

Abstract
Currently, no targeted drug is available for triple-negative breast cancer (TNBC), an aggressive breast cancer that does not express estrogen receptor, progesterone receptor, or HER2. TNBC has high mitotic activity, and since Aurora A and B mitotic kinases drive cell division and are overexpressed in tumors with a high mitotic index, we hypothesized that inhibiting Aurora A and B produces a significant antitumor effect in TNBC. We tested this hypothesis by determining the antitumor effects of KW-2450, a multikinase inhibitor of both Aurora A and B kinases. We observed significant inhibitory activities of KW-2450 on cell viability, apoptosis, colony formation in agar, and mammosphere formation in TNBC cells. The growth of TNBC xenografts was significantly inhibited with KW-2450. In cell cycle analysis, KW-2450 induced tetraploid accumulation followed by apoptosis or surviving octaploid (8N) cells, depending on dose. These phenotypes resembled those of Aurora B knockdown and complete pharmaceutical inhibition of Aurora A. We demonstrated that 8N cells resulting from KW-2450 treatment depended on the activation of mitogen-activated protein kinase kinase (MEK) for their survival. When treated with the MEK inhibitor selumetinib combined with KW-2450, compared with KW-2450 alone, the 8N cell population was significantly reduced and apoptosis was increased. Indeed this combination showed synergistic antitumor effect in SUM149 TNBC xenografts. Collectively, Aurora A and B inhibition had a significant antitumor effect against TNBC, and this antitumor effect was maximized by the combination of selumetinib with Aurora A and B inhibition.
Keywords: KW-2450, triple-negative breast cancer, Aurora A, Aurora B, apoptosis
Introduction
Triple-negative breast cancer (TNBC), which is negative for estrogen receptor (ER), progesterone receptor, and HER2, is clinically and biologically distinct from the other breast cancer subtypes (e.g., ER+, HER2+ breast cancer) (1, 2). From a clinical standpoint, TNBC is one of the most aggressive forms of breast cancer because of its early relapse behavior, high propensity for metastasis, and refractoriness to multiple anticancer drugs. Patients with TNBC cannot be treated with anti-estrogen agents (e.g., tamoxifen, aromatase inhibitors) or anti-HER2 agents (e.g., trastuzumab) because of the lack of expression of ER, progesterone receptor, and HER2. Currently, no molecularly targeted drug is available that has shown a benefit in TNBC (3); therefore, such a drug needs to be developed.
Aurora A and Aurora B are mitosis-related serine/threonine kinases that are overexpressed or genetically amplified in a significant percentage of various tumor types including TNBC (4, 5). A growing number of studies support the development of Aurora A/B inhibitors as anticancer drugs (6–8). Physiologically, Aurora A plays a critical role in centromere duplication and maturation throughout the late G2 to mitotic phases; therefore, Aurora A inhibition delays initiation of mitosis, which in turn causes mitotic accumulation and activates the spindle assembly checkpoint (SAC) (4, 9). In contrast, Aurora B secures the integrity of chromosome segregation at the SAC during metaphase. Between anaphase and telophase, Aurora B executes cytokinesis. Therefore, Aurora B inhibition causes inactivation of the SAC and accelerates mitotic-slippage by destabilizing the cytokinetic process (10–12). Thus, either Aurora A or Aurora B inhibition could evoke cell death at the SAC or after mitotic-slippage (e.g., at the postmitotic G1 checkpoint), respectively, in cancer cells (13, 14).
We previously revealed the tumor suppressive functions of the SAC and postmitotic G1 checkpoint by using cancer cell lines and a transgenic mouse tumor model (15–17). In cultured cell line models, we showed that the SAC was indispensable to induce apoptosis of MCF7 cells with paclitaxel and that the SAC functioned independently from the G2 checkpoint (15); we further showed that the SAC induced mitotic cell death in p53-null HCT116 colon cancer cells treated with cisplatin (16). With use of a mouse mammary pre-neoplastic model, we showed that perturbation of Aurora A function (i.e., overexpressed Aurora A) activated the postmitotic G1 checkpoint and inhibited the transformation to lethal tumors (17). On the basis of these experimental findings, in addition to the clinical significance of Aurora A/B kinases (4–6), we reasoned that we could develop an Aurora A/B inhibitor against TNBC, exploiting the SAC and/or postmitotic G1 checkpoint of TNBC cells. To this end, in the present study we used KW-2450, a multikinase inhibitor that exhibits more robust inhibitory activities against Aurora A and Aurora B, and tested its antitumor effects in vitro and in vivo (18).
We here show that KW-2450 induced cell death in vitro and produced antitumor effects in vivo in a type of TNBC cells (i.e., MDA-MB-468). In contrast, other TNBC cells (i.e., MDA-MB-231, SUM149 cells) were relatively resistant to KW-2450–induced cell death, escaping from the SAC and postmitotic G1 checkpoints, and interestingly entered into the octaploid (8N) phase. We were able to attribute these phenotypes to the inhibition of Aurora A and B. We further discovered that survival of the 8N cells depended on the activation of the mitogen-activated protein kinase kinase (MEK) pathway and that these cells were therefore killed when treated with the MEK inhibitor selumetinib combined with KW-2450. We here propose Aurora A/B inhibition and Aurora A/B inhibition combined with MEK inhibition as promising therapeutic approaches in TNBC.
Materials and Methods
Cell lines
A panel of 11 phenotypically diverse human breast cancer cell lines (SUM149, SUM159, SUM190, MDA-MB-468, MDA-MB231, MCF7, KPL-4, BT-549, HCC70, T47D, and ZR75-1) and HCT116 colon cancer cell lines (which have either p53+/+ or p53−/− genotype) were used. SUM149, SUM159, and SUM190 cells were maintained in Ham’s F12 with 5% FBS, 1X Antibiotic-Antimycotic (AA), 1 μg/mL hydrocortisone, and 5 μg/mL insulin. The remaining cells were maintained in culture media as follows: MDA-MB-468, MDA-MB231, MCF7, and KPL-4 cells in Dulbecco’s Modified Eagle’s Medium/Nutrient Mixture F-12 (F12); BT-549, HCC70, T47D and ZR75-1 cells in RPMI 1640 medium; HCT116 p53+/+ and HCT116 p53−/− colon cancer cells in McCoy’s 5A medium; all media were supplemented with 10% FBS and 1X AA. All materials were provided by Life Technologies (Grand Island, NY).
SUM149, SUM159, and SUM190 were obtained from Asterand (Detroid, MI) in 2011, 2012, and 2011; and authenticated in 2014, 2013, and 2014, respectively. MDA-MB-231, MDA-MB-468, and HCC70 were all obtained from American Type Culture Collection (ATCC) in 2008 and authenticated in 2014. MCF-7 was obtained from ATCC in 2008 and authenticated in 2010. BT-549, T47D and ZR75-1 were all obtained from ATCC in 2008 but have not been authenticated yet. KPL4 was kindly provided by J. Kurebayashi in 2008 but not authenticated yet. HCT116 p53+/+ and HCT116 p53−/− were kindly provided by Dr. G. A. Calin (MD Anderson, Houston TX) under the material transfer agreement between Dr. B. Vogelstein (Ludwig Center at Johns Hopkins, Baltimore MD) and N.T. Ueno in 2013 but not authenticated yet. All authentications were validated by the Characterized Cell Line Core Facility at MD Anderson Cancer Center by using a short tandem repeat method. For all cell lines, TP53 mutation status is available in Supplementary Table S1.
Drugs
KW-2450 was provided by Kyowa Hakko Kirin Co., Ltd. (Tokyo, Japan). Selumetinib was purchased from ChemieTek (Indianapolis, IN). Paclitaxel was purchased from the core facility for experimental supplies at The University of Texas MD Anderson Cancer Center.
