

Abstract
The external granule layer (EGL) is a proliferative region that produces over 90% of the neurons in the cerebellum but can also malignantly transform into a cerebellar tumor called the medulloblastoma (the most common malignant brain tumor in children). Current dogma considers Hedgehog stimulation a potent proliferative signal for EGL neural progenitor cells (NPCs) and medulloblastomas. However, the Hedgehog pathway also acts as a survival signal in the neural tube where it regulates dorsoventral patterning by controlling NPC apoptosis. Here we show Hedgehog stimulation is also a potent survival signal in the EGL and medulloblastomas that produces a massive apoptotic response within hours of signal loss in mice. This toxicity can be produced by numerous Hedgehog antagonists (vismodegib, cyclopamine, and jervine) and is Bax/Bak dependent but p53 independent. Finally, since glucocorticoids can also induce EGL and medulloblastoma apoptosis, we show Hedgehog's effects on apoptosis can occur independent of glucocorticoid stimulation. This effect may play a major role in cerebellar development by directing where EGL proliferation occurs thereby morphologically sculpting growth. It may also be a previously unknown major therapeutic effect of Hedgehog antagonists during medulloblastoma therapy. Results are discussed in terms of their implications for both cerebellar development and medulloblastoma treatment.
Keywords: Sonic Hedgehog, cerebellum, medulloblastoma, apoptosis, external granule layer, neural progenitor cell
Introduction
The external granule layer (EGL) is a proliferative layer producing over 90% of cerebellar neurons which represent over half of the neurons in the entire brain (the cerebellum actually contains more neurons than the cerebrum) (Andersen et al., 1992; Harvey and Napper, 1988). Once produced, granule neurons migrate past the molecular and Purkinje cell layers before populating the internal granule layer (Figure 1A). Since Purkinje cells form synaptic connections with granule neurons, neurogenesis is regulated to maintain a consistent ratio between these cell types (Goldowitz and Hamre, 1998). A critical factor regulating EGL proliferation is the Sonic Hedgehog (Hh) pathway which begins with secretion of Sonic Hh ligand from Purkinje cells onto EGL neural progenitor cells (NPCs) where it binds and inhibits the Patched receptor. Since Patched activation normally represses Smoothened, this diminishes inhibition of Smoothened leading to a downstream activation of Gli transcription factors which are thought to potently increase NPC proliferation (Klein et al., 2001; Wechsler-Reya and Scott, 1999).
Figure 1. Hedgehog pathway antagonism produces selective apoptosis in the external granule layer of the developing cerebellum.
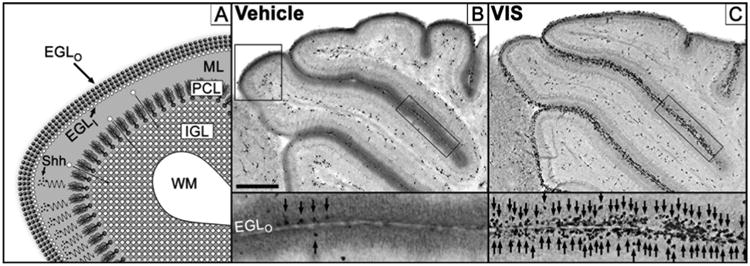
(A) Magnified diagram of upper left boxed region in (B). The outermost layer of the cerebellum is the transient external granule layer (EGL) composed of an outer germinal matrix populated by neural progenitor cells (EGLO; dark spheres) and an inner layer (EGLI; white spheres) where newly formed granule cell neurons congregate and mature. Immature granule cells then migrate (white spheres with arrows) past the molecular (ML) and Purkinje cell layers (PCL) before incorporating into the internal granule layer (IGL) that lies superficial to the cerebellar white matter (WM). Purkinje cells regulate EGL proliferation by secretion of Sonic Hedgehog ligand (Shh; small black dots) that diffuses (squiggly arrows) to the EGL where it stimulates the Hedgehog pathway. (B) Immunolabeling for the apoptotic marker activated caspase-3 (AC3) results in low levels of apoptosis following vehicle treatment while (C) 50 mg/kg vismodegib dramatically increases apoptosis. Insets reflect rectangular black-boxed regions over the EGL with arrows indicating AC3 positive EGL NPCs. Upper left boxed region in (B) diagrammed in (A). Scale Bar: 300 μm. Figure 1A is a derivative work based on a previously published figure by one of the authors (K.K.N.) under Creative Commons-BY license (Noguchi, 2014).
There is convincing evidence that EGL NPCs can become tumor-initiating cells that develop into medulloblastomas (MBs), the most common malignant brain tumor in children. For instance, mice with heterozygous mutations in the Patched gene exhibit disrupted inhibition of Smoothened leading to constitutive Hh activation (Goodrich et al., 1997; Oliver et al., 2005). While the EGL normally disappears by two weeks of age, Patched mice exhibit ectopic preneoplastic EGL remnants that are not a MB but have a high probability of progressing into one. Importantly, similar mutations of the Patched gene spontaneously occur in humans with an analogous increase in MB vulnerability (Oliver et al., 2005). Consistent with this finding, up to 30% of human MBs are driven by excessive Hh stimulation (Leary and Olson, 2012). Thus, animal research mirrors clinical findings suggesting the Hh pathway plays a key role in MB development. This has positioned the Hh pathway as a key area of research for testing MB formation and/or treatment. Much of this interest centers on novel Hedgehog antagonists (HAs) which effectively treat MBs in transgenic mice (Berman et al., 2002; Romer and Curran, 2005). This enthusiasm recently peaked due to the first Food and Drug Administration approval of the HA vismodegib. While vismodegib is currently approved for treating basal-cell carcinoma, several MB clinical trials have reported anti-tumor effects including some with remarkable results (Gajjar et al., 2010; Robinson, 2013; Rudin et al., 2009).
Interestingly, Hh signaling can have different effects in other regions of the nervous system. Sonic Hh ligand is also produced by the notochord and floor plate in early development which directs dorsoventral patterning of the neural tube by acting as a survival signal (Charrier et al., 2001; Thibert et al., 2003). Thus, NPCs proximal to Hh signaling survive and continue proliferating but more distal cells undergo apoptosis if signaling is lost (Guerrero and Ruiz i Altaba, 2003). Interestingly, blocking NPC apoptosis in the neural tube helps but does not completely rescue the effects of Hh inhibition on the neural tube, suggesting that Hh signaling regulates neural tube development through both apoptosis and proliferation (Guerrero and Ruiz i Altaba, 2003). Based on this research, we tested if Hh stimulation analogously acts as a survival signal in the EGL and MBs. Surprisingly, the loss of Hh signaling produced a massive apoptotic response within a few hours of signal loss.
Materials and Methods
Animals and Drugs
All procedures were in accordance with the Institutional Animal Care and Use Committee at Washington University in St. Louis and National Institutes of Health guidelines and used mice from both genders. ICR mice (Harlan, Indianapolis, IN, USA) were used in all experiments unless otherwise indicated. p53 knockout (Stock #2101) and Math1-Cre (Stock#11104) mice were purchased from Jackson Laboratories (Bar Harbor, MA, USA). Bax/Bak mice were a kind gift from Scott Oaks (University of California, San Francisco). p53, Math1-Cre, and Bax/Bak mice were maintained on the C57BL/6 strain. Patched mice (Goodrich et al., 1997) were a kind gift from Jane Johnson (UT Southwestern Medical Center) and were on a mixed C57BL/6/129X1/SvJ background. All experiments used mice from both genders.
