiO2, 80:20 EtOAc/hexanes) afforded the product (0.78 g, 73%) as a white solid. Rf = 0.22 (1:1 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 8.05, 7.84 (2s, 2H; H-2, H-8), 7.35–7.22 (m, 15H; 3×C6H5), 7.06 (s, 1H; N6-H), 6.12 (dd, 3J(1′,2′) = 4.5, 3J(1′,F2′) = 15.6 Hz, 1H; H-1′), 5.85 (ddt, 3J(1′,2′) ≈ 3J(2′,3′) ≈ 4.5, 3J(2′,F3′) = 13.4, 2J(2′,F2′) = 52.2 Hz, 1H; H-2′), 5.44 (dddd, 3J(2′,3′) = 4.4, 3J(3′,4′) = 5.9, 3J(3′,F2′) = 14.3, 2J(3′,F3′) = 52.4 Hz, 1H; H-3′), 4.92–4.89 (m, 1H; 5′-OH), 4.49 (dm, 3J(4′,F3′) = 15.2 Hz, 1H; H-4′), 4.01–3.91 (m, 2H; H-5′/5″); 13C NMR (100 MHz, CDCl3): δ 154.6, 152.6, 148.1, 144.9, 138.9 (d, J = 3.0 Hz), 129.2, 128.2, 127.2, 121.8, 94.6 (dd, 2J(C2′,F3′) = 26.4, 1J(C2′,F2′) = 190.3 Hz), 92.1 (dd, 2J(C3′,F2′) = 25.2, 1J(C2′,F2′) = 192.4 Hz), 86.7 (dd, 3J(C1′,F3′) = 7.2, 2J(C1′,F2′) = 33.6 Hz), 80.7 (dd, 3J(C4′,F2′) = 4.2, 2J(C4′,F3′) = 20.3 Hz), 71.7, 62.3 (d, 3J(C5′,F3′) = 7.5 Hz); 19F NMR (376 MHz, CDCl3): δ −197.8 (dtd, 3J(F2′,F3′) = 10.5, 3J(3′,F2′) = 14.8, 3J(1′,F2′) = 14.9, 2J(2′,F2′) = 52.3 Hz; F-2′), −209.9 (dtd, 3J(F2′,F3′) = 10.1, 3J(2′,F3′) = 13.6, 3J(4′,F3′) = 14.2, 2J(3′,F3′) = 52.3 Hz; F-3′); HRMS (ESI+): m/z calcd for C29H26F2N5O2 [(M + H)+] 514.2049, found 514.2025 (Δ −4.6 ppm).
N6-Trityl-9-(2,3-dideoxy-2,3-difluoro-β-d-ribofuranosyl)adenine (41f)
This was prepared from ditritylated compound 35 (0.81 g, 1.07 mmol) using general procedure 2. Purification by flash chromatography (SiO2, 50:50 EtOAc/hexanes) afforded the product (0.51 g, 93%) as a waxy white solid. Rf = 0.40 (3:1 EtOAc/hexanes); 1H NMR (400 MHz, [D6]DMSO): δ 8.52, 7.94 (2s, 2H; H-2, H-8), 7.65 (s, 1H; N6-H), 7.35–7.19 (m, 15H; 3×C6H5), 6.34 (dd, 3J(1′,2′) = 5.7 Hz, 3J(1′,F2′) = 13.8, 1H; H-1′), 5.95 (ddt, 3J(1′,2′) ≈ 3J(2′,3′) ≈ 5.8, 3J(2′,F3′) = 16.4, 2J(2′,F2′) = 50.2 Hz, 1H; H-2′), 5.53 (dm, 2J(3′,F3′) = 53.1 Hz, 1H; H-3′), 4.76 (br s, 1H; 5′-OH), 4.38 (dm, 3J(4′,F3′) = 23.8 Hz, 1H; H-4′), 3.68 (dd, 3J(5′,4′) = 3.5, 2J(5′,5″) = 12.3 Hz, 1H; H-5′), 3.64 (dd, 3J(5″,4′) = 3.5, 2J(5′,5″) = 12.3 Hz, 1H; H-5″); 13C NMR (100 MHz, CDCl3): δ 154.8, 151.9, 147.4, 144.7, 140.2, 129.1, 128.2, 127.3, 122.6, 91.3 (dd, 2J(C2′,F3′) = 13.0, 1J(C2′,F2′) = 185.5 Hz), 89.6 (dd, 2J(C3′,F2′) = 15.0, 1J(C2′,F2′) = 201.7 Hz), 88.1 (d, 2J(C1′,F2′) = 30.4 Hz), 86.2 (dd, 3J(C4′,F2′) = 3.1, 2J(C4′,F3′) = 21.1 Hz), 71.8, 62.3 (d, 3J(C5′,F3′) = 11.4 Hz); 19F NMR (376 MHz, [D6]DMSO): δ −204.6 (dddd, 3J(F2′,F3′) = 5.0, 3J(2′,F3′) = 16.2, 3J(4′,F3′) = 22.5, 2J(3′,F3′) = 53.4 Hz; F-3′), −212.7 (dm, 2J(2′,F2′) = 50.0 Hz; F-2′); HRMS (ESI+): m/z calcd for C29H26F2N5O2 [(M + H)+] 514.2049, found 514.2075 (Δ 5.0 ppm).
General Procedure 3: Azidation
To a solution of alcohol (1 mmol, 1 equiv) in anhydrous 1,4-dioxane (9 mL) was added DPPA (3 equiv), and DBU (2 equiv) dropwise and the reaction mixture was stirred at rt. The conversion of alcohol to the phosphate intermediate was monitored by TLC, which was usually completed in 1–3 h. NaN3 (10 equiv) and 15-crown-5 (0.1 equiv) were then added and the reaction mixture was refluxed (oil bath at 110 °C) for 1–24 h. The reaction mixture was allowed to cool to rt, filtered through a short pad of silica, and the silica was washed with EtOAc. The combined filtrate was evaporated under reduced pressure. Purification by flash chromatography (SiO2, EtOAc/hexanes gradient) afforded the product.
N6-Trityl-9-[5-azido-2,5-dideoxy-2-fluoro-3-O-(4-methoxybenzyl)-β-d-ribofuranosyl]adenine (42a)
This was prepared from alcohol 41a (0.062 g, 0.098 mmol) using general procedure 3. The reaction mixture was refluxed for 17 h. Purification by flash chromatography (SiO2, 50:50 EtOAc/hexanes) afforded the product (0.053 g, 83%) as a white solid. Rf = 0.45 (1:1 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 8.00, 7.90 (2s, 2H; H-2, H-8), 7.35–7.20 (m, 17H; 3×C6H6, −PMB), 7.00 (s, 1H; N6-H), 6.87 (d, J = 8.6 Hz, 2H; −PMB), 6.12 (dd, 3J(1′,2′) = 1.5, 3J(1′,F2′) = 19.7 Hz, 1H; H-1′), 5.53 (ddd, 3J(1′,2′) = 1.5, 3J(2′,3′) = 3.5, 2J(2′,F2′) = 52.8 Hz, 1H; H-2′), 4.61 (ddd, 3J(2′,3′) = 4.2, 3J(3′,4′) = 8.7, 3J(3′,F2′) = 20.4 Hz, 1H; H-3′), 4.60 (ABq, Δδ = 47.6 Hz, J = 11.3 Hz, 2H; −PMB), 4.26 (ddd, 3J(4′,5′) = 2.9, 3J(4′,5″) = 5.1, 3J(4′,3′) = 8.1 Hz, 1H; H-4′), 3.78 (s, 3H; −PMB), 3.63 (dd, 3J(4′,5′) = 2.9, 2J(5′,5″) = 13.5 Hz, 1H; H-5′), 3.43 (dd, 3J(4′,5″) = 5.1, 2J(5′,5″) = 13.5 Hz, 1H; H-5″); 13C NMR (100 MHz, CDCl3): δ 160.0, 154.4, 152.6, 148.2, 145.0, 139.1, 130.2, 129.2, 129.0, 128.1, 127.2, 121.5, 114.2, 91.4 (d, 1J(C2′,F2′) = 190.2 Hz), 88.4 (d, 2J(C1′,F2′) = 34.5 Hz), 80.2, 75.7 (d, 2J(C3′,F2′) = 16.0 Hz), 72.9, 71.7, 55.5, 51.4; 19F NMR (376 MHz, CDCl3): δ −202.0 (dt, 3J(1′,F2′) = 20.1, 3J(3′,F2′) = 20.1, 2J(2′,F2′) = 52.9 Hz).