Western blot analysis
Cell pellets were lysed as described previously (19). Primary antibodies that we used in this study were rabbit anti-phospho Aurora A (T288), rabbit anti-insulin-like growth factor-I receptor (IGF- IR), rabbit anti-insulin receptor (InsR) β, rabbit anti-phospho-IGF-IR/InsR kinase (Thr202/Tyr204), rabbit anti–extracellular signal-regulated protein kinases (ERKs), rabbit anti–phospho-ERKs, rabbit anti–c-Jun N-terminal kinases (JNKs), rabbit anti–phospho-JNKs, rabbit anti–p38, and rabbit anti–phospho-p38 (all from Cell Signaling Technology, Beverly, MA); and mouse anti-Aurora A (BD Biosciences, San Jose, CA), rabbit anti-cyclin B1 (Santa Cruz Biotechnology, Dallas, TX), and mouse anti–β-actin Ab (diluted at 1: 5000; Sigma-Aldrich, St. Louis, MO). Antibodies were diluted at a ratio of 1:1000, unless noted. Signals were detected with use of an Odyssey IR imaging system (LI-COR, Lincoln, NE).
Immunofluorescence analysis and Aurora A assay
MDA-MB-468 cells were grown on 4-chamber slides (BD Biosciences, San Jose, CA) and treated with DMSO or KW-2450. For immunofluorescent staining, cells were stained with anti-Aurora A pT288 rabbit antibody (1:250) and anti-Aurora A kinase mouse monoclonal antibody (1:250), followed by anti-rabbit Alexa Fluor 488 and anti-mouse Alexa Fluor 594 secondary antibodies. DNA was counterstained with DAPI (VECTA STAIN). Fluorescently labeled cells were visualized with a Nikon FV 1000 fluorescent microscope, and images were captured with a digital camera (Nikon). For the Aurora A assay, kinase activities of Aurora A were determined by measuring pT288 fluorescent intensity (Alexa Fluor 488) in the mitotic cells that had bipolar Aurora A spots and presented as the value relative to Aurora A fluorescent intensity (Alexa Fluor 594). A concentration–inhibition curve was generated by calculating the Aurora A kinase activities in KW-2450–treated samples relative to the DMSO-treated control, and the IC50 for inhibiting Aurora A kinase activity was determined by Prism5 software.
Cell-cycle, mitosis, and Aurora B assays
Cells were plated in 6-cm dishes, cultured for 1–2 days, and then treated with DMSO, KW-2450, or KW-2450 plus selumetinib. In each experiment, DMSO-treated cells were used as the control. Cell-cycle distribution was analyzed by a fluorescence-activated cell sorter (FACS) as described previously (19, 20). For mitotic analysis, cells were stained with anti-phospho-histone H3 (pHisH3) conjugated with FITC, incubated with RNase (100 μg/mL), and stained with PI (25 μg/mL). For the Aurora B assay, 4N pHisH3+ (FITC+) cells were quantified by FACS to measure the kinase activities of Aurora B in KW-2450 treatments (because histone H3 is a kinase substrate of Aurora B). A concentration–inhibition curve was generated, and the IC50 for inhibiting Aurora B kinase activity was determined as in the Aurora A assay.
Growth inhibition assay in vitro
Cell viability was determined by a CellTiter-Blue Cell Viability Assay (Promega Corporation, Madison, WI), which was performed according to the manufacturer’s instructions. Briefly, 1–2 days before treatment, cells were seeded into a 96-well plate at 2,000 cells per well. Cells were then treated with KW-2450 or selumetinib as a single or combinatory treatment for 72 hours. The viability of untreated cells was set as 100% in each experiment, and the percentage of viability in each drug concentration was calculated in reference to the untreated cells. The values of percent inhibition and corresponding drug concentrations were incorporated into the Prism5 software, and IC50s were then calculated according to a sigmoidal algorithm.
siRNA transfection
MDA-MB-468 cells were transfected with a SMART pool (20 nM final) of On-Target plus siRNAs (Thermo Scientific) to Aurora A, Aurora B, IGF-IR, or InsR with use of Lipofectamine RNAi Max reagent (Invitrogen). On-Target plus Non-Targeting siRNAs Pool was used as control siRNAs.
Annexin V assay
An Annexin V: PE Apoptosis Detection Kit 1 was purchased from Life Technologies, and procedures were followed according to the manufacturer’s instructions. We treated cells with KW-2450 or DMSO, collected floating cells in supernatants, and attached cells by trypsinization. After cells were washed with PBS, they were stained with anti-Annexin V conjugated with PE and then analyzed by FACS.
Reactive-oxygen species (ROS) assay
A CellROX Green Reagent Kit for oxidative stress detection was purchased from Life Technologies, and procedures were followed according to the manufacturer’s instructions. TNBC cells were treated with KW-2450 or DMSO for 2 days and then incubated with CellROX Green Reagent (5 μM final concentration) for 30 minutes. After the cells were incubated, we collected floating cells in supernatants and attached cells by trypsinization. After cells were washed with PBS, they were resuspended with PBS, stained with PI (0.5 μg/mL final concentration), and analyzed by FACS. To separate live cells from dead cells, PI+ cells and PI- cells were gated, respectively, and PI- cells (live cells) were used to determine ROS levels in living cells.
CD44+CD24− cancer stem cell assay
SUM149 and MDA-MB-231 cells were plated in 60-mm dishes, cultured for 1–2 days, and then treated with KW-2450 or DMSO. After 48 hours, floating and adherent cells were collected by trypsinization, washed with PBS, stained with anti-CD44-FITC and anti-CD24-PE (both antibodies were from Life Technologies), and then analyzed by FACS. Cellular aggregates were depleted by a proper gating strategy, and singlets were analyzed to determine the percentages of CD44+CD24− and non-CD44+CD24− fractions by using Flowjo software.
Colony formation in agar assay
Colony formation in the agar assay was performed as previously described (21). Briefly, TNBC cells (2 × 103 per well) were compounded in 0.4% agar media, which contained KW-2450 or DMSO, and plated over a base layer of 0.8% agarose media in 6-well plates. After 3 weeks of incubation, plates were stained with p-iodonitrotetrazolium violet (1 mg/mL) for 24 hours at 37°C, and colony numbers were counted by using GelCount (OXFORD OPTRONIX, United Kingdom).
Mammosphere-forming assay
The mammosphere-forming assay was performed as described previously (22). Cells were plated into 6-well low-attachment dishes at a density of 2,000 cells/mL. KW-2450 or DMSO was added when cells were plated. Cells were cultured for 4 or 5 days and then incubated with MTT (50–100 μg/mL) for 1 hour before the mammospheres were counted by GelCount.
Animal xenograft studies
All animal experiments were approved by the Institutional Animal Care and Use Committee at The University of Texas MD Anderson Cancer Center. A total volume of 0.2 mL of cell suspension containing 1× 106 (for MDA-MB-468) or 4 × 106 (for SUM149) cells with 50% Matrigel were injected into the bottom right mammary fat pad of 6-week-old nu/nu mice. After 1–2 months, mice with well-established tumors (tumor volume, approximately 100 mm3) were allocated into the following groups: vehicle (0.5% methylcellulose); 40 or 80 mg/kg of KW-2450; 50 or 100 mg/kg of selumetinib; KW-2450 (40 mg/kg) plus selumetinib (50 or 100 mg/kg); these agents were administered daily by oral gavage for 28 days. Tumor volume was determined twice a week by measuring the tumor diameter in two dimensions with a caliper. Volume (V) was determined by the following equation, for which L was tumor length and W was tumor width: V = (L × W2)*π/6. Tumor volumes at day 0 (the day that drug treatment commenced) were set as 1 (V0), and then tumor volumes at each time point (V) were shown as the ratios to V0 for each tumor (V/V0). The values of V/V0 were compared between the control and treated groups at each time point by the t test.
Combinatory drug treatment in vitro
In combinatory treatments with KW-2450, Selumetinib were added at the double concentrations to those of KW-2450 at each concentration point (described as ×2 in Fig. 6B). Synergism between KW-2450 and selumetinib in terms of growth inhibition in vitro was determined by CalcuSyn software (BIOSOFT, Cambridge, United Kingdom).
Figure 6. KW-2450 and selumetinib synergized in antitumor effect against TNBC in vitro and in vivo.