Since many solvents are neurotoxic in developing brain (Farber et al., 2010; Hanslick et al., 2009; Lau et al., 2012), vismodegib (LC Laboratories, Woburn, MA, USA), cyclopamine (LC Laboratories, Woburn, MA, USA), jervine (Santa Cruz Biotechnology, Dallas, TX, USA), mifepristone (Sigma-Aldrich, St. Louis, MO, USA), itraconazole (Santa Cruz Biotechnology, Dallas, TX, USA), and fluocinolone acetonide (Sigma-Aldrich, St. Louis, MO, USA) were dissolved in palm oil and injected intraperitoneally. Injections of cytosine arabinoside (AraC; Sigma-Aldrich, St. Louis, MO, USA) and dexamethasone sodium phosphate (Voigt Global Distribution LLC, Lawrence, KS, USA) were dissolved in saline. Unless otherwise indicated, animals were perfused six hours postinjection for immunolabeling. Following injection, animals were housed separately from their mothers in a veterinary recovery chamber (Mediheat V1200, Dalton, GA, USA) at an ambient temperature of 30° C until perfusion.
Histology
For immunohistochemistry, animals were deeply anesthetized, transcardially perfused with 4% paraformaldehyde in 0.1 M Tris buffer, and brains sagittally sectioned on a vibratome at 75 μm. Sections were then incubated in a quenching solution (absolute methanol with 3% hydrogen peroxide) for 15 minutes, a blocking solution (PBS with 2% BSA, 0.2% milk, and 0.1% triton X-100) for an hour, and a primary antibody overnight. Finally, sections were placed in secondary antibody for an hour, reacted with ABC reagents, and incubated in either a chromogen (Vector VIP substrate kit, Vector Laboratories, Burlingame, CA, USA) or fluorescent probes. Photocomposites were taken with a Leica DM400B microscope connected to a Leica DFC310FX camera using Surveyor software V7.0.0.6 MT (Objective Imaging, Cambridge, UK). Immunohistochemistry was performed with antibodies raised against activated caspase-3 (Cell Signaling, Danvers, MA, USA), KI-67 (BD Biosciences, San Jose, CA, USA), or Cre (Millipore, Billerica, MA).
Semi-quantitative evaluation of activated caspase-3 labeling in the EGL
We have found semi-quantitative evaluation is a highly sensitive measure of EGL apoptosis (Noguchi et al., 2011, 2008). Briefly, a rater blind to treatment examined several midsagittal sections and semi-quantitatively evaluated AC3 in the EGL by assigning a rating to each animal using the following scale as a guide: 0 = no apoptotic profiles in all EGL regions, 1 = apoptotic profiles in a minority of EGL regions, 2 = apoptotic profiles are seen in a majority of EGL regions, and 3 = apoptotic profiles in all regions of the EGL. The scale was used as a general guide but rating scores could vary continuously (not limited to integers) between 0-3. Previous research has established that t-tests and ANOVAs are appropriate and robust statistical analyses for this type of scoring (Norman, 2010). Unless otherwise indicated, degeneration scores were evaluated using a t-test or one-way ANOVA with Bonferroni post hoc. To evaluate whether HAs dose dependently increased EGL apoptosis, we conducted Pearson's r correlation between dose and degeneration score to determine whether a linear correlation exists. All data were analyzed using Prism software (Version 5.0a; Graphpad Software Inc., San Diego, CA, USA).
Quantification of Activated Caspase-3 in Medulloblastomas
Since MBs vary in size and shape, a semi-quantitative scale was inadequate to quantify apoptosis. Therefore, apoptotic density counts were performed within tumors. At the first indication of tumor burden (tremor/gait disturbance, ataxia, loss of balance, listing to one side), animals were injected with 50 mg/kg vismodegib or vehicle and perfused 6 hours later. Cerebella plus MB were sectioned at 75 μM and stained for AC3. A rater blind to treatment performed imaging and quantification. Cell counts were performed on multiple sagittal sections (at least 3 per animal) within the same tumor and averaged to derive a single cell count per animal. Each section was imaged on a Nikon Eclipse E800 microscope at 10× using a Leica DFC490 digital camera connected to a MacPro (1,1 dual core Intel Xeon 2.66 GHz) with Leica Firecam software (Leica Microsystems, v 3.0.1). For each section, we imaged the central portion of the tumor and avoided any regions with imperfections due to tissue processing. Images were converted to 8-bit using Image J software (National Institutes of Health, v 1.42q), thresholding applied to maximize AC3 staining contours, and cell counts performed using Analyze Particles (Size: 75-Infinity, Circularity 0.0-1.0). A density count was calculated by dividing the cell count for each section by the area of the image (1.13 mm × 0.88 mm = 0.9944 mm2). Finally, density counts for each section were averaged to give a single average density count per animal. Density values between treatment groups were analyzed with a t-test.
Results
Hedgehog antagonists produces EGL apoptosis in a dose dependent manner
If Hh signaling acts as a survival signal, HAs should produce EGL apoptosis. We administered several HAs to postnatal day (PND)7 mice and perfused for activated caspase-3 (AC3) immunolabeling six hours later. Caspase-3 is a protease which is activated after a cell has irreversibly committed to apoptosis (Olney et al., 2004) and AC3 is a sensitive marker of EGL NPC apoptosis (Noguchi et al., 2011, 2008). Following HA exposure, the entire brain was screened for apoptosis but toxicity was only found in the EGL. Therefore, apoptosis was semi-quantitatively measured in the EGL only. Vismodegib is the only HA to receive Food and Drug Administration approval and effectively treats MBs in mice at 50 mg/kg (Wong et al., 2011). Semi-quantitative analysis revealed vismodegib significantly increased EGL apoptosis (F[4,26] = 39.40, p< 0.0001) at 1 mg/kg and above (Figure 1B-C and Figure 2A) in a dose dependent manner (r = 0.633, p< 0.0001). While vismodegib potently increased EGL apoptosis, this toxicity may be due to an off target effect. If true, cyclopamine and jervine (HAs that are structurally unrelated to vismodegib (Rudin, 2012)) would probably not produce the same toxicity. Semi-quantitative analysis revealed that cyclopamine, which effectively inhibits MB growth at 50 mg/kg (Berman et al., 2002; Gould and Missailidis, 2011), significantly increased EGL apoptosis (F[4,25] = 27.78, p< 0.0001) at 37.5 mg/kg and above (Figure 2B) in a dose dependent manner (r = 0.884, p< 0.001). Jervine is related to cyclopamine and derived from the same plant. Semi-quantitative analysis revealed jervine significantly increased EGL apoptosis (F[3,11] = 16.61, p< 0.001) at 50 mg/kg and above (Figure 2C) in a dose dependent manner (r = 0.897, p< 0.0001). Our results indicate structurally unrelated HAs potently induce an identical pattern of EGL apoptosis within hours of administration. Itraconazole is an anti-fungal HA that potently suppresses MB growth in a mouse allograft model (Kim et al., 2010). Unfortunately, since this model transplants MB tissue into the flanks of mice, there are concerns that itraconazole's limited ability to cross the blood brain barrier may limit its efficacy (Kethireddy and Andes, 2007). Semi-quantitative analysis revealed that itraconazole does not increase EGL apoptosis (F[3,16] = 0.72, p> 0.05) at any dose tested (Figure 2D) suggesting blood brain barrier permeability severely curbs its usefulness.