N6-Trityl-9-[5-azido-2,5-dideoxy-2-fluoro-3-O-(4-methoxybenzyl)-β-d-arabinofuranosyl]adenine (42b)
This was prepared from alcohol 41b (0.23 g, 0.364 mmol) using general procedure 3. The reaction mixture was refluxed for 24 h. Purification by flash chromatography (SiO2, 50:50 EtOAc/hexanes) afforded the product (0.14 g, 58%) as a white solid. Rf = 0.63 (1:1 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 8.06 (s, 1H; H-2), 8.04 (d, 5J(8,F2′) = 2.9 Hz, 1H; H-8), 7.37–7.21 (m, 17H; 3×C6H6, −PMB), 6.99 (s, 1H; N6-H), 6.92 (d, J = 8.5 Hz, 2H; −PMB), 6.42 (dd, 3J(1′,2′) = 3.1, 3J(1′,F2′) = 21.2 Hz, 1H; H-1′), 5.11 (ddd, 3J(2′,3′) = 1.5, 3J(1′,2′) = 3.3, 2J(2′,F2′) = 52.3 Hz, 1H; H-2′), 4.60 (ABq, Δδ = 35.2 Hz, J = 11.5 Hz, 2H; −PMB), 4.27 (ddd, 3J(2′,3′) = 1.2, 3J(3′,4′) = 3.9, 3J(3′,F2′) = 17.9 Hz, 1H; H-3′), 4.16 (q, 3J(4′,5′) = 4.7, 3J(4′,5″) = 4.7, 3J(4′,3′) = 4.6 Hz, 1H; H-4′), 3.82 (s, 3H; −PMB), 3.58 (dd, 3J(4′,5′) = 4.5, 2J(5′,5″) = 13.2 Hz, 1H; H-5′), 3.43 (dd, 3J(4′,5″) = 5.3, 2J(5′,5″) = 13.2 Hz, 1H; H-5″); 13C NMR (100 MHz, CDCl3): δ 160.0, 154.3, 152.6, 148.8, 145.1, 139.4 (d, 4J(C8,F2′) = 6.6 Hz), 129.9, 129.2, 128.5, 128.1, 127.1, 120.4, 114.3, 93.2 (d, 1J(C2′,F2′) = 193.0 Hz), 83.1 (d, 2J(C1′,F2′) = 17.1 Hz), 81.8 (d, 2J(C3′,F2′) = 25.6 Hz), 81.2 (d, 3J(C4′,F2′) = 2.1 Hz), 72.7, 71.6, 55.5, 51.6; 19F NMR (376 MHz, CDCl3): δ −196.3 (dddd, 5J(8,F2′) = 3.0, 3J(3′,F2′) = 17.7, 3J(1′,F2′) = 20.8, 2J(2′,F2′) = 51.3 Hz).
N6-Trityl-9-[5-azido-3,5-dideoxy-3-fluoro-2-O-(4-methoxybenzyl)-β-d-xylofuranosyl]adenine (42c)
This was prepared from alcohol 41c (0.70 g, 1.108 mmol) using general procedure 3. Conversion of alcohol to the phosphate intermediate was completed in 1 h. For the conversion of phosphate to azide, the reaction mixture was refluxed for 24 h. Purification by flash chromatography (SiO2, 50:50 EtOAc/hexanes) afforded the product (0.41 g, 57%) as a white solid. Rf = 0.61 (1:1 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 8.07, 7.86 (2s, 2H; H-2, H-8), 7.37–7.21 (m, 17H; 3×C6H5 and −PMB), 6.94 (s, 1H; N6-H), 6.83 (d, J = 8.6 Hz, 2H; −PMB), 6.27 (d, 3J(1′,2′) = 1.7 Hz, 1H; H-1′), 5.06 (ddd, 3J(2′,3′) = 1.1, 3J(3′,4′) = 2.7, 3J(3′,F3′) = 51.0 Hz, 1H; H-3′), 4.71 (ABq, Δδ = 48.8 Hz, J = 11.6 Hz, 2H; −PMB), 4.50 (dt, 3J(2′,3′) = 1.4, 3J(1′,2′) = 1.4, 3J(2′,F3′) = 15.2 Hz, 1H; H-2′), 4.44 (dtd, 3J(3′,4′) = 2.7, 3J(5′,4′) = 6.6, 3J(5″,4′) = 6.6, 3J(4′,F3′) = 28.4 Hz, 1H; H-4′), 3.78 (s, 3H; −PMB), 3.76–3.73 (m, 1H; H-5′), 3.67 (dd, 3J(5′,4′) = 6.4, 2J(5′,5″) = 12.8 Hz, 1H; H-5″); 13C NMR (100 MHz, CDCl3): δ 159.9, 154.2, 152.7, 148.6, 145.1, 137.7 (d, 5J(C8,F3′) = 6.0 Hz), 130.0, 129.2, 128.4, 128.1, 127.1, 121.1, 114.2, 94.0 (d, 1J(C3′,F3′) = 186.0 Hz), 88.2, 85.0 (d, 2J(C2′,F3′) = 27.2 Hz), 80.7 (d, 2J(C4′,F3′) = 19.5 Hz), 72.6, 71.6, 55.5, 48.8 (d, 3J(C5′,F3′) = 9.3 Hz); 19F NMR (376 MHz, CDCl3): δ −202.6 (ddd, 3J(2′,F3′) = 14.7, 3J(4′,F3′) = 28.6, 2J(3′,F3′) = 51.2 Hz); HRMS (ESI+): m/z calcd for C37H34FN8O3 [(M + H)+] 657.2732, found 657.2692 (Δ −6.1 ppm).
N6-Trityl-9-[5-azido-3,5-dideoxy-3-fluoro-2-O-(4-methoxybenzyl)-β-d-ribofuranosyl]adenine (42d)
This was prepared from alcohol 41d (0.38 g, 0.602 mmol) using general procedure 3. Conversion of alcohol to phosphate intermediate was completed in 1 h and the conversion of phosphate to azide was completed in 3 h. Purification by flash chromatography (SiO2, 50:50 EtOAc/hexanes) afforded the product (0.32 g, 81%) as a white solid. Rf = 0.55 (1:1 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 8.00, 7.76 (2s, 2H; H-2, H-8), 7.37–7.22 (m, 15H; 3×C6H5), 7.11 (d, J = 8.6 Hz, 2H; −PMB), 6.95 (s, 1H; N6-H), 6.78 (d, J = 8.6 Hz, 2H; −PMB), 6.02 (d, 3J(1′,2′) = 6.6 Hz, 1H; H-1′), 5.09 (ddd, 3J(2′,3′) = 4.5, 3J(1′,2′) = 6.7, 3J(2′,F3′) = 18.4 Hz, 1H; H-2′), 5.08 (ddd, 3J(3′,4′) = 2.2, 3J(3′,2′) = 4.5, 2J(3′,F3′) = 54.4 Hz, 1H; H-3′), 4.55 (ABq, Δδ = 41.2 Hz, J = 11.5 Hz, 2H; −PMB), 4.45 (dtd, 3J(3′,4′) = 2.2, 3J(5′,4′) = 5.2, 3J(5″,4′) = 5.2, 3J(4′,F3′) = 23.6 Hz, 1H; H-4′), 3.77 (s, 3H; −PMB), 3.68 (dd, 3J(5′,4′) = 5.4, 2J(5′,5″) = 13.1 Hz; H-5′), 3.67 (dd, 3J(5′,4′) = 4.9, 2J(5′,5″) = 13.1 Hz, 1H; H-5″); 13C NMR (100 MHz, CDCl3): δ 159.9, 154.4, 152.5, 148.8, 145.1, 139.6, 129.8, 129.2, 128.7, 128.1, 127.2, 121.9, 114.1, 90.0 (d, 1J(C3′,F3′) = 189.6 Hz), 87.4, 81.6 (d, 2J(C2′,F3′) = 24.3 Hz), 77.2 (d, 2J(C4′,F3′) = 15.5 Hz), 72.9, 71.7, 55.5, 51.6 ppm (d, 3J(C5′,F3′) = 8.6 Hz); 19F NMR (376 MHz, CDCl3): δ −201.8 (ddd, 3J(2′,F3′) = 18.1, 3J(4′,F3′) = 23.4, 2J(3′,F3′) = 54.1 Hz); HRMS (ESI+): m/z calcd for C37H34FN8O3 [(M + H)+] 657.2732, found 657.2731 (Δ −0.2 ppm).
N6-Trityl-9-(5-azido-2,3-difluoro-2,3,5-trideoxy-β-d-xylofuranosyl)adenine (42e)
This was prepared from alcohol 41e (0.76 g, 1.48 mmol) using general procedure 3. Conversion of alcohol to phosphate intermediate was completed in 2 h. For the conversion of phosphonate to the azide product, the reaction mixture was refluxed for 24 h. Purification by flash chromatography (SiO2, 30:70 EtOAc/hexanes) afforded the product (0.58 g, 73%) as a white solid. Rf = 0.57 (1:1 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 8.07, 7.87 (2s, 2H; H-2, H-8), 7.36–7.21 (m, 15H; 3×C6H5), 6.96 (s, 1H; N6-H), 6.38 (d, 3J(1′,F2′) = 20.4 Hz, 1H; H-1′), 5.57 (dd, 3J(2′,F3′) = 10.5, 2J(2′,F2′) = 47.9 Hz, 1H; H-2′), 5.28 (dddd, 4J(3′,5′) = 1.1, 3J(3′,4′) = 3.1, 3J(3′,F2′) = 8.9, 2J(3′,F3′) = 49.7 Hz, 1H; H-3′), 4.48 (dm, 3J(4′,F3′) = 28.3 Hz, 1H; H-4′), 3.80 (ddd, 4J(3′,5′) = 1.1, 3J(5′,4′) = 6.6, 2J(5′,5″) = 13.0 Hz, 1H; H-5′), 3.71 (dd, 3J(5″,4′) = 6.5, 2J(5′,5″) = 12.8 Hz, 1H; H-5″); 13C NMR (100 MHz, CDCl3): δ 154.3, 153.0, 148.7, 145.0, 137.4 (d, J = 6.4 Hz), 129.2, 128.1, 127.2, 121.0, 96.1 (dd, 2J(C2′,F3′) = 31.5, 1J(C2′,F2′) = 186.9 Hz), 92.3 (dd, 2J(C3′,F2′) = 30.4, 1J(C3′,F3′) = 185.9 Hz), 87.5 (d, 2J(C1′,F2′) = 36.7 Hz), 80.5 (d, 2J(C4′,F3′) = 19.7 Hz), 71.7, 48.5 (d, 3J(C5′,F3′) = 9.1 Hz); 19F NMR (376 MHz, CDCl3): δ −192.1 (ddddd, 4J(4′,F2′) = 3.3, 3J(3′,F2′) = 8.9, 3J(F3′,F2′) = 14.8, 3J(1′,F2′) = 20.8, 2J(2′,F2′) = 48.1 Hz; F-2′), −208.7 (dddd, 3J(2′,F3′) = 10.5, 3J(F3′,F2′) = 14.2, 3J(4′,F3′) = 28.1, 2J(3′,F3′) = 49.9 Hz; F-3′); HRMS (ESI+): m/z calcd for C29H24F2N8ONa [(M + Na)+] 561.1933, found 561.1943 (Δ 1.8 ppm).