A, Western blots for phosphorylated forms of MAPKs (i.e., ERKs, JNKs, p38 MAPK) in TNBC cells treated with various concentrations of KW-2450. Paired nonphosphorylated forms are also presented to show basic expressions of those kinases. The samples were collected at 24 hours (for SUM149) or 48 hours (for MDA-MB-231 and MDA-MB-468) of KW-2450 treatments. β-actin is shown as a loading control at the bottom of each. B, Line charts depict growth inhibition curves of MDA-MB-231 and SUM149 cells, treated with sequent doses of KW-2450, selumetinib, or KW-2450 plus selumetinib. The x-axis shows the dose series of KW-2450. At each dose, selumetinib was used with a double dose of KW-2450, described as ×2 in parentheses next to selumetinib. For example, at the 1 μM dose point for KW2450, the dose is 2 μM for selumetinib monotherapy and for combination with KW-2450. We applied CalcuSyn software to determine whether there is synergistic, additional, or antagonistic effects between these two drugs at 50%, 75%, and 90% of effective doses: ED50, ED75, and ED90, respectively. CI values from CalcuSyn are shown at the bottom of each bar chart. CI values <0.1 indicate very strong synergism; 0.10–0.30, strong synergism; 0.30–0.70, synergism; 0.70–0.90 moderate to slight synergism; 0.90–1.10, nearly additive; >1.10, antagonism. Asterisks indicate the dose points where we saw significant differences by t test in both comparisons: combination vs KW-2450, combination vs selumetinib. Statistical significance was set at P < 0.05 (marked with *). C, Each colored bar depicts the relative proportions of cell cycle phases (subG1, 2N, S1, 4N, S2, or 8N) in each treatment: control, KW-2450, KW-2450 plus selumetinib. SUM149 and MDA-MB-231 cells were treated for 2 days and then fixed with 70% ethanol, stained with propidium iodide (PI), and analyzed by FACS. Values are expressed as percentages of total counted cells. Data represent means ± s.e.m. SubG1, dead cells; 2N, diploid or G0/G1; S1, DNA-synthesis after G1; 4N, tetraploid or G2/M; S2, DNA synthesis after mitosis-failure; 8N, octaploid. *, P < 0.05; **, P < 0.005. D, Line charts depict growth curves of SUM149 xenografts treated with vehicle, single treatments, or combinatory treatments for 4 weeks. V/V0, volume at any treatment time point/volume at day 0. Each dot shows mean ± s.e.m. Volumes of tumors treated with vehicle were compared with each single treatment at each time point, and when statistically significant difference (P < 0.05) was seen in every comparison, # was noted along with the dots of the vehicle line. Volumes of tumors treated with combination therapies were compared with the corresponding paired single treatments, and when a statistically significant difference (P < 0.05) was seen in both comparisons,* it was noted along with the dots of the combinatory treatment lines. E, Model of cell fates of TNBC cells treated with Aurora A/B inhibitor and relevant molecules to those cell fates; apoptosis or surviving 8N cells.
Statistical analyses
Statistical analyses were conducted by R software. Data were presented as means ± s.e.m. Means for all data were compared by an unpaired t test. P values of <0.05 were considered statistically significant.
Results
KW-2450 inhibits kinase activities of Aurora A, Aurora B, and IGF-IR at various concentrations
The chemical structure of KW-2450 is shown in Fig. 1A. We first tested KW-2450’s inhibitory activity against Aurora A kinase in cell based assay. We measured the inhibitory effect of KW-2450 on Aurora A activity at various concentrations in MDA-MB-468 cells, which was probed with the antibody against Aurora A autophosphorylation on threonine-288 (pT288) (Fig. 1B). Threonine-288 is an autophosphorylation site of Aurora A and thus a marker of Aurora A activity (4, 13). To accumulate the cells in the mitotic phase, we pretreated them with paclitaxel at 100 nM as previously described (15) for 20 hours and then treated them with KW-2450 at various concentrations for 4 hours. In control cells (DMSO added to paclitaxel pretreatment), pT288 was detected only in mitotic cells and was localized to the spindle centers, likely representing centrosomes (4). Treatment with KW-2450 at 0.2 and 1 μM inhibited Aurora A autophosphorylation on T288 in MDA-MB-468 cells (Fig. 1B). Total Aurora A was still present with KW-2450 treatments at these concentrations, demonstrating that the decreased pT288 staining was due to inhibition of phosphorylation but not to Aurora A degradation. The concentration and inhibition curve was drawn to determine the potency of KW-2450 against Aurora A kinase activity in mitotic MDA-MB-468 cells by quantifying the ratio of immunofluorescent intensities of pT288 to those of pan-Aurora A (Fig. 1C). KW-2450 inhibited Aurora A kinase activity within 4 hours with an IC50 value of 0.215 μM (95% CI = 0.173–0.268 μM) in MDA-MB-468 cells.
Figure 1. KW-2450 inhibits Aurora A and B kinases.

A, Chemical structure of KW-2450. B, Immunofluorescent images of MDA-MB-468 cells treated with DMSO or KW-2450 at various concentrations for 4 hours. Cells were pre-arrested by treating with paclitaxel (100 nM, for 20 hours). Arrowheads indicate phosphorylated Aurora A on Thr-288 with DMSO treatment. Scale bar, 10 μm. C, An Aurora A pT288 autophosphorylation assay was used to measure inhibition of Aurora A by KW-2450 in MDA-MB-468 cells. The concentration–inhibition curve was generated by calculating the decrease in Aurora A pT288 fluorescent intensity in KW-2450–treated samples relative to the DMSO-treated controls. D, Representative flow cytometry dot plots of MDA-MB-468 cells, pre-arrested with paclitaxel (100 nM, for 20 hours) and followed by treatments with DMSO or KW-2450 at various concentrations for 4 hours. Cells were stained with phosphorylated histone H3 (Ser-10) (pHisH3) mouse monoclonal antibody (y-axis) and propidium iodide (x-axis). Percentages are proportions of pHisH3+ tetraploid cells (gated areas) of whole cell fractions. E, An Aurora B Ser-10 histone H3 phosphorylation assay was used to measure the inhibition of Aurora B by KW-2450 in MDA-MB-468 cells. Concentration–inhibition curves were generated by calculating the decrease in pHisH3+ tetraploid cells in KW-2450–treated samples relative to the DMSO-treated controls. (C and E) The points represent averages of eight (for C) and three (for E) replicate samples ± s.e.m. (bars). The IC50’s were determined by Prism5 software and are shown with 95% confidence intervals (CIs).
We next tested the inhibitory effect of KW-2450 on Aurora B activity in MDA-MB-468, SUM149, and MDA-MB-231 cells, which were probed with antibody against the histone H3 phosphorylated on Ser-10 (pHisH3). Histone H3 is a kinase substrate of Aurora B and a marker of Aurora B kinase activation (13, 23). We pretreated the cells with paclitaxel for 20 hours and then treated them with KW-2450 at various concentrations for 4 hours under continuous paclitaxel treatment, and pHisH3-positive (pHisH3+) cells were quantified using FACS analysis (Fig. 1D). Treatment with KW-2450 at 0.5 and 10 μM reduced pHisH3+ cells, which is indicative of Aurora B inhibition in MDA-MB-468 cells (Fig. 1D). The concentration and inhibition curve was drawn to determine the potency of KW-2450 against Aurora B kinase activity by quantifying pHisH3+ mitotic cells (Fig. 1E). KW-2450 inhibited Aurora B within 4 hours with IC50s of 0.478 μM (95% CI = 0.368–0.620 μM) in MDA-MB-468 cells (Fig. 1E), 0.272 μM (95% CI = 0.212–0.350 μM) in SUM149 cells, and 0.066 μM (95% CI = 0.055–0.081 μM) in MDA-MB-231 cells.
KW-2450 was initially developed as a kinase inhibitor against IGF-IR and InsR. We evaluated the effect of KW-2450 on IGF-IR kinase activity by Western blotting for IGF-IR autophosphorylation (Tyr1135/1136), which is a functional marker of IGF-IR kinase activity in mammalian cells (Supplementary Fig. S1). In both MDA-MB-468 and SUM149 cells, which endogenously overexpressed IGF-IR, KW-2450 (0.002 μM) reduced kinase activities of IGF-IR by about 25% compared with the DMSO control (which showed 100% activity). This indicates that KW-2450’s IC50 for inhibiting IGF-IR in the cell-based assay was less than 0.002 μM. Taken together, these data suggest that KW-2450 is able to inhibit Aurora A and Aurora B kinases at a higher concentration and IGF-IR kinase at a lower concentration.