Figure 2. Characterization of EGL apoptosis produced by Hedgehog antagonists.
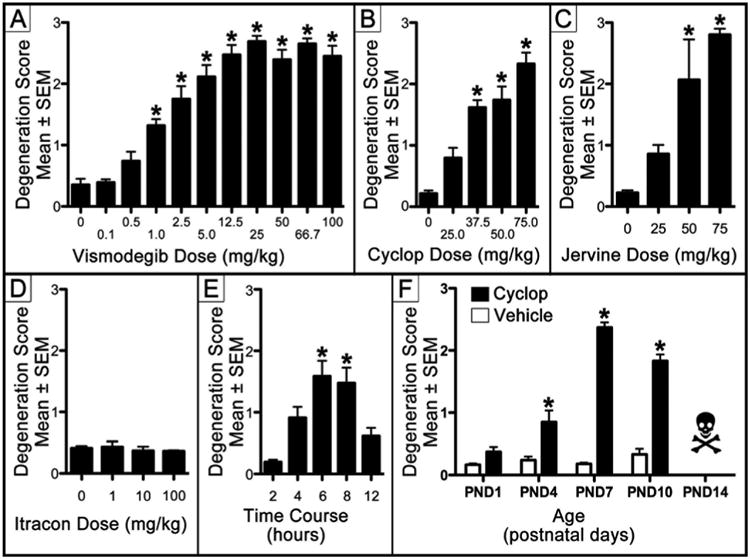
(A-D) To determine if the loss of Hedgehog signaling produces apoptosis, four HAs were administered on PND7 and the EGL screened for NPC apoptosis. Analysis revealed (A) vismodegib, (B) cyclopamine (Cyclop), and (C) jervine significantly increased EGL apoptosis at 1.0, 37.5, and 50 mg/kg and above respectively. (D) While itraconazole (Itracon) is a Hedgehog antagonist, it has limited brain blood barrier permeability and did not increase apoptosis at any dose. (E) To examine the time course of AC3 expression we administered 50 mg/kg Cyclop and perfused for apoptosis at different postinjection intervals. Cyclop significantly increased EGL apoptosis 6-8 hours postinjection. (F) To determine the window of vulnerability for HA-induced apoptosis we administered 50 mg/kg Cyclop on PNDs 1, 4, 7, and 10 (Note: Cyclop administration on PND14 was lethal). Cyclop significantly increased EGL apoptosis between PND4-PND10. * = p < 0.001.
Time Course and Window of Vulnerability
We next examined the time course of HA-induced AC3 activation. Cyclopamine (50 mg/kg) was administered to PND7 ICR mice perfused for AC3 at 2, 4, 6, 8, or 12 hours after injection. Semi-quantitatively evaluation of EGL apoptosis revealed a statistically significant difference between groups (F[4,29]= 8.673, p< 0.0001). We previously established that it takes more than 2 hours for EGL NPCs to become AC3 positive (Noguchi et al., 2008). Therefore, Bonferroni posthoc versus the 2-hour postinjection period was performed. Results revealed that cyclopamine significantly increased AC3 immunolabeling between 6-8 hours postinjection (Figure 2E).
We next examined the window of vulnerability for cyclopamine-induced apoptosis. Cyclopamine (50 mg/kg) or vehicle was administered on PNDs 1, 4, 7, 10, or 14. Surprisingly, all of the PND14 cyclopamine treated mice died within two hours of injection. To examine this further, we ran a similar study using cyclopamine from a different bottle and found that mice treated on PND14 all died while all treated on PND9 lived. Interestingly, 50 mg/kg vismodegib on PND14 was not similarly lethal (since the EGL naturally disappears around this age, apoptosis was not quantified). Therefore, we conclude that cyclopamine has age dependent lethal toxicity that was previously unrecognized. It is possible PND14 mice more efficiently metabolize cyclopamine leading to toxic metabolites. Cerebella from the younger groups were processed for AC3 and EGL degeneration semi-quantitatively evaluated. A two-way ANOVA revealed a significant main effect of age (F[3,56] = 45.72, p< 0.0001) and treatment (F[1,56] = 14.74, p< 0.0001) in addition to an interaction (F[3,56] = 41.79, p< 0.0001). A subsequent one-way ANOVA planned comparison between treatment groups at the same age was significant (F[7,56] = 81.33, p< 0.0001) and a Bonferroni posthoc revealed cyclopamine significantly increased EGL apoptosis on PNDs 4, 7, and 10 (Figure 2F).
Intracellular signaling mechanisms regulating HA-induced apoptosis
Bax and Bak are key intracellular signaling proteins that regulate apoptosis semi-redundantly. Bak knockouts appear normal but Bax/Bak double knockouts are markedly resistant to numerous apoptotic insults and, since apoptosis is critical for development, exhibit high mortality rates (Lindsten et al., 2000). Therefore, we created Cre/lox mice harboring a global knockout of Bak and a conditional knockout of a floxed Bax allele in the EGL using the Math1-Cre transgene (Math1-Cre+/0:Baxf/f:Bak-/-; henceforth called a Bax/Bak CKO mice). Since Math1-Cre knocks out the Bax allele selectively in the EGL, apoptotic resistance is limited and mice are healthy and viable. Bax/Bak littermates that were Cre negative (Math1-Cre0/0:Baxf/f:Bak-/-; henceforth called Bax/Bak Cre- mice) were used as controls since they display normal apoptosis. We previously found glucocorticoids (GCs) produce Bax/Bak dependent EGL apoptosis (Noguchi et al., 2008). Therefore, we validated these mice by administering 3 mg/kg dexamethasone (DEX; a synthetic GC) before perfusion for AC3 immunolabeling. As expected, exposure potently increased EGL apoptosis in Bax/Bak Cre- but not Bax/Bak CKO mice (t[6]= 11.81, p< 0.0001; Figure 3A). Next, both genotypes were administered 50 mg/kg cyclopamine and perfused six hours later. Cre immunolabeling was performed to confirm expression only in the EGL of Bax/Bak CKO mice (Figure 3I). Semi-quantitative evaluation of AC3 revealed high amounts of EGL apoptosis in Bax/Bak Cre-mice that was absent in Bax/Bak CKO mice (Figure 3B and Figure 3I; t[10] = 7.441, p < 0.0001). Next we examined if HA-induced apoptosis was p53 dependent.p53 screens the genome for errors and, if present, activates DNA repair proteins. However, if DNA damage is irreparable, apoptosis is initiated to prevent further cell proliferation. p53 knockout mice were first validated by injecting 25 mg/kg cytosine arabinoside, a genotoxin that produces p53 dependent EGL apoptosis (Noguchi et al., 2008). As predicted, p53 heterozygous mice exhibited dramatic increases in EGL apoptosis which was absent in p53 knockouts (t[15] = 52.63, p< 0.0001; Figure 3C and Figure 3J). Finally, we administered 50 mg/kg vismodegib and found that p53 knockout had no effect on apoptosis (t[14] = 0.2078, p > 0.05; Figure 3D). Thus, we conclude HA-induced apoptosis is Bax/Bak dependent but p53 independent.