N6-Trityl-9-(5-azido-2,3-difluoro-2,3,5-trideoxy-β-d-ribofuranosyl)adenine (42f)
This was prepared from alcohol 41f (0.41 g, 0.798 mmol) using general procedure 3. Conversion of alcohol to phosphate intermediate was completed in 2 h and the conversion of phosphate to azide was completed in 1 h. Purification by flash chromatography (SiO2, 30:70 EtOAc/hexanes) afforded the product (0.40 g, 93%) as a colorless liquid. Rf = 0.54 (1:1 EtOAc/hexanes); 1H NMR (400 MHz, [D6]DMSO): δ 8.52, 7.96 (2s, 2H; H-2, H-8), 7.65 (s, 1H; N6-H), 7.34–7.19 (m, 15H; 3×C6H5), 6.38 (dd, 3J(1′,2′) = 4.8, 3J(1′,F2′) = 15.2 Hz, 1H; H-1′), 6.15 (ddt, 3J(1′,2′) ≈ 3J(2′,3′) ≈ 4.8, 3J(2′,F3′) = 12.8, 2J(2′,F2′) = 50.0 Hz, 1H; H-2′), 5.53 (dm, 2J(3′,F3′) = 52.4 Hz, 1H; H-3′), 4.49 (dm, 3J(4′,F3′) = 21.2 Hz, 1H; H-4′), 3.73 (dd, 3J(5′,4′) = 5.6, 2J(5′,5″) = 13.2 Hz, 1H; H-5′), 3.52 (dd, 3J(5″,4′) = 4.4, 2J(5′,5″) = 13.2 Hz, 1H; H-5″); 13C NMR (100 MHz, [D6]DMSO): δ 153.7, 151.5, 148.2, 144.7, 140.8, 128.6, 127.7, 126.6, 120.8, 89.4 (dd, 2J(C2′,F3′) = 14.2, 1J(C2′,F2′) = 192.2 Hz), 88.8 (dd, 2J(C3′,F2′) = 13.7, 1J(C3′,F3′) = 188.4 Hz), 84.8 (dd, 3J(C1′,F3′) = 2.7, 2J(C1′,F2′) = 31.6 Hz), 79.9 (dd, 3J(C4′,F2′) = 2.5, 2J(C4′,F3′) = 23.6 Hz), 70.4, 50.3 (d, 3J(C5′,F3′) = 6.3 Hz); 19F NMR (376 MHz, CDCl3): δ −209.2 (dt, 3J(2′,F3′) = 13.8, 3J(4′,F3′) = 13.8, 2J(3′,F3′) = 51.3 Hz; F-3′), −210.5 (ddd, 3J(3′,F2′) = 8.6, 3J(1′,F2′) = 18.3, 2J(2′,F2′) = 52.0 Hz; F-2′); HRMS (ESI+): m/z calcd for C29H25F2N8O [(M + H)+] 539.2114, found 539.2159 (Δ 8.3 ppm).
N6-Trityl-9-[2,5-dideoxy-2-fluoro-3-O-(4-methoxybenzyl)-5-(N-sulfamoyl)amino-β-d-ribofuranosyl]adenine (43a)
To a solution of azide 42a (0.053 g, 0.081 mmol) in CH2Cl2 (2 mL) was added zinc turnings (0.081 g, 1.24 mmol, 15 equiv) and AcOH (43 μL, 0.751 mmol, 9.3 equiv), and the reaction mixture was stirred at rt for 1 h. The reaction mixture was filtered through a short pad of Celite, and the residue was washed with MeOH (3 × 5 mL). The combined filtrates were concentrated and dissolved in EtOAc (5 mL). The EtOAc layer was washed with sat. NaHCO3, brine, dried (MgSO4), and evaporated to obtain the crude 5′-aminonucleoside. To a solution of the 5′-aminonucleoside in 1,4-dioxane (2 mL) was added sulfamide (0.024 g, 0.250 mmol, 3.0 equiv) and the mixture was refluxed for 2 h. The reaction mixture was evaporated, then partitioned between CH2Cl2 (5 mL) and H2O (5 mL). The phases were separated and the organic phase was washed with brine, dried (Na2SO4) and evaporated under reduced pressure. Purification by flash chromatography (SiO2, gradient: hexanes to 70% EtOAc in hexanes) afforded the product (0.051 g, 89%) as a white solid. Rf = 0.52 (3:1 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 8.57 (dd, 3J(5″,NH) = 3.4 Hz, 3J(5′,NH) = 8.2, 1H; C5′-NH), 8.06, 7.78 (2s, 2H; H-2, H-8), 7.34–7.20 (m, 17H; 3×C6H6, −PMB), 7.01 (s, 1H; N6-H), 6.87 (d, J = 8.5 Hz, 2H; −PMB), 6.05 (dd, 3J(1′,2′) = 5.5, 3J(1′,F2′) = 13.7 Hz, 1H; H-1′), 5.59 (dt, 3J(2′,3′) = 5.3, 3J(1′,2′) = 5.3, 2J(2′,F2′) = 51.5 Hz, 1H; H-2′), 4.64 (ABq, Δδ = 72.0 Hz, J = 11.0 Hz, 2H; −PMB), 4.56–4.37 (m, 4H; H-3′, H-4′, −NH2), 3.79 (s, 3H; −PMB), 3.38–3.35 (m, 2H; H-5′/5″); 13C NMR (100 MHz, CDCl3): δ 159.9, 154.7, 152.5, 147.6, 144.8, 139.7, 130.1, 129.4, 129.2, 128.2, 127.2, 122.0, 114.2, 91.2 (d, 1J(C2′,F2′) = 196.4 Hz), 88.4 (d, 2J(C1′,F2′) = 31.7 Hz), 82.9 (d, 3J(C4′,F2′) = 2.5 Hz), 76.5 (d, 2J(C3′,F2′) = 14.0 Hz), 73.5 (d, 4J(PMB-CH2-,F2′) = 2.8 Hz), 71.7, 55.5, 44.7; 19F NMR (376 MHz, CDCl3): δ = −210.0 (dm, 2J(2′,F2′) = 51.8 Hz); HRMS (ESI+): m/z calcd for C37H36FN7O5SNa [(M + Na)+] 732.2375, found 732.2407 (Δ 4.4 ppm).
N6-Trityl-9-[2,5-dideoxy-2-fluoro-3-O-(4-methoxybenzyl)-5-(N-sulfamoyl)amino-β-d-arabinofuranosyl]adenine (43b)
To a solution of azide 42b (0.17 g, 0.259 mmol) in CH2Cl2 (7 mL) was added zinc turnings (0.26 g, 3.978 mmol, 15 equiv) and AcOH (0.14 mL, 2.445 mmol, 9.5 equiv), and the reaction mixture was stirred at rt for 1 h. The reaction mixture was filtered through a short pad of Celite and the residue was washed with MeOH (3 × 5 ml). The combined filtrates were concentrated and dissolved in EtOAc (10 mL). The EtOAc layer was washed with sat. NaHCO3, brine, dried (MgSO4), and evaporated to obtain the crude 5′-aminonucleoside. To a solution of the 5′-aminonucleoside in 1,4-dioxane (5 mL) was added sulfamide (0.072 g, 0.749 mmol, 2.9 equiv) and the mixture was refluxed for 2 h. The reaction mixture was evaporated then partitioned between CH2Cl2 (10 mL) and water (10 mL). The phases were separated, and the organic layer was washed with brine, dried (Na2SO4) and evaporated under reduced pressure. Purification by flash chromatography (SiO2, gradient: hexanes to 70% EtOAc in hexanes) afforded the product (0.15 g, 83%) as a white solid. Rf = 0.39 (3:1 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 8.00 (s, 1H; H-2), 7.99 (d, 5J(8,F2′) = 2.0 Hz, 1H; H-8), 7.34–7.19 (m, 17H; 3×C6H6, −PMB), 7.15–7.10 (m, 1H; C5′-NH), 6.95 (s, 1H; N6-H), 6.88 (d, J = 8.4 Hz, 2H; −PMB), 6.34 (dd, 3J(1′,2′) = 4.7, 3J(1′,F2′) = 14.1 Hz, 1H; H-1′), 5.18 (dt, 3J(2′,3′) = 4.0, 3J(1′,2′) = 4.0, 2J(2′,F2′) = 52.2 Hz, 1H; H-2′), 4.80 (s, 2H; −NH2), 4.61 (ABq, Δδ = 30.8 Hz, J = 11.3 Hz, 2H; −PMB), 4.47 (ddd, 3J(2′,3′) = 3.5, 3J(3′,4′) = 5.6, 3J(3′,F2′) = 18.1 Hz, 1H; H-3′), 4.08 (q, 3J(4′,5′) = 4.3, 3J(4′,5″) = 4.3, 3J(4′,3′) = 4.3 Hz, 1H; H-4′), 3.78 (s, 3H; −PMB), 3.40–3.38 (m, 2H; H-5′/5″); 13C NMR (100 MHz, CDCl3): δ 159.8, 154.1, 152.5, 148.6, 145.0, 139.8 (d, J = 3.9 Hz), 130.2, 129.2, 128.7, 128.1, 127.1, 120.1, 114.3, 94.3 (d, 1J(C2′,F2′) = 196.4 Hz), 82.9 (d, 2J(C1′,F2′) = 17.5 Hz), 80.4 (d, 2J(C3′,F2′) = 23.7 Hz), 79.9 (d, 3J(C4′,F2′) = 5.4 Hz), 72.7, 71.7, 55.5, 43.7; 19F NMR (376 MHz, CDCl3): δ −195.0 (dt, 3J(3′,F2′) = 16.0, 3J(1′,F2′) = 16.0, 2J(2′,F2′) = 52.3 Hz); HRMS (ESI+): m/z calcd for C37H36FN7O5SNa [(M + Na)+] 732.2375, found 732.2412 (Δ 5.0 ppm).