KW-2450 shows in vitro antitumor effects in TNBC cells
The effects of KW-2450 on cell viability of TNBC cells were tested by the CellTiter-Blue Cell Viability Assay (Promega Corporation). Concentration–inhibition curves were drawn as shown in Figure 2A, and IC50s were calculated among a panel of cell lines (Table 1). IC50 values for inhibiting cell viability in TNBC cells were almost the same as the IC50S for inhibiting Aurora A (IC50 = 0.22 μM) and Aurora B (IC50 = 0.48 μM). In contrast, KW-2450’s IC50 values for inhibiting cell viability in ER+ breast cancer cells were lower than those in TNBC cells (P = 0.011; Table 1).
Figure 2. KW-2450 has in vitro antitumor activities against TNBC cells.

A, Growth inhibition assays of KW-2450 in vitro were performed by using the CellTiter-Blue assay. The viabilities of TNBC cells treated with KW-2450 were determined in reference to the control treatment, and the concentration-inhibition curve was drawn. Each point represents an average ± s.e.m. (bars) from quadruplicate. B, Colony formation in agar assay in TNBC cells treated with KW-2450 or DMSO. Histogram depicts percentage of colony formations of MDA-MB-231 or SUM149 cells treated with KW-2450 (0.05, 0.1, 0.5 μM) in relation to DMSO treatment, which was set at 100%. Representative images of colony formation in agar of SUM149 and MDA-MB-231 cells are shown at the bottom of histograms. C, Mammosphere-forming assay in TNBC cells treated with KW-2450 or DMSO. Histogram depicts percentage of mammosphere formation of SUM149 and MDA-MB-231 cells treated with KW-2450 (0.05, 0.1, 0.5 μM) in relation to DMSO treatment, which was set at 100%. Representative images of mammospheres of SUM149 and MDA-MB-231 cells are shown at the bottom of histograms (Scale bar, 100 μm). B, C, Error bars, standard error of mean (s.e.m.). *, P < 0.05; **, P < 0.01.
Table 1.
Growth inhibition (IC50) across various breast cancer cell lines by KW-2450
| Cell line | Subtype | IC50 [95% CI] |
|---|---|---|
| MDA-MB-468 | TN | 0.36 [0.31 — 0.41] |
| MDA-MB-231 | TN | 0.49 [0.44 – 0.55] |
| SUM149 | TN | 0.30 [0.29 – 0.32] |
| SUM159 | TN | 0.38 [0.36 – 0.41] |
| BT-549 | TN | 0.52 [0.48 – 0.57] |
| HCC70 | TN | 0.27 [0.25 – 0.29] |
| SUM190 | HER2+ | 0.22 [0.09 – 0.55] |
| KPL4 | HER2+ ER+ | 0.67 [0.61 – 0.75] |
| T47D | ER+ | 0.26 [0.22 – 0.32] |
| ZR75-1 | ER+ | 0.11 [0.08 – 0.16] |
| MCF7 | ER+ | 0.011 [0.006 – 0.021] |
TN, triple-negative; HER2+, HER2/neu receptor-positive; ER+, estrogen receptor-positive; CI, confidence interval. IC50 values of the TN group were compared with those of the ER+ group by t test and the difference was statistically significant (P = 0.011).
We previously showed in TNBC and glioblastoma multiforme mouse models that cancer stem cells (CSCs) have a quiescent trait and overexpress IGF-I, which was a key molecule for making CSCs quiescent in these models (20, 24). On the basis of these findings, we assumed that letting CSCs exit from the quiescent state and stay in mitosis by inhibiting IGF-IR and Aurora A/B may work as CSC-targeted therapy in TNBC. We therefore treated TNBC cells with KW-2450 at various concentrations and assessed anti-CSC activities of KW-2450 by the following assays: colony formation in agar assay, mammosphere-forming assay, and CD44+CD24− CSC markers labeling assay (22, 24, 25). KW-2450 dose-dependently inhibited colony formation in agar and mammosphere-forming in floating culture in SUM149 and MDA-MB-231 cells (Fig. 2B, C). The CD44+CD24− CSC populations were also significantly reduced, depending on the concentrations of KW-2450 in both cell lines (Supplementary Fig. S2). Overall, KW-2450 showed antitumor and anti-CSC activities in in vitro cell assays.
KW-2450 induces 4N and 8N accumulations and apoptosis in TNBC cells
The effect of KW-2450 on the cell cycle was evaluated by FACS analysis (Fig. 3A). In all three TNBC cell types, KW-2450 dose-dependently induced tetraploid (4N), 8N, and subG1 (cell death) phases. It is known that Aurora A inhibition induces the delay in mitotic entry, which causes 4N, and that Aurora B inhibition induces mitotic-slippage, which causes 8N in mammalian cells (9, 10). Both of these outcomes, 4N or 8N, could cause cell death after all (13, 14). Based on this evidence, Aurora A and/or B could be primarily inhibited at higher concentrations of KW-2450 (0.5, 1 μM) in TNBC cells. Meanwhile, we tested the significance of inhibiting IGF-IR or InsR using siRNAs in these TNBC cells (Supplementary Fig. S3). We then confirmed that neither of the siRNAs to IGF-IR or InsR caused 4N, 8N, and subG1 phenotypes, suggesting that neither IGF-IR nor InsR inhibition is responsible for these phenotypes.
Figure 3. KW-2450 dose-dependently induces 4N, 8N accumulations and apoptosis in TNBC cells.

A, DNA profiles of TNBC cells treated with DMSO or KW-2450 at various concentrations for 48 hours. 2N, 4N, and 8N reflect relative DNA contents and represent diploid, tetraploid, and octaploid cells, respectively. SubG1 reflects dead cells. S1 and S2 reflect the DNA synthesis phases of diploid cells and tetraploid cells, respectively. Each value of the histogram represents an average ± s.e.m. from triplicate. B, C, FACS analysis of Annexin V+ cells in MDA-MB-468 cells treated with KW-2450 (0.5 μM). B, representative FACS dot plots of MDA-MB-468 cells treated with KW-2450 (0.5 μM) or DMSO for 48 hours. C, Line chart depicts the time-lapse percentage change in Annexin V+ cells in MDA-MB-468 cells treated with KW-2450 (0.5 μM) or DMSO. D, E, Reactive-oxygen-species (ROS) assay in TNBC cells treated with KW-2450 (0.5, 1 μM) or DMSO for 48 hours. Samples were stained with oxidative stress reagent (GFP) and PI, and ROS+ (GFP+) cells and PI+ (dead cells) were quantified by flow cytometry. D, Flow cytometry histograms depict the representative staining patterns of oxidized-green-fluorescein in TNBC cells treated with KW-2450 or DMSO. F, Time-lapse cell cycle analyses of MDA-MB-468 and MDA-MB-231 cells treated with KW-2450 (0.5 μM). FACS cell-cycle histograms derived from 0 to 72 hours of KW-2450 treatment (0.5 μM) are shown, and for 72 hours of KW-2450 treatments, the percentages of subG1 fraction (dead cells) are shown.
In MDA-MB-468 cells, compared with both MDA-MD-231 and SUM149 cells, we observed more subG1 cells at higher concentrations of KW-2450 (0.5, 1 μM). To determine whether this cell death could be attributed to apoptosis, we performed an Annexin V assay using MDA-MB-468 cells (Fig. 3B, 3C). We treated MDA-MB-468 cells with 0.5 μM KW-2450 and monitored the amount of Annexin V+ cells undergoing apoptosis during the treatment. The number of Annexin V+ cells substantially increased within 24 to 48 hours of KW-2450 treatment compared with the DMSO control, suggesting ongoing apoptosis (Fig. 3C).