Figure 3. Mechanism of Hedgehog induced EGL apoptosis.
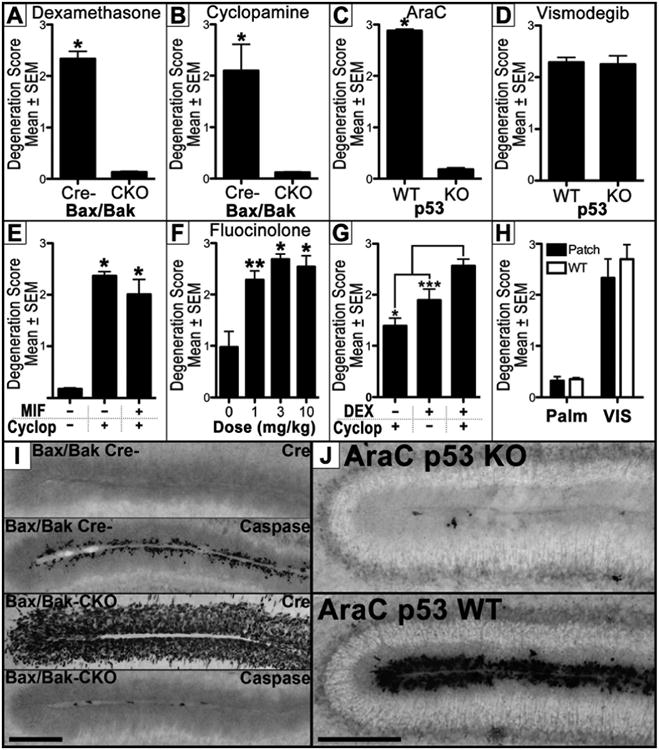
(A-B) Administration of (A) dexamethasone and (B) cyclopamine to Bax/Bak conditional knockout (CKO) and Bax/Bak Cre negative (Cre-) mice show the EGL apoptosis produced by both is Bax/Bak dependent. (C) Alternatively, administration of the genotoxin cytosine arabinoside (AraC) or (D) the Hedgehog antagonist (HA) vismodegib to p53 knockout (KO) mice show that only AraC is p53 dependent. (E) Pretreatment with the glucocorticoid (GC) antagonist mifepristone (MIF) shows cyclopamine (Cyclop) induced apoptosis can occur independent of GC stimulation. (F) Administration of fluocinolone acetonide (a GC that potentiates the Hedgehog pathway) reveals that GCs do not produce EGL apoptosis by inhibiting the Hedgehog pathway. (G) Co-administration of dexamethasone (a synthetic GC; DEX) and cyclopamine significantly increases EGL apoptosis when compared to each drug alone. (H) Vismodegib treatment to both Patched and WT mice increases EGL apoptosis on PND7. (I) BaxBak Cre negative (BaxBak Cre-; top two panels) express no Cre leading to preserved Bax gene function and EGL apoptosis following 50 mg/kg vismodegib. Alternatively, Bax/Bak conditional knockout mice (Bax/Bak CKO; bottom two panels) express Cre selectively in the EGL leading to conditional Bax knockout and no EGL apoptosis following vismodegib exposure. Scale Bar: 150 μm. (J) Apoptosis produced by the genotoxin cytosine arabinoside (AraC) is potently suppressed in p53 knockout (KO; top) mice when compared to p53 wild-type (WT; bottom) mice. Scale Bar: 150 μm. * = p < 0.001, ** = p < 0.01, *** = p < 0.05
Hedgehog antagonists produce EGL apoptosis independent of glucocorticoid stimulation
Next we tested whether HAs were producing EGL apoptosis by regulating GC stimulation. In previous research, we discovered GCs potently increase apoptosis selectively in the EGL which may regulate its natural disappearance once neurogenesis is complete (as reviewed previously [Noguchi, 2014]). In the neonatal mouse brain, the 11β-hydroxysteroid dehydrogenase Type 2 (HSD2) enzyme is almost exclusively located in the EGL where it metabolizes endogenous GCs and protects against GC-induced EGL apoptosis (Heine and Rowitch, 2009). HSD2 protection is so important that its inhibition dramatically increases EGL apoptosis due to endogenous GC stimulation. As a result, this apoptosis can be completely blocked by pretreatment with the GC antagonist mifepristone (Noguchi et al., 2011). Importantly, Hh stimulation potently increases HSD2 expression which protects against GC-induced EGL apoptosis (Heine et al., 2011). Thus, it is possible HAs inhibit HSD2 resulting in increased GC-induced EGL apoptosis. If true, pretreatment with mifepristone should analogously block HA toxicity. Therefore, we injected three groups of mice with either 100 mg/kg mifepristone 15 minutes before cyclopamine exposure, vehicle 15 minutes before cyclopamine, or two vehicle injections separated by 15 minutes. Semi-quantitative analysis revealed mifepristone had no significant effect on cyclopamine-induced EGL apoptosis (Figure 3E; F[2,19] = 95.20, p < 0.0001). We therefore conclude cyclopamine can produce EGL apoptosis independent of GC stimulation.
Glucocorticoid-induced EGL apoptosis is not mediated through Hedgehog Inhibition
While we have found GCs potently increase EGL apoptosis (Noguchi et al., 2008), others have suggested GCs affect cerebellar development by directly inhibiting the Hh pathway (Heine and Rowitch, 2009). Unfortunately, this was in vitro research that the authors could not replicate (Heine et al., 2011) or reproduce in vivo (Heine et al., 2010). Of further concern, two independent groups discovered select GCs stimulate or potentiate (rather than inhibit) the Hh pathway (Wang et al., 2011, 2010, 2012). This suggests Hh inhibition is not an inherent downstream effect of GC signaling. Nevertheless, if this research is true, it is possible GCs inhibit the Hh pathway resulting in EGL apoptosis. Fluocinolone acetonide (FA) is a GC that potentiates the Hh pathway (Wang et al., 2010) and, as a result, cannot increase EGL apoptosis due to Hh inhibition. Therefore, we administered FA and perfused for AC3 immunolabeling six hours later. Semi-quantitative analysis revealed FA significantly increased EGL apoptosis (F[3,12] = 13.69, p < 0.001) at all doses tested in dose dependent manner (r = 0.5025, p < 0.05; Figure 3F), indicating that GC-induced EGL apoptosis is not produced by Hh inhibition.
Hedgehog antagonist-induced apoptosis is potentiated by glucocorticoids
The ability of HAs to induce EGL apoptosis independent of GCs suggests co-administration may have an additive effect. We found a 3 mg/kg DEX injection potently increases EGL apoptosis (Noguchi et al., 2011) comparable to HAs over the same time course. Therefore, PND7 mice were administered two injections 15 minutes apart of either 50 mg/kg cyclopamine and vehicle, 3 mg/kg DEX and vehicle, or 50 mg/kg cyclopamine and 3 mg/kg DEX. AC3 immunolabeling and semi-quantitative analysis revealed co-administration significantly increased EGL apoptosis compared to each drug alone (F[2,17]= 13.41, p < 0.001; Figure 3G). This indicates that GCs can potentiate toxicity produced by HAs.