N6-Trityl-9-[3,5-dideoxy-3-fluoro-2-O-(4-methoxybenzyl)-5-(N-sulfamoyl)amino-β-d-xylofuranosyl]adenine (43c)
To a solution of azide 42c (0.54 g, 0.822 mmol) in CH2Cl2 (23 mL) was added zinc turnings (0.81g, 12.393 mmol, 15 equiv) and AcOH (0.35 mL, 6.165 mmol, 7.5 equiv), and the reaction mixture was stirred at rt for 1 h. The reaction mixture was filtered through a short pad of Celite, and the residue was washed with MeOH (3 × 10 mL). The combined filtrates were concentrated and dissolved in EtOAc (10 mL). The EtOAc layer was washed with sat. NaHCO3, brine, dried (MgSO4), and evaporated to obtain the crude 5′-aminonucleoside. To a solution of the 5′-aminonucleoside in 1,4-dioxane (19 mL) was added sulfamide (0.23 g, 2.393 mmol, 2.9 equiv) and the mixture was refluxed for 2 h. The reaction mixture was evaporated then partitioned between CH2Cl2 (10 mL) and water (10 mL). The phases were separated and the CH2Cl2 layer was washed with brine, dried (Na2SO4) and evaporated under reduced pressure. Purification by flash chromatography (SiO2, gradient: hexanes to 70% EtOAc in hexanes) afforded the product (0.50 g, 86%) as a white solid. Rf = 0.39 (3:1 EtOAc/hexanes); 1H NMR (400 MHz, [D6]DMSO): δ 8.19, 7.94 (2s, 2H; H-2, H-8), 7.52 (s, 1H; N6-H), 7.35–7.20 (m, 17H; 3×C6H5, −PMB), 6.89 (t, 3J(5′/5″, NH) = 6.1 Hz, 1H; C5′-NH), 6.85 (d, J = 8.6 Hz, 2H; −PMB), 6.65 (s, 2H; −NH2), 6.09 (d, 3J(1′,2′) = 2.2 Hz, 1H; H-1′), 5.34 (dd, 3J(3′,4′) = 2.6, 2J(3′,F3′) = 51.1 Hz, 1H; H-3′), 4.79 (dd, 3J(1′,2′) = 3.0, 3J(2′,F3′) = 15.8 Hz, 1H; H-2′), 4.65 (s, 2H; −PMB), 4.42 (dtd, 3J(3′,4′) = 2.8, 3J(5′,4′) = 6.5, 3J(5″,4′) = 6.6, 3J(4′,F3′) = 29.7 Hz, 1H; H-4′), 3.71 (s, 3H; −PMB), 3.35 (dt, 3J(5″,NH) = 6.5, 3J(5″,4′) = 6.6, 2J(5″,5′) = 13.3 Hz, 1H; H-5′), 3.26 (dt, 3J(5″,4′) = 6.5, 3J(5″,NH) = 6.5, 2J(5″,5′) = 13.3 Hz, 1H; H-5″); 13C NMR (100 MHz, [D6]DMSO): δ 159.0, 153.5, 151.4, 148.2, 144.8, 138.9 (d, 5J(C8,F3′) = 4.8 Hz), 129.6, 128.9, 128.6, 127.7, 126.6, 120.6, 113.7, 93.4 (d, 1J(C3′,F3′) = 183.4 Hz), 87.4, 84.2 (d, 2J(C2′,F3′) = 27.6 Hz), 80.3 (d, 2J(C4′,F3′) = 19.3 Hz), 71.4, 70.3, 55.0, 40.5 (d, 3J(C5′,F3′) = 10.7 Hz); 19F NMR (376 MHz, [D6]DMSO): δ −200.8 (ddd, 3J(2′,F3′) = 15.6, 3J(4′,F3′) = 29.7, 2J(3′,F3′) = 50.8 Hz); 19F NMR (376 MHz, CDCl3): δ −201.8 (ddd, 3J(2′,F3′) = 16.2, 3J(4′,F3′) = 23.2, 2J(3′,F3′) = 52.5 Hz); HRMS (ESI+): m/z calcd for C37H37FN7O5S [(M + H)+] 710.2555, found 710.2576 (Δ 2.9 ppm).
N6-Trityl-9-[3,5-dideoxy-3-fluoro-2-O-(4-methoxybenzyl)-5-(N-sulfamoyl)amino-β-d-ribofuranosyl]adenine (43d)
To a solution of azide 42d (0.29 g, 0.442 mmol) in CH2Cl2 (12 mL) was added zinc turnings (0.43g, 6.624 mmol, 15 equiv) and AcOH (0.19 mL, 3.312 mmol, 7.5 equiv) and the reaction mixture was stirred at rt for 1 h. The reaction mixture was filtered through a short pad of Celite washing with MeOH (3 × 10 mL). The combined filtrates were concentrated and dissolved in EtOAc (10 mL). The EtOAc layer was washed with sat. NaHCO3, brine, dried (MgSO4), and evaporated to obtain the crude 5′-aminonucleoside. To a solution of the 5′-aminonucleoside in 1,4-dioxane (9 mL) was added sulfamide (0.123 g, 1.284 mmol, 2.9 equiv) and the mixture was refluxed for 2 h. The reaction mixture was evaporated then partitioned between CH2Cl2 (10 mL) and water (10 mL). The phases were separated and the CH2Cl2 layer was washed with brine, dried (Na2SO4) and evaporated under reduced pressure. Purification by flash chromatography (SiO2, gradient: hexanes to 70% EtOAc in hexanes) afforded the product (0.26 g, 84%) as a white solid. Rf = 0.51 (3:1 EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 9.32 (d, 3J(NH,5′) = 10.0 Hz, 1H; C5′-NH), 8.03, 7.73 (2s, 2H; H-2, H-8), 7.36–7.22 (m, 15H; 3×C6H5), 7.05 (s, 1H; N6-H), 7.04 (d, J = 8.6 Hz, 2H; −PMB), 6.73 (d, J = 8.6 Hz, 2H; −PMB), 5.85 (d, 3J(1′,2′) = 8.3 Hz, 1H; H-1′), 5.12 (dd, 3J(3′,2′) = 4.3, 2J(3′,F3′) = 54.4 Hz, 1H; H-3′), 4.90 (ddd, 3J(2′,3′) = 4.7, 3J(1′,2′) = 8.3, 3J(2′,F3′) = 24.2 Hz, 1H; H-2′), 4.58 (dt, 3J(5′,4′) = 2.3, 3J(5″,4′) = 2.3, 3J(4′,F3′) = 28.7 Hz, 1H; H-4′), 4.45 (ABq, Δδ = 31.6 Hz, J = 11.2 Hz, 2H; −PMB), 4.34 (s, 2H; −NH2), 3.74 (s, 3H; −PMB), 3.48–3.35 (m, 2H; H-5′/5″); 13C NMR (100 MHz, [D6]DMSO): δ 158.9, 153.8, 151.1, 147.9, 144.8, 141.3, 129.2, 128.9, 128.6, 127.7, 126.6, 121.3, 113.5, 90.4 (d, 1J(C3′,F3′) = 183.0 Hz), 86.2, 82.1 (d, 2J(C2′,F3′) = 23.4 Hz), 76.8 (d, 2J(C4′,F3′) = 15.6 Hz), 71.1, 70.4, 50.0, 43.7 (d, 3J(C5′,F3′) = 10.8 Hz); 19F NMR (376 MHz, CDCl3): δ −196.1 (ddd, 3J(2′,F3′) = 24.1, 3J(4′,F3′) = 28.7, 2J(3′,F3′) = 53.6 Hz); 19F NMR (376 MHz, [D6]DMSO): δ −196.6 (ddd, 3J(2′,F3′) = 24.0, 3J(4′,F3′) = 27.6, 2J(3′,F3′) = 53.0 Hz); HRMS (ESI+): m/z calcd for C37H36FN7O5SNa [(M + Na)+] 732.2375, found 732.2421 (Δ 6.2 ppm).