Reactive oxygen species (ROS) is a key mediator of apoptosis and accumulates in the cells by activation of mitochondrial pathways (26, 27). We quantified intracellular ROS levels among three types of TNBC cells treated with DMSO or KW-2450 (0.5 or 1 μM). In MDA-MB-468 cells treated with KW-2450, we observed considerably higher accumulation of ROS compared with levels in the DMSO control (Fig. 3D, 3E). In contrast, in MDA-MB-231 cells and SUM149 cells treated with KW-2450, we noted less ROS accumulation than in MDA-MB-468 cells. These levels of ROS accumulation were proportional to the percentage of dead cells, with more dead MDA-MB-468 cells and fewer dead MDA-MB-231 and SUM149 cells, suggesting the biological association between ROS accumulation and apoptotic outcome. We concluded that KW-2450 induced apoptosis presumably by utilizing mitochondrial (or oxidative) pathways in TNBC cells.
KW-2450 induces apoptosis, senescence, or cell cycle arrest at 8N in TNBC cells
To gain insight into how KW-2450 induces apoptosis in TNBC cells, we next analyzed the kinetics of the cell cycle associated with KW-2450 treatment (0.5 μM) in MDA-MB-468 and MDA-MB-231 cells (Fig. 3F). We chose a concentration of 0.5 μM because we had observed a difference in the rates of apoptosis between MDA-MB-468 cells and the other two cell types (i.e., MDA-MB-231 and SUM149 cells) at this concentration, which we thought could give us insight at a molecular level into the sensitivity to KW-2450. At 12 and 24 hours of KW-2450 treatment, we observed accumulation of 4N in both MDA-MB-468 and MDA-MB-231 cells (Fig. 3F). At 48 hours of treatment, we observed a significant amount of subG1 and a few 8N fractions in MDA-MB-468, which was the reciprocal to that observed in MDA-MB-231 cells: a few subG1 and a significant amount of 8N. What intrinsic factors in TNBC cells produced this difference in terms of the response to KW-2450? To answer this question, we focused on cell cycle checkpoints, which are the SAC and the post-mitotic G1 checkpoint. First, we abrogated the SAC function by knocking down Mad2 or BudR1, both which are indispensable functional components of the SAC, and treated with KW-2450 (0.5 μM) (Supplementary Fig. S4). Depletion of Mad2 or BudR1 did not reduce the amount of apoptotic cells caused by KW2450, suggesting that SAC activation is not responsible for cell death caused by KW-2450 in MDA-MB-468 cells.
Next, we focused on the checkpoint that occurs just after cells exit mitosis: the post-mitotic G1 checkpoint. At this checkpoint, p53 plays a crucial role in letting the cells undergo apoptosis or arrest. To test whether p53 determines the response to KW-2450 at the post-mitotic G1 checkpoint, we used p53 genotype-matched HCC116 colon cancer cells (p53+/+ vs p53−/−) (Supplementary Fig. S5). With KW-2450 treatment (0.5, 1 μM), HCT116 (p53−/−) cells showed significant accumulation of 8N compared with HCT116 (p53+/+) cells, suggesting that abrogated p53 contributed to the 8N accumulation. We also treated MCF7 and ZR 75-1 breast cancer cells, which have wild-type p53, with KW-2450 and confirmed that neither cell type accumulated at the 8N phase, even at higher concentrations of KW-2450 (0.5, 1 μM) (Supplementary Fig. S6), suggesting again that p53 at post-mitotic G1 is a determinant of the 8N phenotype with KW-2450 treatment.
Independent of cell cycle checkpoints, we also sought to determine whether senescence causes resistance to KW-2450 in TNBC cells, as reported by others (28, 29). We did not see significant senescence induction in any TNBC cells treated with 0.5 μM KW-2450 for 5 days; however, we did see induction of senescence at higher concentrations (1, 4 μM) and with longer exposure to KW-2450 (for 10 days) (Supplementary Fig. S7). Collectively, 8N cells caused by KW-2450 (0.5 μM) did not undergo apoptosis or senescence, thus arresting at the 8N phase as surviving cells.
Inhibition of Aurora A and B is responsible for 4N, 8N, and apoptosis caused by KW-2450
Next, by using MDA-MB-468 and MDA-MB-231 cells, we determined which inhibition against Aurora A or Aurora B was mainly responsible for 4N or 8N accumulation coupled with apoptosis. To this end, we performed knockdown experiments using siRNAs to Aurora A or B in MDA-MB-468 and MDA-MB-231 cells (Fig. 4). When Aurora B was knocked down in MDA-MB-468 and MDA-MB-231 cells, 4N accumulations were observed in both MDA-MB-468 and MDA-MB-231 cells and in a small fraction of 8N in MDA-MB-231 cells (Fig. 4A, B). Treatment with Aurora B siRNA also induced a substantial amount of cell death (subG1) in MDA-MB-468 cells but less in MDA-MB-231 cells, whose trend was similar to the one observed in KW-2450 treatment. In contrast, treatment with Aurora A siRNA did not cause significant changes in cell cycle profiles of MDA-MB-468 and MDA-MB-231 cells. However, inhibition of Aurora A is known to cause 4N accumulation; we therefore further inhibited Aurora A kinase by using Aurora A–specific inhibitor MLN8054 in MDA-MB-468 cells (Fig. 4C–E). With use of 0.05 and 0.1 μM MLN8054, Aurora A kinase activities were inhibited by about 30% and 0%, respectively (Fig. 4D), which caused accumulations of 4N and subG1 phases depending on the dose (Fig. 4E). Especially at 0.1 μM MLN8054, which achieved complete inhibition of Aurora A, MLN8054 dramatically induced accumulation of 4N and subG1 phases. These data suggest that complete inhibition of Aurora A kinase does induce 4N and apoptosis in MDA-MB-468 cells.
Figure 4. Pharmacological inhibition of Aurora A or siRNA knock down of Aurora B induced 4N and apoptosis as seen in KW-2450 treatment.

A, B, Knockdown experiments with siRNAs that target Aurora A or Aurora B in MDA-MB-468 cells (A) and MDA-MB-231 cells (B). Left of A or B, Knocked down efficiencies are shown by RT-PCR analyses using PCR primers that were designed to amplify the partial sequence of Aurora A or Aurora B gene. Right of A or B, Representative histograms depict DNA profiles of MDA-MB-468 and MDA-MB-231 cells treated with siRNAs to Aurora A or to Aurora B for 48 or 72 hours. The DNA profiles treated with control siRNA are overlapped with the corresponding sample histogram and shown as gray lines. C, D, E, Pharmacological inhibitions of Aurora A with Aurora A–specific inhibitor MLN8054 in MDA-MB-468 cells. C, Immunofluorescent images of MDA-MB-468 cells probed with antibodies to p-Aurora A (pT288) and pan-Aurora A. Nuclei were counterstained with DAPI. Scale bar, 10 μm. D, Histograms depicts percentage of Aurora A kinase activities in MDA-MB-468 cells treated with DMSO or MLN8054 (0.05, 0.1 μM). E, Cell cycle histograms of MDA-MB-468 cells treated with DMSO or MLN8054 (0.05, 0.1 μM).
In summary, using two types of TNBC cells, we showed that either specific inhibition of Aurora A or Aurora B could cause 4N and/or 8N accumulation and apoptosis in TNBC cells, depending on the type of cells. Therefore, we cannot conclude that inhibition of either Aurora A or Aurora B is responsible for these phenotypes caused by KW-2450, but presumably inhibition of either produces these results.
KW-2450 single treatment inhibits tumor growth of TNBC cells in vivo
We next evaluated in vivo antitumor activity of KW-2450 by using MDA-MB-468 tumors grown as xenografts in nude mice (Fig. 5). KW-2450 was administered orally at 80 mg/kg once a day for 28 days for MDA-MB-468 tumor-bearing mice. For control tumor-bearing mice, the same amount of vehicle was administered on the same schedule. From days 14 to 28, we observed significant tumor growth inhibition in MDA-MB-468 tumors treated with KW-2450 (80 mg/kg) compared with vehicle control (Fig. 5A). We monitored the body weights of MDA-MB-468 tumor-bearing mice during the treatments and observed that KW-2450-treated mice had 11.5 ± 4.9 (average ± s.e.m.) % more body weight at the end of the KW-2450 treatment compared with vehicle-treated mice, although this difference did not reach statistical significance (Supplementary Fig. S8).