Hedgehog antagonists also produce apoptosis in medulloblastomas
There is convincing evidence that the EGL sometimes fails to naturally disappear and its remnants proliferate out of control to form a MB (Goodrich et al., 1997; Oliver et al., 2005). Thus, it is important to determine if the EGL maintains a vulnerability to HA-induced apoptosis as it transitions to a MB. To test this possibility, we obtained mice that harbor a heterozygous knockout of the Patched receptor gene (Patched mice) leading to constitutive Hh activation and MB vulnerability through a haploinsufficient mechanism (Goodrich et al., 1997; Oliver et al., 2005). While the EGL normally disappears by PND14, it can often persist in Patched mice and malignantly transform into a MB. While the Patched mutation leads to increased Hh signaling, vismodegib should still antagonize the Hedgehog pathway by inhibiting Smoothened lying downstream of the Patched receptor. Therefore, PND7 Patched mice and their wild-type littermates were injected with 50 mg/kg vismodegib or vehicle and perfused 6 hours later for AC3. Semi-quantitative scoring of apoptosis was analyzed via 2-way ANOVA and revealed a main effect of treatment (F[1,13] = 258.6, p < 0.0001) but not genotype (F[1,14] = 2.171, p > 0.05) and no interaction (F[1,13] = 1.536, p > 0.05). A subsequent one-way ANOVA planned comparison between treatment groups within the same genotype (F[3,13] = 87.45, p < 0.0001) and a Bonferroni posthoc revealed vismodegib increased EGL apoptosis in both genotypes (Figure 3H). Therefore, we conclude vismodegib can produce EGL apoptosis in neonatal Patched mice.
Some Patched mice were then allowed to age and perfused at the first sign of tumor burden (tremor/gait disturbance, ataxia, loss of balance, listing to one side). Animals exhibited large MBs positive for the proliferative cell marker KI-67 (Figure 4A). We next tested if vismodegib induces apoptosis in MBs. Patched mice displaying tumor burden were administered 50 mg/kg vismodegib or vehicle and perfused six hours later. Since MBs vary in size and shape, semi-quantitative scoring was impractical. Thus, we performed density cell counts in tumors to quantify AC3 positive apoptotic cells. Abundant amounts of apoptosis was detected in MBs with no treatment consistent with previous research (DeSouza et al., 2014) (Figure 4B and Figure 4C). However, vismodegib significantly increased tumor density counts compared to vehicle, (t[11] = 4.126, p < 0.01; Figure 4B and Figure 4C-D) indicating vulnerability to HA-induced apoptosis is maintained as EGL NPCs malignantly transform into tumors.
Figure 4. Hedgehog antagonist-induced apoptosis in medulloblastoma prone Patched mice.

(A) A Patched mouse with medulloblastoma (white asterisk) reveals high levels of KI-67 (proliferative cell marker; green) immunolabeling in the tumor while DAPI (nuclear stain; blue) highlights normal internal granule layer cells. Scale Bar: 1000 μm. (B) Vismodegib (VIS) significantly increased apoptotic density counts (measured with activated caspase-3) in Patched mice with medulloblastomas when compared to vehicle treatment. (C) While medulloblastomas in vehicle treated Patched mice still display high levels of physiological apoptosis, this toxicity is significantly elevated following VIS exposure (D). Scale Bar: 250 μm.
Discussion
Implications for cerebellar development
Hh signaling is intensely studied in cerebellar development due to its robust effects on the EGL. However Hh signaling has traditionally been thought of as a potent mitogen that increases EGL proliferation (Wechsler-Reya and Scott, 1999). Alternatively, the discovery that this pathway acts as a survival signal for the EGL may fundamentally change our understanding of its role in cerebellar development. Firstly, since NPC apoptosis dramatically decreases neurogenesis (Depaepe et al., 2005; Haydar et al., 1999), much (or even a major) proportion of reduced proliferation caused by HAs may be due to the dramatic loss of NPCs (Figure 1B-C). Predictably, confusing changes in progenitor cell apoptosis with its effects on proliferation has happened for other signaling pathways. For instance, it was once thought Bcl-2 (an anti-apoptotic protein) enhanced proliferation but later research discovered that it actually reduced progenitor cell apoptosis (Cory and Adams, 2002). Secondly, since Hh signaling morphologically sculpts the neural tube by spatially regulating NPC apoptosis (Charrier et al., 2001; Thibert et al., 2003), this pathway may play a similar role in the developing cerebellum. For instance, laminin, Notch2, and SDF-1α are present in the outer EGL and increase Hh signaling (Wechsler-Reya, 2003). However, this same pathway is inhibited in the inner EGL by vitronectin and the internal granule layer by Notch1 (Fan et al., 2004; Wechsler-Reya, 2001). Therefore, despite the fact that Sonic Hh ligand becomes less concentrated at distances farther away from Purkinje cells (its source), Gli transcription (the read-out of the Hh pathway) is most highly expressed in the outer EGL (Vaillant and Monard, 2009; Wechsler-Reya, 2003). As a result, strong Hh signaling would prevent progenitors appropriately located in the outer EGL from apoptotic death. Alternatively, if NPCs became displaced or the EGL expanded to regions too distant from the outer EGL, the loss of Hh signaling would initiate apoptosis. This suggests the Hh pathway may act as a developmental signal to regionally isolate NPCs and morphologically sculpt cerebellar shape by controlling where proliferation occurs. Finally, while there is a tremendous focus on the neurotrophic theory (neuronal apoptosis) (la Rosa and De Pablo, 2000), a majority of neurodevelopmental apoptosis occurs in proliferative cells (Blaschke et al., 1996; Depaepe et al., 2005; Kuida et al., 1996) and very few regulators of NPC apoptosis have been discovered so far (Haydar et al., 1999; la Rosa and De Pablo, 2000). As a result, this may be one of only a few regulators of NPC apoptosis that is currently known.
Paradoxically, while we have found Hh antagonism potently increases EGL apoptosis in vivo, loss of Hh signaling in vitro produces differentiation (Wechsler-Reya and Scott, 1999). Interestingly, previous research may provide insight into this inconsistency. While we found HA-induced apoptosis is Bax/Bak dependent, these proteins normally need to be activated before signaling apoptotic death. Importantly, around PND7, strong endogenous Hh stimulation transitions EGL NPCs to a “primed for death” state in which progenitors become loaded with the activated form of Bax, but Bcl-xL (an anti-apoptotic protein in the BCL2 family) is preventing it from producing apoptosis (Crowther et al., 2013) (Figure 5; circles A, C, and D). Thus, these “primed” NPCs will undergo apoptosis only when a BH3-only sensitizer (a family of proteins that regulate apoptosis) inhibits Bcl-xL and allows activated Bax to produce apoptosis (Figure 5; circles E-F). We suggest Hh stimulation may also inhibit a BH3-only sensitizer allowing Bcl-xL to prevent apoptosis (Figure 5; circle G). Thus, injection of a HA (Figure 5; circle B) may trigger BH3-only sensitizer activation that inhibits Bcl-xL enough to allow loaded activated Bax to initiate apoptosis. Alternatively, when EGL NPCs differentiate (rather than undergo apoptosis) in vitro, they are cultured in Hh-free medium (Lin and Bulleit, 1996; Wechsler-Reya and Scott, 1999). As a result, there is no initial strong Hh stimulation to load activated Bax (priming them for death) before the abrupt loss of Hh signaling. This effect may be even more important in MBs driven by Hh stimulation where large numbers of proliferating cells are “primed” for death. Ultimately, this suggests the fate of EGL NPCs can vary dramatically dependent on Hh signaling over time. This will be an important line of research to pursue in future research.