N6-Trityl-9-[2,3,5-trideoxy-2,3-difluoro-5-(N-sulfamoyl)amino-β-d-xylofuranosyl]adenine (43e)
To a solution of azide 42e (0.15 g, 0.278 mmol) in MeOH (7 mL) was added 10% Pd/C (0.030 g, 55% wet with water), and the reaction mixture was stirred under H2 (1 atm) for 1 h. The reaction mixture was filtered through a short pad of Celite washing with MeOH (3 × 5 mL). The combined filtrates were concentrated to obtain the crude 5′-aminonucleoside. To a solution of the crude 5′-aminonucleoside in 1,4-dioxane (5 mL) was added sulfamide (0.09 g, 0.94 mmol) and the mixture was refluxed for 2 h. The reaction mixture was evaporated under reduced pressure. Purification by flash chromatography (SiO2, gradient: hexanes to 70% EtOAc in hexanes) afforded the product (0.14 g, 84%) as a white solid. Rf = 0.32 (3:1 EtOAc/hexanes); 1H NMR (400 MHz, [D6]DMSO): δ 8.22 (s, 1H), 7.95 (s, 1H), 7.59 (s, 1H; N6-H), 7.35–7.20 (m, 15H; 3×C6H5), 6.96 (t, 3J(5′/5″,NH) = 6.1 Hz, 1H; C5′-NH), 6.67 (s, 2H; −NH2), 6.29 (dd, 3J(1′,2′) = 2.1 Hz, 3J(1′,F2′) = 19.7, 1H; H-1′), 5.99 (ddt, 3J(2′,3′) ≈ 3J(1′,2′) ≈ 1.7, 3J(2′,F3′) = 12.7, 2J(2′,F2′) = 47.8 Hz, 1H; H-2′), 5.56 (dddd, 3J(2′,3′) = 1.4, 3J(3′,4′) = 3.2, 3J(3′,F2′) = 10.2, 2J(3′,F3′) = 49.6 Hz, 1H; H-3′), 4.53 (dm, 3J(4′,F3′) = 28.7 Hz, 1H; H-4′), 3.41–3.25 (m, 2H; H-5′/5″); 13C NMR (100 MHz, [D6]DMSO): δ 153.6, 151.6, 148.2, 144.8, 139.0 (d, J = 4.5 Hz), 128.6, 127.7, 126.6, 120.5, 95.5 (dd, 2J(C2′,F3′) = 32.0, 1J(C2′,F2′) = 182.8 Hz), 92.3 (dd, 2J(C3′,F2′) = 29.6, 1J(C3′,F3′) = 182.9 Hz), 86.3 (d, 2J(C1′,F2′) = 36.1 Hz), 79.9 (d, 2J(C4′,F3′) = 19.5 Hz), 70.3, 40.3 (d, 3J(C5′,F3′) = 10.6 Hz); 19F NMR (376 MHz, [D6]DMSO): δ −193.8 (ddtd, 4J(F2′,4′) = 2.7, 3J(F2′,3′) = 10.4, 3J(F2′,F3′) = 10.4, 3J(1′,F2′) = 20.7, 2J(2′,F2′) = 47.8 Hz; F-2′), −207.0 (ddt, 3J(F2′,F3′) = 11.6, 3J(2′,F3′) = 11.6, 3J(4′,F3′) = 28.7, 2J(3′,F3′) = 50.7 Hz; F-3′); HRMS (ESI+): m/z calcd for C29H27F2N7O3SNa [(M + Na)+] 614.1756, found 614.1786 (Δ 4.9 ppm).
N6-Trityl-9-[2,3,5-trideoxy-2,3-difluoro-5-(N-sulfamoyl)amino-β-d-ribofuranosyl]adenine (43f)
To a solution of azide 42f (0.32 g, 0.59 mmol) in MeOH (10 mL) was added 10% Pd/C (0.06g, 55% wet with water) and the reaction mixture was stirred under H2 (1 atm) for 1 h. The reaction mixture was filtered through a short pad of Celite washing with MeOH (3 × 10 mL). The combined filtrates were concentrated to obtain the crude 5′-aminonucleoside. To a solution of the 5′-aminonucleoside in 1,4-dioxane (9 mL) was added sulfamide (0.18 g, 1.87 mmol, 3.1 equiv) and the mixture was refluxed for 2 h. The reaction mixture was evaporated under reduced pressure. Purification by flash chromatography (SiO2, gradient: hexanes to 70% EtOAc in hexanes) afforded the product (0.34 g, 94%) as a white solid. Rf = 0.30 (3:1 EtOAc/hexanes); 1H NMR (400 MHz, [D6]DMSO): δ 8.50 (s, 1H), 7.93 (s, 1H), 7.71 (s, 1H; N6-H), 7.41 (t, 3J(5′/5″,NH) = 6.4 Hz, 1H; C5′-NH), 7.35–7.20 (m, 15H; 3×C6H5), 6.72 (s, 2H; −NH2), 6.37 (dd, 3J(1′,2′) = 6.0 Hz, 3J(1′,F2′) = 13.2, 1H; H-1′), 6.00 (dddd, 3J(2′,3′) = 4.8, 3J(1′,2′) = 5.6, 3J(2′,F3′) = 16.4, 2J(2′,F2′) = 49.6 Hz, 1H; H-2′), 5.51 (dm, 2J(3′,F3′) = 52.8 Hz, 1H; H-3′), 4.51 (dm, 3J(4′,F3′) = 24.4 Hz, 1H; H-4′), 3.27–3.24 (m, 2H; H-5′/5″); 13C NMR (100 MHz, CDCl3): δ 154.7, 152.3, 147.4, 144.6, 140.1, 129.1, 128.2, 127.3, 122.2, 90.4 (dd, 2J(C2′,F3′) = 12.3, 1J(C2′,F2′) = 185.0 Hz), 88.4 (dd, 2J(C3′,F2′) = 15.1, 1J(C3′,F3′) = 201.3 Hz), 87.5 (d, 2J(C1′,F2′) = 29.7 Hz), 82.9 (dd, 3J(C4′,F2′) = 2.5, 2J(C4′,F3′) = 24.5 Hz), 71.7, 44.5 (d, 3J(C5′,F3′) = 10.4 Hz); 19F NMR (376 MHz, [D6]DMSO): δ −202.6 (dddd, 3J(F2′,F3′) = 5.1, 3J(2′,F3′) = 16.6, 3J(4′,F3′) = 22.6, 2J(3′,F3′) = 53.1 Hz; F-3′), −213.9 (dm, 2J(2′,F2′) = 50.0 Hz; F-2′); HRMS (ESI+): m/z calcd for C29H27F2N7O3SNa [(M + Na)+] 614.1756, found 614.1779 (Δ 3.7 ppm).
General Procedure 4: Coupling and Deprotection
To a solution of sulfamide (1.0 mmol, 1.0 equiv) in DMF (20 mL) cooled to 0 °C was added NHS ester 44 (2.0 mmol, 2.0 equiv) followed by Cs2CO3 (3.0 mmol, 3 equiv) and reaction mixture was stirred at that temperature for 30 min. The reaction mixture was allowed to warm to rt and stirred for 15 h. The reaction mixture was filtered and evaporated under reduced pressure to obtain the crude coupled product. To the crude material was added 80% aqueous TFA (15 mL) and the mixture was stirred for 1 h. The reaction mixture was evaporated and re-evaporated with 5% MeOH in CH2Cl2 (×2) under reduced pressure. Purification of the residue by flash chromatography (84:15:1 EtOAc/MeOH/Et3N) afforded the product with some contamination. Additional purification was performed by preparative reverse-phase HPLC using a Phenomenex Gemini 10 μm C18 110Å (250 × 21.2 mm) column at a flow rate of 21 mL/min with a gradient from 5% to 30% MeCN in 20 mM aqueous triethylammonium bicarbonate (pH 7.5) over 5 min, followed by isocratic elution with 30% MeCN for 15 min. The appropriate fractions containing the product were pooled and lyophilized to afford the final product.
9-[2,5-Dideoxy-2-fluoro-5-{N-(N-2-hydroxybenzoyl)sulfamoyl}amino-β-d-ribofuranosyl]adenine Triethylammonium Salt (12)
This was prepared from sulfamide 43a (0.100 g, 0.141 mmol) using general procedure 4 to afford the title compound (0.033 g, 38%) as a white solid. 1H NMR (400 MHz, [D6]DMSO): δ 8.33, 8.14 (2s, 2H; H-2, H-8), 7.77 (dd, J = 1.8, 7.7 Hz; 1H), 7.34 (br s, 2H; C6-NH2), 7.22 (ddd, J = 1.9, 7.3, 8.3 Hz; 1H), 6.71 (m, 2H), 6.19 (dd, 3J(1′,2′) = 3.5, 3J(1′,F2′) = 17.0 Hz, 1H; H-1′), 6.12 (t, 3J(5′/5″,NH) = 7.0 Hz, 1H; 5′-NH), 5.76 (d, 3J(3′,OH) = 5.8 Hz, 1H; 3′-OH′), 5.51 (dt, 3J(2′,3′) ≈ 3J(1′,2′) = 4.0, 2J(2′,F2′) = 52.9 Hz, 1H; H-2′), 4.48 (dm, 3J(3′,F2′) = 16.1 Hz, 1H; H-3′), 4.04 (q, 3J(3′,4′) ≈ 3J(5′,4′) ≈ 3J(5″,4′) = 5.2 Hz, 1H; H-4′), 3.16 (ddd, 3J(5′,4′) = 3.8, 3J(5′, NH) = 6.5, 2J(5′, 5″) = 13.2 Hz, 1H; H-5′), 3.03 (ddd, 3J(5″,4′) = 5.9, 3J(5″,NH) = 7.6, 2J(5′, 5″) = 13.3 Hz, 1H; H-5″), 2.80 (q, J = 7.2 Hz, 6H, Et3N), 1.06 (t, J = 7.2 Hz, 9H, Et3N); 13C NMR (100 MHz, [D6]DMSO): δ 170.0, 160.5, 156.1, 152.8, 148.8, 139.7, 132.1, 129.4, 120.4, 119.2, 117.2, 116.5, 92.7 (d, 1J(C2′,F2′) = 187.9 Hz), 85.7 (d, 2J(C1′,F2′) = 32.8 Hz), 82.2, 69.7 (d, 2J(C3′,F2′) = 15.6 Hz), 45.8, 44.8, 9.1; 19F NMR (376 MHz, [D6]DMSO): δ −208.5 (dt, 3J(3′,F2′) = 16.3, 3J(1′,F2′) = 16.3, 2J(2′,F2′) = 52.9 Hz); HRMS (ESI−): m/z calcd for C17H17FN7O6S [(M – Et3NH)−] 466.0951, found 466.0988 (Δ −7.9 ppm).