Figure 5. KW-2450 inhibits tumor growth of TNBC cells in vivo.
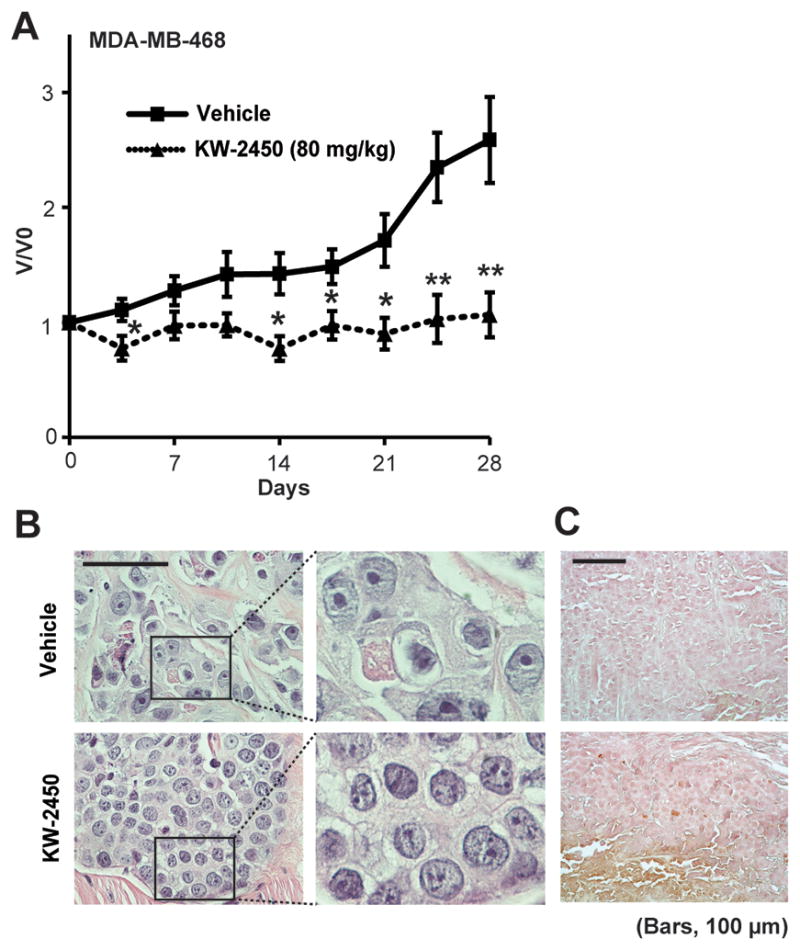
A, Line charts depict growth curves of MDA-MB-468 xenografts treated with vehicle or KW-2450 (80 mg/kg/day) for 4 weeks. The tumor volumes at day 0 (the date treatments commenced) were set to 1 to determine the relative volumes in each tumor during treatments. V/V0, volume at any treatment time point/volume at day 0. Each dot shows mean ± s.e.m. Volumes of tumors treated with KW-2450 were compared with vehicle control at each time point, and statistical differences are indicated with the following marks: *, P < 0.05; **, P < 0.01. B, Hematoxylin and eosin–stained section of MDA-MB-468 xenografts treated with vehicle or KW-2450 (80 mg/kg). Boxed areas in left images are shown to the right. Scale bars, 100 μm. C, Apoptotic cells (brown) were detected by TUNEL assay in MDA-MB-468 tumor treated with KW-2450. Nuclei were counterstained with hematoxylin (purple). Shown are representative fields in vehicle-treated control (upper) and KW-2450-treated tumor (lower). Scale bars, 100 μm.
We next examined the mitotic features of MDA-MB-468 tumors treated with KW-2450 (80 mg/kg) or vehicle (Fig. 5B). Tumors treated with KW-2450 had significantly more multinucleated cells than did vehicle-treated tumors, suggesting that 80 mg/kg of KW-2450 reached the pharmacodynamic level needed to inhibit Aurora A and/or Aurora B in these tumors. Accumulation of apoptotic cells was also observed in the MDA-MB-468 tumors treated with KW-2450, suggesting that 80 mg/kg of KW-2450 induced apoptosis by inhibiting Aurora A and/or Aurora B in vivo (Fig. 5C).
KW-2450 and MEK inhibitor synergize in inducing cell death in TNBC cells
We observed diverse susceptibilities to cell death among cultured TNBC cells treated with 0.5 μM KW-2450 (Fig. 3A, 3F). As described above, abrogation of p53 can induce 8N accumulation under KW-2450 treatment, which is a resistance phenotype against KW-2450. Given TNBC cells frequently have mutated p53 (Supplementary Table S1), we need to overcome this resistance mechanism derived from abrogated p53 in TNBC cells. We focused on mitogen-activated protein kinases (MAPKs) (e.g., ERK1/2, p38 MAPK, JNKs), which are already targetable with existing kinase inhibitors. MAPKs could prevent cell death in KW-2450 treatment similar to the way in which these MAPK pathways respond to anticancer drugs (30–33). We first examined the dose effect of KW-2450 on the activation of MAPK pathways by detecting the phosphorylation of ERK1/2, p38 MAPK, and JNKs (Fig. 6A). We observed activation in ERK1/2 as a result of dose escalation of KW-2450 but not in p38 MAPK or JNKs, suggesting that the activation of MEK1/2 (upstream kinases of ERK1/2) and of ERK1/2 may play a role in protecting TNBC cells treated with KW-2450. To test this hypothesis, we added the MEK1/2 inhibitor selumetinib (34, 35) to KW-2450 in SUM149 and MDA-MB-231 cells and evaluated its synergistic effects by using a cell viability assay and cell cycle analysis (Fig. 6B, C). In testing various concentrations, we observed synergism between KW-2450 and selumetinib in terms of cell growth inhibition (Fig. 6B). This synergism was also observed in the cell cycle analysis. We observed that selumetinib (1 μM) plus KW-2450 (0.5 μM) significantly increased subG1 populations in both MDA-MB-231 and SUM149 cells compared with KW-2450 alone (Fig. 6C). In parallel, 8N cells were significantly reduced with selumetinib plus KW-2450 compared with KW-2450 alone in both MDA-MB-231 and SUM149 cells. These findings suggest that activated ERK1/2 protects TNBC cells treated with KW-2450 from cell death at an 8N phase.
Finally, we tested whether this synergistic effect would be observed in vivo (Fig. 6D). To answer this question, we used SUM149 cells, which had shown relative resistance to cell death when treated with KW-2450 alone; cell death was induced in vitro, however, by adding selumetinib onto KW-2450. For treatment with selumetinib, either of 50 or 100 mg/kg per day of selumetinib was used for single and combination therapies with KW-2450 as previously described. For treatment with KW-2450, 80 or 40 mg/kg of KW-2450 was used for single and combination therapies (only with 40 mg/kg) with selumetinib. Every single-agent treatment showed significant growth inhibition compared with the vehicle-treated group from day 3 through day 28. Furthermore, either of the combination therapies showed significant growth inhibition compared with either matched single treatments throughout the treatment courses.
Discussion
In the present study, we concluded that Aurora A and B were primary targets of KW-2450 in the antitumor effects seen in TNBC. TNBC cell lines relatively resistant to KW-2450 underwent cell death when treated in a combination with the MEK inhibitor selumetinib.