Figure 5. Hedgehog signaling primes EGL NPCs for apoptosis.
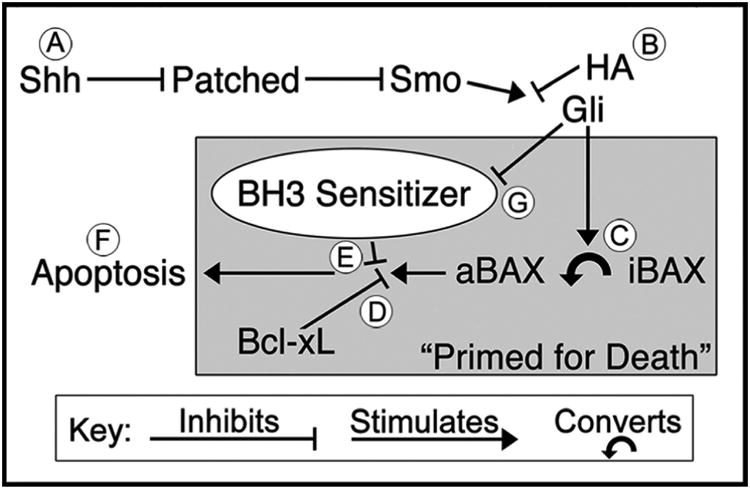
(A) The Sonic Hedgehog (Hh) pathway begins with the release of the Sonic Hh ligand from Purkinje cells onto EGL NPCs where it binds and inhibits the Patched receptor. Since activation of the Patched receptor normally represses Smoothened (Smo) activity, this reduces inhibition of Smo leading to a downstream activation of Gli transcription factors. (B) The Hh antagonists (HAs) vismodegib, cyclopamine, and jervine antagonize the Hh pathway at the level of Smo. (C) Hh stimulation transitions EGL NPCs into a “Primed for Death” state that converts inactivated Bax (iBAX) into an activated form (aBAX) that can produce apoptosis. (D) However, aBAX is prevented from producing apoptosis by the Bcl-xL anti-apoptotic protein. (E) Since Bcl-xL is the only thing preventing apoptosis, its inhibition by a BH3 sensitizer allows aBAX to initiate apoptosis (F). (G) We hypothesize that Hedgehog stimulation acts as a survival signal by inhibiting the production of a BH3-only sensitizer that would otherwise block Bcl-xL and allow aBAX to produce apoptosis.
Translational Implications
While there is intense interest in HA therapies for MBs (Robinson, 2013), their anti-tumor effects are traditionally thought to be regulated by reductions in proliferation not increased apoptosis (Berman et al., 2002; Romer and Curran, 2005). Interestingly, some have acknowledged that the treatment regimens used are chronic which makes it difficult to detect apoptotic cells that are quickly removed in rodents (Romer and Curran, 2005). Indeed, we found AC3 positivity occurs within a limited window of 6-8 hours after HA exposure. The obvious translational implication is that strong Hh signaling would reduce apoptosis, thereby promoting neoplastic transformation and enhancing malignancy. While a major downstream effect of NPC apoptosis is decreased proliferation, it is tempting to dismiss these findings as unimportant. However, the effect of HAs on apoptosis (rather than proliferation) has unique translational implications. Firstly, since apoptosis involves particular signaling pathways distinct from proliferation this research may provide novel insight into MB resistance. For instance, Hh-driven MBs in children over 3 years of age tend to harbor frequent p53 mutations (Kool et al., 2014). Since we found HA-induced apoptosis is p53 independent, this therapy may be particularly effective in these types of MBs. Secondly, since Hh signaling can also serve as a survival signal in MB cells, this toxicity may prevent metastasis by killing tumor cells as they spread to other regions where this signal is absent. If true, tumor cells need to develop a cell intrinsic resistance to this toxicity in order to spread. Thirdly, since we found HAs induce apoptosis independent of GC signaling, co-administration of both drugs may be a more effective treatment than each alone. Studies in mice and humans report that vismodegib treatment can cause somatic missense mutations in the Smoothened allele that block this drug's binding target (Metcalfe and de Sauvage, 2011; Yauch et al., 2009). However, GCs are unaffected by this mutation and are also able to increase MB apoptosis, reduce tumor size, and enhance survival in mice (Heine et al., 2010). As a result, co-administration of both drugs may lead to a treatment less prone to MB resistance. Fourthly, while up to 30% of MBs are driven by excessive Hh signaling, another 25% (called Group 3 MBs) may also be of EGL origin (Kawauchi et al., 2012). If Hh signaling is truly a survival signal, then HAs may effectively treat this group by selectively increasing tumor apoptosis. Finally, this toxicity can also be used as a sensitive in vivo model for rapidly screening novel HAs for their therapeutic potential in the intact animal. For instance, previous research has found vismodegib can inhibit Gli1 mRNA down to a dose of approximately 1 mg/kg (Wong et al., 2011). This correlates well with our finding that this same drug increased EGL apoptosis at 1 mg/kg and above. In addition, while itraconazole is an HA, it has a limited ability to cross the blood brain barrier (Kethireddy and Andes, 2007). Consistent with this finding, we found this drug has no effect on EGL apoptosis at any dose tested.
Conclusions
While the Hh pathway is important for both cerebellar and MB development, its effects were primarily thought to involve proliferation and/or differentiation. Here we show that Hh signaling may also serve as a survival signal that morphologically sculpts the cerebellum and plays an important role in MB development, metastasis, and treatment.
Acknowledgments
Research in this paper was supported by NIH grants MH083046, HD052664, & HD055365 and the Intellectual and Developmental Disabilities Research Center at Washington University (NIH/NICHD P30 HD062171).
Abbreviations
- HSD2
11β-hydroxysteroid dehydrogenase Type 2
- AC3
activated caspase-3
- DEX
dexamethasone
- EGL
external granule layer
- FA
fluocinolone acetonide
- GCs
glucocorticoids
- Hh
Hedgehog
- HA
Hedgehog antagonist
- MBs
medulloblastomas
- NPCs
neural progenitor cells
Footnotes
The authors disclose no conflicts of interest.
Contributor Information
Omar Hoseá Cabrera, Email: ohcm97@umsl.edu.
Brant S. Swiney, Email: swineyb@psychiatry.wustl.edu.
Patricia Salinas-Contreras, Email: psc000193@gmail.com.
Julie Kathryn Smith, Email: juliek.smith85@gmail.com.
Nuri B. Farber, Email: farbern@psychiatry.wustl.edu.