9-[2,5-Dideoxy-2-fluoro-5-{N-(N-2-hydroxybenzoyl)sulfamoyl}amino-β-d-arabinofuranosyl]adenine Triethylammonium Salt (13)
This was prepared from sulfamide 43b (0.100 g, 0.141 mmol) using general procedure 4 to afford the title compound (0.033 g, 41%) as a white solid. 1H NMR (400 MHz, [D6]DMSO): δ 8.27 (d, 5J(8,F2′) = 2.4 Hz, 1H; H-8), 8.15 (s, 1H; H-2), 7.83 (dd, J = 1.8, 8.2 Hz; 1H), 7.35 (br s, 2H; C6-NH2), 7.23 (td, J = 1.9, 7.4, 7.5 Hz; 1H), 6.74–6.71 (m, 2H), 6.38 (dd, 3J(1′,2′) = 3.7, 3J(1′,F2′) = 19.0 Hz, 1H; H-1′), 5.98 (d, 3J(3′,OH) = 4.6 Hz, 1H; 3′-OH′), 5.91 (t, 3J(5′/5″,NH) = 6.8 Hz, 1H; 5′-NH), 5.06 (dt, 3J(2′,3′) = 2.9, 3J(1′,2′) = 2.9, 2J(2′,F2′) = 52.0 Hz, 1H; H-2′), 4.42 (dq, 3J(2′,3′) = 3.0, 3J(3′,4′) = 3.2, 3J(3′,OH) = 3.2, 3J(3′,F2′) = 16.4 Hz, 1H; H-3′), 4.01 (dt, 3J(3′,4′) = 4.2, 3J(5′,4′) = 4.3, 3J(5″,4′) = 7.9 Hz, 1H; H-4′), 3.15 (ddd, 3J(5′,4′) = 4.7, 3J(5′, NH) = 7.5, 2J(5′, 5″) = 12.4 Hz, 1H; H-5′), 3.04 (dt, 3J(5″,NH) = 6.8, 3J(5″,4′) = 6.8, 2J(5′, 5″) = 13.0 Hz, 1H; H-5″), 2.77 (q, J = 7.2 Hz, 6 H, Et3N), 1.05 (t, J = 7.2 Hz, 9 H, Et3N); 13C NMR (100 MHz, [D6]DMSO): δ 170.1, 160.5, 156.0, 152.8, 149.0, 139.6 (d, 4J(C8,F2′) = 4.2 Hz), 132.1, 129.4, 120.3, 118.2, 117.3, 116.5, 94.7 (d, 1J(C2′,F2′) = 191.0 Hz), 82.6 (d, 3J(C4′,F2′) = 3.0 Hz), 82.3 (d, 2J(C1′,F2′) = 16.5 Hz), 74.2 (d, 2J(C3′,F2′) = 23.7 Hz), 45.7, 44.9, 10.1; 19F NMR (376 MHz, [D6]DMSO): δ −197.6 ppm (dt, 3J(3′,F2′) = 17.7, 3J(1′,F2′) = 17.7, 2J(2′,F2′) = 51.9 Hz); HRMS (ESI+): m/z calcd for C17H18FN7O6SNa [(M – Et3N + Na)+] 490.0916, found 490.0905 (Δ −2.2 ppm).
9-[3,5-Dideoxy-3-fluoro-5-{N-(N-2-hydroxybenzoyl)sulfamoyl}amino-β-d-xylofuranosyl]adenine Triethylammonium Salt (14)
This was prepared from sulfamide 43c (0.48 g, 0.676 mmol) using general procedure 4 to afford the title compound (0.15 g, 39%) as a white solid. 1H NMR (400 MHz, [D6]DMSO): δ 8.15, 8.05 (2s, 2H; H-2, H-8), 7.81 (dd, J = 1.9, 8.0 Hz, 1H), 7.29 (br s, 2H; C6-NH2), 7.23 (td, J = 1.9, 7.4 Hz, 1H), 6.74–6.71 (m, 2H), 6.29 (d, 3J(2′,OH) = 4.6 Hz, 1H; 2′-OH), 5.95 (t, 3J(5′/5″,NH) = 6.7 Hz, 1H; 5′-NH), 5.90 (d, 3J(1′,2′) = 2.3 Hz, 1H; H-1′), 5.08 (ddd, 3J(3′,2′) = 1.3, 3J(3′,4′) = 2.8, 2J(3′,F3′) = 51.9 Hz, 1H; H-3′), 4.72 (dm, 3J(2′,F3′) = 16.1 Hz, 1H; H-2′), 4.34 (dtd, 3J(3′,4′) = 3.1, 3J(5′,4′) = 6.2, 3J(5″,4′) = 6.6, 3J(4′,F3′) = 29.1 Hz, 1H; H-4′), 3.26–3.21 (m, 1H; H-5′), 3.13 (dt, 3J(5″,NH) = 6.7, 3J(5″,4′) = 6.7, 2J(5′,5″) = 13.2 Hz, 1H; H-5″), 2.99 (q, J = 7.2 Hz, 6 H, Et3N), 1.13 (t, J = 7.2 Hz, 9 H, Et3N); 13C NMR (100 MHz, [D6]DMSO): δ 170.2, 160.5, 156.0, 152.7, 149.1, 138.2 (d, 5J(C8,F3′) = 5.4 Hz), 132.1, 129.4, 120.3, 118.8, 117.2, 116.5, 95.7 (d, 1J(C3′,F3′) = 183.9 Hz), 88.9, 80.2 (d, 2J(C2′,F3′) = 18.9 Hz), 77.6 (d, 2J(C4′,F3′) = 26.5 Hz), 45.7, 41.5 (d, 3J(C5′,F3′) = 10.1 Hz), 9.2; 19F NMR (376 MHz, [D6]DMSO): δ −200.2 (ddd, 3J(2′,F3′) = 15.2, 3J(4′,F3′) = 28.9, 2J(3′,F3′) = 51.6 Hz); HRMS (ESI+): m/z calcd for C17H18FN7O6SNa [(M – Et3N + Na)+] 490.0916, found 490.0898 (Δ −3.7 ppm).
9-[3,5-Dideoxy-3-fluoro-5-{N-(N-2-hydroxybenzoyl)sulfamoyl}amino-β-d-ribofuranosyl]adenine Triethylammonium Salt (15)
This was prepared from sulfamide 43d (0.23 g, 0.324 mmol) using general procedure 4 to afford the title compound (0.08 g, 45%) as a white solid. 1H NMR (400 MHz, [D6]DMSO): δ 8.37, 8.17 (2s, 2H; H-2, H-8), 7.78 (dd, J = 1.8, 7.7 Hz; 1H), 7.32 (br s, 2H; C6-NH2), 7.21 (ddd, J = 1.9, 7.2, 8.2 Hz;1H), 6.77 (t, J = 7.0 Hz, 1H; 5′-NH), 6.70 (td, J = 1.2, 9.3 Hz; 2H), 5.90 (d, 3J(2′,OH) = 5.7 Hz, 1H; 2′-OH), 5.88 (d, 3J(1′,2′) = 8.1 Hz, 1H; H-1′), 5.10 (dd, 3J(3′,2′) = 4.1, 2J(3′,F3′) = 55.6 Hz, 1H; H-3′), 5.08 (dd, 3J(1′,2′) = 8.5, 3J(2′,F3′) = 26.0 Hz, 1H; H-2′), 4.34 (ddd, 3J(5′,4′) = 4.1, 3J(5″,4′) = 5.1, 3J(4′,F3′) = 27.8 Hz, 1H; H-4′), 3.13–3.10 (m, 2H, H-5′/5″), 2.91 (q, J = 7.2 Hz, 7.8 H, Et3N), 1.10 (t, J = 7.2 Hz, 11.7 H, Et3N); 13C NMR (100 MHz, [D6]DMSO): δ 169.9, 160.6, 156.1, 152.7, 149.3, 140.4, 132.0, 129.4, 120.5, 119.4, 117.1, 116.4, 93.2 (d, 1J(C3′,F3′) = 181.2 Hz), 86.6, 82.0 (d, 2J(C2′,F3′) = 22.7 Hz), 70.9 (d, 2J(C4′,F3′) = 16.4 Hz), 45.7, 44.3 (d, 3J(C5′,F3′) = 11.3 Hz), 9.4; 19F NMR (376 MHz, [D6]DMSO): δ −197.0 (dt, 3J(2′,F3′) = 27.4, 3J(4′,F3′) = 27.4, 2J(3′,F3′) = 55.3 Hz); HRMS (ESI+): m/z calcd for C17H18FN7O6SNa [(M – Et3N + Na)+] 490.0916, found 490.0939 (Δ 4.7 ppm).