Historically, the development of Aurora kinase inhibitors started with pan-Aurora kinase inhibitors such as VX-680, and several Aurora A/B kinase dual inhibitors followed (7, 8, 27, 36–38). The development of Aurora A/B kinase inhibitors led to the recognition that inhibition of Aurora B kinase is mainly responsible for the antitumor effects of Aurora A/B dual kinase inhibitors (6). KW-2450 is considered a dual Aurora A/B kinase inhibitor, with similar IC50 values against Aurora A and Aurora B kinases (39). Consistent with previous studies of dual kinase inhibitors of Aurora A and B (14, 38), KW-2450 dose-dependently induced 4N, 8N, and apoptosis at various rates, depending on the type of TNBC cells (Fig. 6E). However, at the highest concentrations (1.0 μM), 8N became the dominant population in all TNBC cells, which is a sign that Aurora B inhibition overtook Aurora A inhibition. Moreover, we observed accumulations of mitotic cells (shown with pHisH3+ or increased cyclin B1), which is a well-known outcome of Aurora A inhibition, in MDA-MB-468 and SUM149 cells (Supplementary Fig. S9). Therefore, at 0.5 μM KW-2450 we saw growth inhibition or apoptosis in TNBC cells (Fig. 2, 3), and inhibition of both Aurora A and B could be contributing to these antitumor effects in vitro. Since specific inhibitors to Aurora A and B are under development, testing all of these compounds may reveal further molecular background into the diverse responses to KW-2450 treatment, such as 8N accumulation or apoptosis.
Our finding of the tumor inhibitory effects of KW-2450 according to the subtype of breast cancer was also of interest. We showed that IC50 values for inhibiting cell viability in TNBC were higher than those for ER+ breast cancer. This finding suggests two possibilities: 1) ER+ breast cancer cells are more sensitive to Aurora A and B inhibition than are TNBC cells, or 2) targeted kinases other than Aurora A and B are responsible for these lower IC50 values in ER+ breast cancer. If the latter is the case, IGF-IR could be one such targeted kinase in ER+ breast cancer. In fact, in another study that tested IGF-IR inhibitor NVP-AEW541 for breast cancer cells, MCF7 cells showed the highest sensitivity to IGF-IR inhibition among several breast cancer cells including TNBC cells, which is exactly the same trend that we saw in the present study. These findings, together with our data, suggest that IGF-IR could be a legitimate target in ER+ breast cancer but not in TNBC cells.
In contrast to our conclusion, several studies support targeting of the IGF-IR pathway in TNBC cells. Lee’s group initially showed the prognostic significance of the IGF-IR gene signature to predict poor breast cancer prognosis (40), which was also correlated with the ER-negative subtype. They further developed this concept in a preclinical study. With use of BMS-754807, a dual IGF-IR/InsR inhibitor, they showed the antitumor effects of BMS-754807 in vitro and in vivo (41). BMS-754807 is a reversible inhibitor to both IGF-IR and InsR, which is very potent with the Ki value of <2 nM. In an IGF-IR kinase inhibitory assay with BMS-754807, a concentration of 3 to 10 nM was enough to reduce IGF-IR kinase activity by less than half of that of the DMSO-treated control, meaning that IC50 to inhibit IGF-IR kinase activity was less than 3 to 10 nM, which is close to our value for KW-2450 (<2 nM). Despite this potent inhibitory activity toward IGF-IR, the BMS-754807 IC50 values to inhibit cell growth in MDA-MB-468 and MDA-MB-231 cells were much higher than those of KW-2450 (IC50s of BMS-754807: 2.91 and 2.27 μM for MDA-MB-468 and MDA-MB-231 cells, respectively). These facts suggest that BMS-754807 could be inhibiting not only IGF-IR but also other off-targets with IC50s. According to the original report of BMS-754807, the 51 top-ranked potential targets included all Aurora kinase family proteins (Aurora A, B, C) and another mitotic kinase, PLK4. These findings, together with our data, suggest that IGF-IR and/or InsR inhibition may not be enough to inhibit the growth of TNBC cells and that other targets need to be inhibited. Among these potential targets, Aurora A and B kinases are the leading candidates to be inhibited to have antitumor effects in the context of TNBC.
In conclusion, we showed the antitumor effects of KW-2450, focusing on Aurora A and B kinase inhibition in TNBC, and discovered a novel combinatorial opportunity with Aurora A/B inhibitor and MEK inhibitor for those cells with less sensitivity to KW-2450 (Fig. 6E). In the future, we will need to answer the questions “how does the MEK-ERKs pathway get activated in response to Aurora A/B inhibition?” and “Why is there diversity in terms of the response to Aurora A/B inhibition (e.g., activation level of MEK)?” Delineating the molecular background and marker development in regard to this combinatory therapy is worth pursuing in TNBC, which currently does not have any molecularly targeted therapies in a standard care setting.
Supplementary Material
Acknowledgments
Financial Information: This research was supported by Kyowa Hakko Kirin Co., Ltd. (Tokyo, Japan) (N.T. Ueno) and in part by the U.S. National Institutes of Health/National Cancer Institute under award number P30CA016672 (R. A. DePinho) and by the Nellie B. Connally Breast Cancer Research Fund (D. Tripathy). K. Kai was supported by the program “Strategic Young Researcher Overseas Visits Program for Accelerating Brain Circulation” by the Ministry of Education, Culture, Sports, Science and Technology, Japan.
We thank Dr. Jangsoon Lee, Mr. Dionysios N Giannoukos, and Mr. Parth U Sawhney for their help in animal experiments and Ms. Tamara K. Locke (The University of Texas MD Anderson Cancer Center) for her help in editing this manuscript.
Footnotes
Disclosure of Potential Conflicts of Interest: MD Anderson received a research fund from Kyowa Hakko Kirin Co., Ltd. (Tokyo, Japan) (PI, Naoto T. Ueno).
References
- 1.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363:1938–48. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 2.Carey L, Winer E, Viale G, Cameron D, Gianni L. Triple-negative breast cancer: disease entity or title of convenience? Nat Rev Clin Oncol. 2010;7:683–92. doi: 10.1038/nrclinonc.2010.154. [DOI] [PubMed] [Google Scholar]
- 3.Pal SK, Childs BH, Pegram M. Triple negative breast cancer: unmet medical needs. Breast Cancer Res Treat. 2011;125:627–36. doi: 10.1007/s10549-010-1293-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marumoto T, Zhang D, Saya H. Aurora-A - a guardian of poles. Nat Rev Cancer. 2005;5:42–50. doi: 10.1038/nrc1526. [DOI] [PubMed] [Google Scholar]
- 5.Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keen N, Taylor S. Aurora-kinase inhibitors as anticancer agents. Nat Rev Cancer. 2004;4:927–36. doi: 10.1038/nrc1502. [DOI] [PubMed] [Google Scholar]
- 7.Katayama H, Sen S. Aurora kinase inhibitors as anticancer molecules. Biochim Biophys Acta. 2010;1799:829–39. doi: 10.1016/j.bbagrm.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kitzen JJ, de Jonge MJ, Verweij J. Aurora kinase inhibitors. Crit Rev Oncol Hematol. 2010;73:99–110. doi: 10.1016/j.critrevonc.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 9.Marumoto T, Honda S, Hara T, Nitta M, Hirota T, Kohmura E, et al. Aurora-A kinase maintains the fidelity of early and late mitotic events in HeLa cells. J Biol Chem. 2003;278:51786–95. doi: 10.1074/jbc.M306275200. [DOI] [PubMed] [Google Scholar]