References
- Andersen BB, Korbo L, Pakkenberg B. A quantitative study of the human cerebellum with unbiased stereological techniques. J Comp Neurol. 1992;326:549–560. doi: 10.1002/cne.903260405. [DOI] [PubMed] [Google Scholar]
- Berman DM, Karhadkar SS, Hallahan AR, Pritchard JI, Eberhart CG, Watkins DN, Chen JK, Cooper MK, Taipale J, Olson JM, Beachy PA. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science. 2002;297:1559–61. doi: 10.1126/science.1073733. [DOI] [PubMed] [Google Scholar]
- Blaschke AJ, Staley K, Chun J. Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development. 1996;122:1165–74. doi: 10.1242/dev.122.4.1165. [DOI] [PubMed] [Google Scholar]
- Charrier JB, Lapointe F, Le Douarin NM, Teillet MA. Anti-apoptotic role of Sonic hedgehog protein at the early stages of nervous system organogenesis. Development. 2001;128:4011–20. doi: 10.1242/dev.128.20.4011. [DOI] [PubMed] [Google Scholar]
- Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–56. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- Crowther AJ, Gama V, Bevilacqua A, Chang SX, Yuan H, Deshmukh M, Gershon TR. Tonic activation of Bax primes neural progenitors for rapid apoptosis through a mechanism preserved in medulloblastoma. J Neurosci. 2013;33:18098–108. doi: 10.1523/JNEUROSCI.2602-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depaepe V, Suarez-Gonzalez N, Dufour A, Passante L, Gorski JA, Jones KR, Ledent C, Vanderhaeghen P. Ephrin signalling controls brain size by regulating apoptosis of neural progenitors. Nature. 2005;435:1244–1250. doi: 10.1038/nature03651. [DOI] [PubMed] [Google Scholar]
- DeSouza RM, Jones BRT, Lowis SP, Kurian KM. Pediatric medulloblastoma - update on molecular classification driving targeted therapies. Front Oncol. 2014;4:176. doi: 10.3389/fonc.2014.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Mikolaenko I, Elhassan I, Ni X, Wang Y, Ball D, Brat DJ, Perry A, Eberhart CG. Notch1 and notch2 have opposite effects on embryonal brain tumor growth. Cancer Res. 2004;64:7787–93. doi: 10.1158/0008-5472.CAN-04-1446. [DOI] [PubMed] [Google Scholar]
- Farber NB, Creeley CE, Olney JW. Alcohol-induced neuroapoptosis in the fetal macaque brain. Neurobiol Dis. 2010;40:200–6. doi: 10.1016/j.nbd.2010.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajjar AJ, Stewart CF, Ellison DW, Curran P, Phillips P, Goldman S, Packer R, Kun LE, Boyett JM, Gilbertson RJ. A phase I pharmacokinetic trial of sonic hedgehog (SHH) antagonist GDC-0449 in pediatric patients with recurrent or refractory medulloblastoma: A Pediatric Brain Tumor Consortium study (PBTC 25) J Clin Oncol. 2010;28 CRA9501. [Google Scholar]
- Goldowitz D, Hamre K. The cells and molecules that make a cerebellum. Trends Neurosci. 1998;21:375–82. doi: 10.1016/s0166-2236(98)01313-7. [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Milenković L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–13. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- Gould A, Missailidis S. Targeting the hedgehog pathway: the development of cyclopamine and the development of anti-cancer drugs targeting the hedgehog pathway. Mini Rev Med Chem. 2011;11:200–13. doi: 10.2174/138955711795049871. [DOI] [PubMed] [Google Scholar]
- Guerrero I, Ruiz i Altaba A. Development Longing for ligand: hedgehog, patched, and cell death. Science. 2003;301:774–6. doi: 10.1126/science.1088625. [DOI] [PubMed] [Google Scholar]
- Hanslick JL, Lau K, Noguchi KK, Olney JW, Zorumski CF, Mennerick S, Farber NB. Dimethyl sulfoxide (DMSO) produces widespread apoptosis in the developing central nervous system. Neurobiol Dis. 2009;34:1–10. doi: 10.1016/j.nbd.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey RJ, Napper RM. Quantitative study of granule and Purkinje cells in the cerebellar cortex of the rat. J Comp Neurol. 1988;274:151–157. doi: 10.1002/cne.902740202. [DOI] [PubMed] [Google Scholar]
- Haydar TF, Kuan CY, Flavell RA, Rakic P. The role of cell death in regulating the size and shape of the mammalian forebrain. Cereb Cortex. 1999;9:621–626. doi: 10.1093/cercor/9.6.621. [DOI] [PubMed] [Google Scholar]
- Heine VM, Griveau A, Chapin C, Ballard PL, Chen JK, Rowitch DH. A small-molecule smoothened agonist prevents glucocorticoid-induced neonatal cerebellar injury. Sci Transl Med. 2011;3:105ra104. doi: 10.1126/scitranslmed.3002731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine VM, Priller M, Ling J, Rowitch DH, Schüller U. Dexamethasone destabilizes Nmyc to inhibit the growth of hedgehog-associated medulloblastoma. Cancer Res. 2010;70:5220–5225. doi: 10.1158/0008-5472.CAN-10-0554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine VM, Rowitch DH. Hedgehog signaling has a protective effect in glucocorticoid-induced mouse neonatal brain injury through an 11betaHSD2-dependent mechanism. J Clin Invest. 2009;119:267–277. doi: 10.1172/JCI36376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi D, Robinson G, Uziel T, Gibson P, Rehg J, Gao C, Finkelstein D, Qu C, Pounds S, Ellison DW, Gilbertson RJ, Roussel MF. A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell. 2012;21:168–80. doi: 10.1016/j.ccr.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kethireddy S, Andes D. CNS pharmacokinetics of antifungal agents. Expert Opin Drug Metab Toxicol. 2007;3:573–581. doi: 10.1517/17425255.3.4.573. [DOI] [PubMed] [Google Scholar]
- Kim J, Tang JY, Gong R, Kim J, Lee JJ, Clemons KV, Chong CR, Chang KS, Fereshteh M, Gardner D, Reya T, Liu JO, Epstein EH, Stevens DA, Beachy PA. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell. 2010;17:388–99. doi: 10.1016/j.ccr.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RS, Rubin JB, Gibson HD, DeHaan EN, Alvarez-Hernandez X, Segal RA, Luster AD. SDF-1{alpha} induces chemotaxis and enhances Sonic hedgehog-induced proliferation of cerebellar granule cells. Development. 2001;128:1971–1981. doi: 10.1242/dev.128.11.1971. [DOI] [PubMed] [Google Scholar]