9-[2,3,5-Trideoxy-2,3-difluoro-5-{N-(N-2-hydroxybenzoyl)sulfamoyl}amino-β-d-xylofuranosyl]adenine Triethylammonium Salt (16)
This was prepared from sulfamide 43e (0.360 g, 0.608 mmol) using general procedure 4 to afford the title compound (0.141 g, 50%) as a white solid. 1H NMR (400 MHz, [D6]DMSO): δ 8.15, 8.07 (2s, 2H; H-2, H-8), 7.79 (dd, J = 1.9, 8.1 Hz, 1H), 7.35 (br s, 2H; C6-NH2), 7.22 (ddd, J = 2.0, 8.0, 9.2 Hz, 1H), 6.73–6.69 (m, 2H), 6.24 (dd, 3J(1′,2′) = 2.1, 3J(1′,F2′) = 19.4 Hz, 1H; H-1′), 5.99 (t, J = 6.7 Hz, 1H; C5′-NH), 5.87 (ddt, 3J(2′,3′) = 1.6, 3J(1′,2′) = 1.7, 3J(2′,F3′) = 12.9, 2J(2′,F2′) = 47.7 Hz, 1H; H-2′), 5.52 (dddd, 3J(2′,3′) = 1.4, 3J(3′,4′) = 3.2, 3J(3′,F2′) = 10.2, 2J(3′,F3′) = 49.6 Hz, 1H; H-3′), 4.52 (dm, 3J(4′,F3′) = 28.9 Hz, 1H; H-4′), 3.28 (ddd, 3J(5′,4′) = 5.8, 3J(5′,NH) = 7.4, 2J(5′,5″) = 13.2 Hz, 1H; H-5′), 3.18–3.12 (m, 1H; H-5″) 2.92 (q, J = 7.2 Hz, 6 H, Et3N), 1.10 (t, J = 7.2 Hz, 9 H, Et3N); 13C NMR (100 MHz, [D6]DMSO): δ 170.2, 160.5, 156.1, 152.9, 149.0, 138.0 (d, J = 4.3 Hz), 132.2, 129.4, 120.3, 128.8, 127.3, 116.5, 95.7 (dd, 2J(C2′,F3′) = 32.0, 1J(C2′,F2′) = 182.9 Hz), 92.5 (dd, 2J(C3′,F2′) = 29.6, 1J(C3′,F3′) = 182.7 Hz), 86.1 (d, 2J(C1′,F2′) = 35.8 Hz), 80.2 (d, 2J(C4′,F3′) = 19.3 Hz), 45.7, 41.2 (d, 3J(C5′,F3′) = 9.9 Hz), 9.2; 19F NMR (376 MHz, [D6]DMSO): δ −193.82 (ddtd, 4J(4′,F2′) = 2.6, 3J(F2′,F3′) = 10.3, 3J(3′,F2′) = 10.3, 3J(1′,F2′) = 18.8, 2J(2′,F2′) = 47.7 Hz; F-2′), −206.98 (ddt, 3J(F2′,F3′) = 11.6, 3J(2′,F3′) = 11.6, 3J(4′,F3′) = 28.9, 2J(3′,F3′) = 50.9 Hz; F-3′); HRMS (ESI+): m/z calcd for C17H17F2N7O5SNa [(M – Et3N + Na)+] 492.0872, found 492.0893 (Δ 4.2 ppm).
9-[2,3,5-Trideoxy-2,3-difluoro-5-{N-(N-2-hydroxybenzoyl)sulfamoyl}amino-β-d-ribofuranosyl]adenine Triethylammonium Salt (17)
This was prepared from sulfamide 43f (0.240 g, 0.406 mmol) using general procedure 4 to afford the title compound (0.097 g, 44%) as a white solid. 1H NMR (400 MHz, [D6]acetone): δ 8.34, 8.30 (2s, 2H; H-2, H-8), 7.92 (dd, J = 1.8, 7.8 Hz, 1H), 7.22 (ddd, J = 1.8, 7.1, 8.2 Hz, 1H), 6.75–6.70 (m, 2H), 6.33 (ddd, 4J(1′,F3′) = 1.4, 3J(1′,2′) = 6.6, 3J(1′,F2′) = 12.3 Hz, 1H; H-1′), 6.13 (dddd, 3J(2′,3′) = 4.4, 3J(1′,2′) = 6.6, 3J(2′,F3′) = 18.0, 2J(2′,F2′) = 50.3 Hz, 1H; H-2′), 5.57 (dm, 2J(3′,F3′) = 53.6 Hz, 1H; H-3′), 4.58 (dm, 3J(4′,F3′) = 25.8 Hz, 1H; H-4′), 3.42 (dd, 3J(5′,4′) = 4.1, 2J(5′,5″) = 13.9 Hz, 1H; H-5′), 3.35 (dd, 3J(5″,4′) = 4.4, 2J(5′,5″) = 13.9 Hz, 1H; H-5″), 2.89 (q, J = 7.2 Hz, 6H, Et3N), 1.15 (t, J = 7.2 Hz, 9H, Et3N); 1H NMR (400 MHz, [D6]DMSO): δ 14.02 (s, 1H), 8.41, 8.17 (2s, 2H; H-2, H-8), 7.78 (dd, J = 1.8, 7.7 Hz, 1H), 7.41 (br s, 2H; C6-NH2), 7.22 (ddd, J = 1.9, 7.3, 8.5 Hz, 1H), 6.73–6.69 (m, 2H), 6.58 (t, J = 7.0 Hz, 1H; C5′-NH), 6.30 (ddd, 4J(1′,F3′) = 1.6, 3J(1′,2′) = 6.7, 3J(1′,F2′) = 12.6 Hz, 1H; H-1′), 6.07 (dddd, 3J(2′,3′) = 4.2, 3J(1′,2′) = 6.5, 3J(2′,F3′) = 18.2, 2J(2′,F2′) = 50.0 Hz, 1H; H-2′), 5.52 (dtd, 3J(3′,4′) = 2.1, 3J(3′,F2′) = 3.9, 3J(2′,3′) = 3.9, 2J(3′,F3′) = 53.8 Hz, 1H; H-3′), 4.45 (dtt, 4J(4′,F2′) = 2.3, 3J(3′,4′) = 2.3, 3J(5′,4′) = 4.5, 3J(5″,4′) = 4.5, 3J(4′,F3′) = 24.9 Hz, 1H; H-4′), 3.18–3.14 (m, 2H; H-5′/5″) 3.05 (q, J = 7.3 Hz, 6H, Et3N), 1.15 (t, J = 7.3 Hz, 9H, Et3N); 13C NMR (100 MHz, [D6]DMSO): δ 170.0, 160.6, 156.2, 153.0, 149.0, 140.0, 132.1, 129.4, 120.4, 119.2, 117.2, 116.5, 90.03 (dd, 2J(C3′,F2′) = 13.4, 1J(C3′,F3′) = 184.0 Hz), 89.2 (dd, 2J(C2′,F3′) = 14.7, 1J(C2′,F2′) = 194.4 Hz), 84.3 (d, 2J(C1′,F2′) = 29.9 Hz), 81.3 (dd, 3J(C4′,F2′) = 3.4, 2J(C4′,F3′) = 22.4 Hz), 45.8, 43.9 (d, 3J(C5′,F3′) = 9.0 Hz), 8.8; 19F NMR (376 MHz, [D6]DMSO): δ −202.03 (dddd, 3J(F2′,F3′) = 5.5, 3J(2′,F3′) = 18.2, 3J(4′,F3′) = 24.4 Hz, 2J(3′,F3′) = 54.0 Hz; F-3′), −215.45 (dddt, 4J(4′,F2′) = 3.1, 3J(3′,F2′) = 3.1, 3J(F2′,F3′) = 5.7, 3J(1′,F2′) = 12.0, 1J(2′,F2′) = 49.9 Hz; F-2′); HRMS (ESI+): m/z calcd for C17H17F2N7O5SNa [(M – Et3N + Na)+] 492.0872, found 492.0899 (Δ 5.4 ppm).
Supplementary Material
Figure 3.

Mean plasma concentration versus time curves after single p.o. (25 mg.kg−1) and i.v. (2.5 mg.kg−1) bolus administration of 10 and 12-17 to rats. Error bars represent standard deviation of the mean (n = 3).
ACKNOWLEDGMENT
We dedicate this manuscript in memory of the late Professor Kyo Watanabe for his pioneering work in nucleoside chemistry and fluorinated nucleosides. This work was supported by a grant from the NIH (AI070219 to C.C.A.) and the Intramural Research Program of the NIAID, NIH (C.E.B.).