- 10.Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, et al. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol. 2003;161:281–94. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ditchfield C, Johnson VL, Tighe A, Ellston R, Haworth C, Johnson T, et al. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol. 2003;161:267–80. doi: 10.1083/jcb.200208091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Girdler F, Gascoigne KE, Eyers PA, Hartmuth S, Crafter C, Foote KM, et al. Validating Aurora B as an anti-cancer drug target. J Cell Sci. 2006;119:3664–75. doi: 10.1242/jcs.03145. [DOI] [PubMed] [Google Scholar]
- 13.Manfredi MG, Ecsedy JA, Meetze KA, Balani SK, Burenkova O, Chen W, et al. Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proc Natl Acad Sci U S A. 2007;104:4106–11. doi: 10.1073/pnas.0608798104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gizatullin F, Yao Y, Kung V, Harding MW, Loda M, Shapiro GI. The Aurora kinase inhibitor VX-680 induces endoreduplication and apoptosis preferentially in cells with compromised p53-dependent postmitotic checkpoint function. Cancer Res. 2006;66:7668–77. doi: 10.1158/0008-5472.CAN-05-3353. [DOI] [PubMed] [Google Scholar]
- 15.Sudo T, Nitta M, Saya H, Ueno NT. Dependence of paclitaxel sensitivity on a functional spindle assembly checkpoint. Cancer Res. 2004;64:2502–8. doi: 10.1158/0008-5472.can-03-2013. [DOI] [PubMed] [Google Scholar]
- 16.Nitta M, Kobayashi O, Honda S, Hirota T, Kuninaka S, Marumoto T, et al. Spindle checkpoint function is required for mitotic catastrophe induced by DNA-damaging agents. Oncogene. 2004;23:6548–58. doi: 10.1038/sj.onc.1207873. [DOI] [PubMed] [Google Scholar]
- 17.Zhang D, Hirota T, Marumoto T, Shimizu M, Kunitoku N, Sasayama T, et al. Cre-loxP-controlled periodic Aurora-A overexpression induces mitotic abnormalities and hyperplasia in mammary glands of mouse models. Oncogene. 2004;23:8720–30. doi: 10.1038/sj.onc.1208153. [DOI] [PubMed] [Google Scholar]
- 18.Dickson M, LoRusso L, Sausville E, Rao N, Kobayashi E, Kurman M, et al. Open-Label, Sequential, Ascending, Multi-Dose, Phase 1 Study of KW-2450 as Monotherapy in Subjects with Previously Treated Advanced Solid Tumors. 2011 ASCO Annual Meeting; June 3 to 7, 2011; Chicago, IL. 2011. p. Abstract #77711. [Google Scholar]
- 19.Zhang D, LaFortune TA, Krishnamurthy S, Esteva FJ, Cristofanilli M, Liu P, et al. Epidermal growth factor receptor tyrosine kinase inhibitor reverses mesenchymal to epithelial phenotype and inhibits metastasis in inflammatory breast cancer. Clin Cancer Res. 2009;15:6639–48. doi: 10.1158/1078-0432.CCR-09-0951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kai K, Iwamoto T, Kobayashi T, Arima Y, Takamoto Y, Ohnishi N, et al. Ink4a/Arf(−/−) and HRAS(G12V) transform mouse mammary cells into triple-negative breast cancer containing tumorigenic CD49f(−) quiescent cells. Oncogene. 2014;33:440–8. doi: 10.1038/onc.2012.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee J, Bartholomeusz C, Mansour O, Humphries J, Hortobagyi GN, Ordentlich P, et al. A class I histone deacetylase inhibitor, entinostat, enhances lapatinib efficacy in HER2-overexpressing breast cancer cells through FOXO3-mediated Bim1 expression. Breast Cancer Res Treat. 2014;146:259–72. doi: 10.1007/s10549-014-3014-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kai K, Nagano O, Sugihara E, Arima Y, Sampetrean O, Ishimoto T, et al. Maintenance of HCT116 colon cancer cell line conforms to a stochastic model but not a cancer stem cell model. Cancer Sci. 2009;100:2275–82. doi: 10.1111/j.1349-7006.2009.01318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crosio C, Fimia GM, Loury R, Kimura M, Okano Y, Zhou H, et al. Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Mol Cell Biol. 2002;22:874–85. doi: 10.1128/MCB.22.3.874-885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Osuka S, Sampetrean O, Shimizu T, Saga I, Onishi N, Sugihara E, et al. IGF1 receptor signaling regulates adaptive radioprotection in glioma stem cells. Stem Cells. 2012;31:627–40. doi: 10.1002/stem.1328. [DOI] [PubMed] [Google Scholar]
- 25.Kai K, Arima Y, Kamiya T, Saya H. Breast cancer stem cells. Breast Cancer. 2010;17:80–5. doi: 10.1007/s12282-009-0176-y. [DOI] [PubMed] [Google Scholar]
- 26.Mates JM, Segura JA, Alonso FJ, Marquez J. Oxidative stress in apoptosis and cancer: an update. Arch Toxicol. 2012;86:1649–65. doi: 10.1007/s00204-012-0906-3. [DOI] [PubMed] [Google Scholar]
- 27.Li M, Jung A, Ganswindt U, Marini P, Friedl A, Daniel PT, et al. Aurora kinase inhibitor ZM447439 induces apoptosis via mitochondrial pathways. Biochem Pharmacol. 2010;79:122–9. doi: 10.1016/j.bcp.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 28.Tentler JJ, Ionkina AA, Tan AC, Newton TP, Pitts TM, Glogowska MJ, et al. p53 Family Members Regulate Phenotypic Response to Aurora Kinase A Inhibition in Triple-Negative Breast Cancer. Mol Cancer Ther. 2015;14:1117–29. doi: 10.1158/1535-7163.MCT-14-0538-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diamond JR, Eckhardt SG, Tan AC, Newton TP, Selby HM, Brunkow KL, et al. Predictive biomarkers of sensitivity to the aurora and angiogenic kinase inhibitor ENMD-2076 in preclinical breast cancer models. Clin Cancer Res. 2013;19:291–303. doi: 10.1158/1078-0432.CCR-12-1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boldt S, Weidle UH, Kolch W. The role of MAPK pathways in the action of chemotherapeutic drugs. Carcinogenesis. 2002;23:1831–8. doi: 10.1093/carcin/23.11.1831. [DOI] [PubMed] [Google Scholar]
- 31.Stadheim TA, Xiao H, Eastman A. Inhibition of extracellular signal-regulated kinase (ERK) mediates cell cycle phase independent apoptosis in vinblastine-treated ML-1 cells. Cancer Res. 2001;61:1533–40. [PubMed] [Google Scholar]
- 32.Fan M, Chambers TC. Role of mitogen-activated protein kinases in the response of tumor cells to chemotherapy. Drug Resist Updat. 2001;4:253–67. doi: 10.1054/drup.2001.0214. [DOI] [PubMed] [Google Scholar]
- 33.Bacus SS, Gudkov AV, Lowe M, Lyass L, Yung Y, Komarov AP, et al. Taxol-induced apoptosis depends on MAP kinase pathways (ERK and p38) and is independent of p53. Oncogene. 2001;20:147–55. doi: 10.1038/sj.onc.1204062. [DOI] [PubMed] [Google Scholar]
- 34.Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13:1576–83. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- 35.Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26:2139–46. doi: 10.1200/JCO.2007.14.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, et al. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med. 2004;10:262–7. doi: 10.1038/nm1003. [DOI] [PubMed] [Google Scholar]
- 37.Gorgun G, Calabrese E, Hideshima T, Ecsedy J, Perrone G, Mani M, et al. A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood. 2010;115:5202–13. doi: 10.1182/blood-2009-12-259523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soncini C, Carpinelli P, Gianellini L, Fancelli D, Vianello P, Rusconi L, et al. PHA-680632, a novel Aurora kinase inhibitor with potent antitumoral activity. Clin Cancer Res. 2006;12:4080–9. doi: 10.1158/1078-0432.CCR-05-1964. [DOI] [PubMed] [Google Scholar]
- 39.Umehara H, Koizumi F, Maekawa Y, Shimizu M, Nakamura H, Ota T, et al. KW-2450, a novel IGF-1R/IR inhibitor, enhances the antitumor effect of lapatinib, letrozole or 4-hydroxy-tamoxifen in breast cancer cells. AACR 104th Annual Meeting; 2013-- Apr 6–10; Washington, DC. 2013. [Google Scholar]
- 40.Creighton CJ, Casa A, Lazard Z, Huang S, Tsimelzon A, Hilsenbeck SG, et al. Insulin-like growth factor-I activates gene transcription programs strongly associated with poor breast cancer prognosis. J Clin Oncol. 2008;26:4078–85. doi: 10.1200/JCO.2007.13.4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Litzenburger BC, Creighton CJ, Tsimelzon A, Chan BT, Hilsenbeck SG, Wang T, et al. High IGF-IR activity in triple-negative breast cancer cell lines and tumorgrafts correlates with sensitivity to anti-IGF-IR therapy. Clin Cancer Res. 2011;17:2314–27. doi: 10.1158/1078-0432.CCR-10-1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.