- Kool M, Jones DTW, Jäger N, Northcott PA, Pugh TJ, Hovestadt V, Piro RM, Esparza LA, Markant SL, Remke M, Milde T, Bourdeaut F, Ryzhova M, Sturm D, Pfaff E, Stark S, Hutter S, Seker-Cin H, Johann P, Bender S, Schmidt C, Rausch T, Shih D, Reimand J, Sieber L, Wittmann A, Linke L, Witt H, Weber UD, Zapatka M, König R, Beroukhim R, Bergthold G, van Sluis P, Volckmann R, Koster J, Versteeg R, Schmidt S, Wolf S, Lawerenz C, Bartholomae CC, von Kalle C, Unterberg A, Herold-Mende C, Hofer S, Kulozik AE, von Deimling A, Scheurlen W, Felsberg J, Reifenberger G, Hasselblatt M, Crawford JR, Grant GA, Jabado N, Perry A, Cowdrey C, Croul S, Zadeh G, Korbel JO, Doz F, Delattre O, Bader GD, McCabe MG, Collins VP, Kieran MW, Cho YJ, Pomeroy SL, Witt O, Brors B, Taylor MD, Schüller U, Korshunov A, Eils R, Wechsler-Reya RJ, Lichter P, Pfister SM. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014;25:393–405. doi: 10.1016/j.ccr.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- La Rosa EJ, De Pablo F. Cell death in early neural development: beyond the neurotrophic theory. Trends Neurosci. 2000;23:454–458. doi: 10.1016/s0166-2236(00)01628-3. [DOI] [PubMed] [Google Scholar]
- Lau K, Swiney BS, Reeves N, Noguchi KK, Farber NB. Propylene glycol produces excessive apoptosis in the developing mouse brain, alone and in combination with phenobarbital. Pediatr Res. 2012;71:54–62. doi: 10.1038/pr.2011.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leary SES, Olson JM. The molecular classification of medulloblastoma: driving the next generation clinical trials. Curr Opin Pediatr. 2012;24:33–9. doi: 10.1097/MOP.0b013e32834ec106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Bulleit RF. Cell intrinsic mechanisms regulate mouse cerebellar granule neuron differentiation. Neurosci Lett. 1996;220:81–4. doi: 10.1016/s0304-3940(96)13214-6. [DOI] [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, Chen Y, Wei M, Eng VM, Adelman DM, Simon MC, Ma A, Golden JA, Evan G, Korsmeyer SJ, MacGregor GR, Thompson CB. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–99. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalfe C, de Sauvage FJ. Hedgehog fights back: mechanisms of acquired resistance against Smoothened antagonists. Cancer Res. 2011;71:5057–61. doi: 10.1158/0008-5472.CAN-11-0923. [DOI] [PubMed] [Google Scholar]
- Noguchi KK. Glucocorticoid Induced Cerebellar Toxicity in the Developing Neonate: Implications for Glucocorticoid Therapy during Bronchopulmonary Dysplasia. Cells. 2014;3:36–52. doi: 10.3390/cells3010036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi KK, Lau K, Smith DJ, Swiney BS, Farber NB. Glucocorticoid receptor stimulation and the regulation of neonatal cerebellar neural progenitor cell apoptosis. Neurobiol Dis. 2011;43:356–363. doi: 10.1016/j.nbd.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi KK, Walls KC, Wozniak DF, Olney JW, Roth KA, Farber NB. Acute neonatal glucocorticoid exposure produces selective and rapid cerebellar neural progenitor cell apoptotic death. Cell Death Differ. 2008;15:1582–1592. doi: 10.1038/cdd.2008.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman G. Likert scales, levels of measurement and the “laws” of statistics. Adv Health Sci Educ Theory Pract. 2010;15:625–32. doi: 10.1007/s10459-010-9222-y. [DOI] [PubMed] [Google Scholar]
- Oliver TG, Read TA, Kessler JD, Mehmeti A, Wells JF, Huynh TTT, Lin SM, Wechsler-Reya RJ. Loss of patched and disruption of granule cell development in a pre-neoplastic stage of medulloblastoma. Development. 2005;132:2425–39. doi: 10.1242/dev.01793. [DOI] [PubMed] [Google Scholar]
- Olney J, Young C, Wozniak DF, Jevtovic-Todorovic V, Ikonomidou C. Do pediatric drugs cause developing neurons to commit suicide? Trends Pharmacol Sci. 2004;25:135–9. doi: 10.1016/j.tips.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Robinson GW. Impact of tumor location on medulloblastoma subtyping and treatment. Pediatr Blood Cancer. 2013;60:1393–4. doi: 10.1002/pbc.24549. [DOI] [PubMed] [Google Scholar]
- Romer J, Curran T. Targeting medulloblastoma: small-molecule inhibitors of the Sonic Hedgehog pathway as potential cancer therapeutics. Cancer Res. 2005;65:4975–8. doi: 10.1158/0008-5472.CAN-05-0481. [DOI] [PubMed] [Google Scholar]
- Rudin CM. Vismodegib. Clin Cancer Res. 2012;18:3218–22. doi: 10.1158/1078-0432.CCR-12-0568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, Holcomb T, Stinson J, Gould SE, Coleman B, LoRusso PM, Von Hoff DD, de Sauvage FJ, Low JA. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009;361:1173–8. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibert C, Teillet MA, Lapointe F, Mazelin L, Le Douarin NM, Mehlen P. Inhibition of neuroepithelial patched-induced apoptosis by sonic hedgehog. Science. 2003;301:843–6. doi: 10.1126/science.1085405. [DOI] [PubMed] [Google Scholar]
- Vaillant C, Monard D. SHH pathway and cerebellar development. Cerebellum. 2009;8:291–301. doi: 10.1007/s12311-009-0094-8. [DOI] [PubMed] [Google Scholar]
- Wang J, Barak LS, Mook RA, Chen W. Glucocorticoid hedgehog agonists in neurogenesis. Vitam Horm. 2011;87:207–15. doi: 10.1016/B978-0-12-386015-6.00030-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Lu J, Bond MC, Chen M, Ren XR, Lyerly HK, Barak LS, Chen W. Identification of select glucocorticoids as Smoothened agonists: potential utility for regenerative medicine. Proc Natl Acad Sci U S A. 2010;107:9323–8. doi: 10.1073/pnas.0910712107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Davidow L, Arvanites AC, Blanchard J, Lam K, Xu K, Oza V, Yoo JW, Ng JMY, Curran T, Rubin LL, McMahon AP. Glucocorticoid compounds modify smoothened localization and hedgehog pathway activity. Chem Biol. 2012;19:972–82. doi: 10.1016/j.chembiol.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler-Reya RJ. Analysis of gene expression in the normal and malignant cerebellum. Recent Prog Horm Res. 2003;58:227–48. doi: 10.1210/rp.58.1.227. [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya RJ. Caught in the matrix: how vitronectin controls neuronal differentiation. Trends Neurosci. 2001;24:680–682. doi: 10.1016/S0166-2236(00)02058-0. [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22:103–14. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- Wong H, Alicke B, West KA, Pacheco P, La H, Januario T, Yauch RL, de Sauvage FJ, Gould SE. Pharmacokinetic-pharmacodynamic analysis of vismodegib in preclinical models of mutational and ligand-dependent Hedgehog pathway activation. Clin Cancer Res. 2011;17:4682–92. doi: 10.1158/1078-0432.CCR-11-0975. [DOI] [PubMed] [Google Scholar]
- Yauch RL, Dijkgraaf GJP, Alicke B, Januario T, Ahn CP, Holcomb T, Pujara K, Stinson J, Callahan CA, Tang T, Bazan JF, Kan Z, Seshagiri S, Hann CL, Gould SE, Low JA, Rudin CM, de Sauvage FJ. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science. 2009;326:572–4. doi: 10.1126/science.1179386. [DOI] [PMC free article] [PubMed] [Google Scholar]