Footnotes
Supporting Information: 1H, 13C, and 19F NMR spectra for all compounds, HPLC traces and conformational analysis of 12–17, and a complete description of the rat pharmacokinetic studies, MbtA inhibition assay, and M. tuberculosis MIC assay. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.Schaible UE, Kaufmann SH. E. Nat. Rev. Microbiol. 2004;2:946. doi: 10.1038/nrmicro1046. [DOI] [PubMed] [Google Scholar]
- 2.Miethke M, Marahiel MA. Microbiol. Mol. Biol. Rev. 2007;71:413. doi: 10.1128/MMBR.00012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a Quadri LE, Sello J, Keating TA, Weinreb PH, Walsh CT. Chem. Biol. 1998;5:631. doi: 10.1016/s1074-5521(98)90291-5. [DOI] [PubMed] [Google Scholar]; b De Voss JJ, Rutter K, Schroeder BG, Su H, Zhu Y, Barry CE., 3rd Proc. Natl. Acad. Sci. USA. 2000;97:1252. doi: 10.1073/pnas.97.3.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Reddy PV, Puri RV, Chauhan P, Kar R, Rohilla A, Khera A, Tyagi AK. J. Infect. Dis. 2013;208:1255. doi: 10.1093/infdis/jit250. [DOI] [PubMed] [Google Scholar]
- 4.a Ferreras JA, Ryu JS, Lello FD, Tan DS, Quadri LEN. Nat. Chem. Biol. 2005;1:29. doi: 10.1038/nchembio706. [DOI] [PubMed] [Google Scholar]; b Miethke M, Bisseret P, Beckering CL, Vignard D, Eustache J, Marahiel MA. FEBS J. 2006;273:409. doi: 10.1111/j.1742-4658.2005.05077.x. [DOI] [PubMed] [Google Scholar]; c Somu RV, Boshoff H, Qiao C, Bennett EM, Barry CE, III, Aldrich CC. J. Med. Chem. 2006;49:31. doi: 10.1021/jm051060o. [DOI] [PubMed] [Google Scholar]; d Engelhart CA, Aldrich CC. J. Org. Chem. 2013;78:7470. doi: 10.1021/jo400976f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duckworth BP, Nelson KM, Aldrich CC. Curr. Top. Med. Chem. 2012;12:766. doi: 10.2174/156802612799984571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duckworth BP, Wilson DJ, Nelson KM, Boshoff H, Barry CE, III, Aldrich CC. ACS Chem. Biol. 2012;7:1653. doi: 10.1021/cb300112x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qiao C, Gupte A, Boshoff HI, Wilson DJ, Bennett EM, Somu RV, Barry CE, III, Aldrich CC. J. Med. Chem. 2007;50:6080. doi: 10.1021/jm070905o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lun S, Guo H, Adamson J, Cisar JS, Davis TD, Chavadi SS, Warren JD, Quadri LEN, Tan DS, Bishai WR. Antimicrob. Agents Chemother. 2013;57:5138. doi: 10.1128/AAC.00918-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Codington JF, Doerr I, Praag DV, Bendich A, Fox JJ. J. Am. Chem. Soc. 1961;83:5030. [Google Scholar]
- 10.Thibaudeau C, Plavec J, Chattopadhyaya JJ. Org. Chem. 1998;63:4967. [Google Scholar]
- 11.Barchi JJ, Jr., Jeong L-S, Siddiqui SA, Marquez VE. J. Biochem. Biophys. Methods. 1997;34:11. doi: 10.1016/s0165-022x(96)00032-2. [DOI] [PubMed] [Google Scholar]
- 12.a Lee K, Choi Y, Gumina G, Zhou W, Schinazi RF, Chu CK. J. Med. Chem. 2002;45:1313. doi: 10.1021/jm010418n. [DOI] [PubMed] [Google Scholar]; b Chong Y, Gumina G, Mathew JS, Schinazi RF, Chu CK. J. Med. Chem. 2003;46:3245. doi: 10.1021/jm0300274. [DOI] [PubMed] [Google Scholar]; c Zhou W, Gumina G, Chong Y, Wang J, Schinazi RF, Chu CK. J. Med. Chem. 2004;47:3399. doi: 10.1021/jm040027j. [DOI] [PubMed] [Google Scholar]
- 13.Biffinger JC, Kim HW, DiMagno SG. ChemBioChem. 2004;5:622. doi: 10.1002/cbic.200300910. [DOI] [PubMed] [Google Scholar]
- 14.a May JJ, Kessler N, Marahiel MA, Stubbs MT. Proc. Natl. Acad. Sci. USA. 2002;99:12120. doi: 10.1073/pnas.182156699. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Drake EJ, Duckworth BP, Neres J, Aldrich CC, Gulick AM. Biochemistry. 2010;49:9292. doi: 10.1021/bi101226n. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Neres J, Engelhart CA, Drake EJ, Wilson DJ, Fu P, Boshoff HI, Barry CE, III, Gulick AM, Aldrich CC. J. Med. Chem. 2013;56:2385. doi: 10.1021/jm301709s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.For reviews on the synthesis of fluorinated nucleosides, see: Pankiewicz KW. Carbohydr. Res. 2000;327:87. doi: 10.1016/s0008-6215(00)00089-6. Qiu X-L, Xu X-H, Qing F-L. Tetrahedron. 2010;66:789. Liu P, Sharon A, Chu CK. Journal of Fluorine Chemistry. 2008;129:743. doi: 10.1016/j.jfluchem.2008.06.007.
- 16.a Takaku H, Kamaike K. Chem. Lett. 1982;11:189. [Google Scholar]; b Benito D, Matheu MI, Morère A, Díaz Y, Castillón S. Tetrahedron. 2008;64:10906. [Google Scholar]
- 17.Herdewijn P, Aerschot AV, Kerremans L. Nucleosides Nucleotides. 1989;8:65. [Google Scholar]
- 18.Altona C, Sundaralingam MJ. Am. Chem. Soc. 1973;95:2333. doi: 10.1021/ja00788a038. [DOI] [PubMed] [Google Scholar]
- 19.Robins MJ, Samano V, Johnson MD. J. Org. Chem. 1990;55:410. [Google Scholar]
- 20.Pankiewicz KW, Krzeminski J, Ciszewski LA, Ren W-Y, Watanabe KA. J. Org. Chem. 1992;57:553. [Google Scholar]
- 21.Charafeddine A, Dayoub W, Chapuis H, Strazewski P. Chem. Eur. J. 2007;13:5566. doi: 10.1002/chem.200700058. [DOI] [PubMed] [Google Scholar]
- 22.Huang J-T, Chen L-C, Wang L, Kim M-H, Warshaw JA, Armstrong D, Zhu Q-Y, Chou T-C, Watanabe KA, Matulic-Adamic J, Su T-L, Fox JJ, Polsky B, Baron PA, Gold JWM, Hardy WD, Zuckerman EJ. Med. Chem. 1991;34:1640. doi: 10.1021/jm00109a017. [DOI] [PubMed] [Google Scholar]
- 23.Robins MJ, Fouron Y, Mengel R. J. Org. Chem. 1974;39:1564. doi: 10.1021/jo00924a025. [DOI] [PubMed] [Google Scholar]
- 24.Chu CK, Matulic-Adamic J, Huang J-T, Chu T-C, Burchenal JH, Fox JJ, Watanabe KA. Chem. Pharm. Bull. 1989;37:336. doi: 10.1248/cpb.37.336. [DOI] [PubMed] [Google Scholar]
- 25.Mikhailopulo IA, Poopeiko NE, Pricota TI, Sivets GG, Kvasyuk EI, Balzarini J, De Clercq E. J. Med. Chem. 1991;34:2195. doi: 10.1021/jm00111a040. [DOI] [PubMed] [Google Scholar]
- 26.a Mikhailopulo IA, Pricota TI, Sivets GG, Altona C. J. Org. Chem. 2003;68:5897. doi: 10.1021/jo0340859. [DOI] [PubMed] [Google Scholar]; b Sivets GG, Kalinichenko EE, Mikhailopulo IA. Helvetica Chimica Acta. 2007;90:1818. [Google Scholar]
- 27.Maltese M. J. Org. Chem. 2001;66:7615. doi: 10.1021/jo0156971. [DOI] [PubMed] [Google Scholar]
- 28.Liu F, Austin DJ. J. Org. Chem. 2001;66:8643. doi: 10.1021/jo015800m. [DOI] [PubMed] [Google Scholar]
- 29.a Takai H, Obase H, Nakamizo N, Teranishi M, Kubo K, Shuto K, Hashimoto T. Chem. Pharm. Bull. 1985;33:1104. doi: 10.1248/cpb.33.1104. [DOI] [PubMed] [Google Scholar]; b Huie EM, Kirshenbaum MR, Trainor GL. J. Org. Chem. 1992;57:4569. [Google Scholar]; c Lu X, Zhang H, Tonge PJ, Tan DS. Bioorg. Med. Chem. Lett. 2008;18:5963. doi: 10.1016/j.bmcl.2008.07.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neres J, Labello NP, Somu RV, Boshoff HI, Wilson DJ, Vannada J, Chen L, Barry CE, III, Bennett EM, Aldrich CC. J. Med. Chem. 2008;51:5349. doi: 10.1021/jm800567v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hendrickx PMS, Martins JC. Chem. Cent. J. 2008;2:20. doi: 10.1186/1752-153X-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barchi JJ, Jr., Karki RG, Nicklaus MC, Siddiqui SA, George C, Mikhailopulo IA, Marquez VE. J. Am. Chem. Soc. 2008;130:9048. doi: 10.1021/ja800964g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.a Uesugi S, Miki H, Ikehara M, Iwahashi H, Kyogoku Y. Tetrahedron Lett. 1979;42:4073. [Google Scholar]; b Ford H, Jr., Dai F, Mu L, Siddiqui MA, Nicklaus MC, Anderson L, Marquez VE, Barchi J., Jr. J. Biochemistry. 2000;39:2581. doi: 10.1021/bi992112c. [DOI] [PubMed] [Google Scholar]
- 34.Nelson KM, Viswanathan K, Dawadi S, Duckworth BP, Boshoff HI, Barry CE, 3rd, Aldrich CC. J. Med. Chem. doi: 10.1021/acs.jmedchem.5b00391. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.a Schmelz S, Naismith JH. Curr. Opin. Struct. Biol. 2009;19:666. doi: 10.1016/j.sbi.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gulick AM. ACS Chem. Biol. 2009;4:811. doi: 10.1021/cb900156h. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hedberg C, Itzen A. ACS Chem. Biol. 2015;10:12. doi: 10.1021/cb500854e. [DOI] [PubMed] [Google Scholar]
- 36.Borda EJ, Sigurdsson S. Th. Nucl. Acids Res. 2005;33(3):1058. doi: 10.1093/nar/gki237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.