

Abstract
B cell lymphomas (BCL) are characterized by widespread deregulation of gene expression when compared with their normal B cell counterparts. Recent epigenomic studies defined cis-regulatory elements (REs) whose activities are altered in BCL to drive some of these pathogenic expression changes. During transformation, multiple mechanisms are employed to alter RE activities, including perturbations in the function of chromatin modifiers, which can lead to revision of the B cell epigenome. Inherited and somatic variants also alter RE function via disruption of TF binding. Aberrant expression of non-coding RNAs deregulates genes involved in B cell differentiation via direct repression and post-transcriptional targeting. These discoveries have established epigenetic etiologies for B cell transformation that are being exploited by novel therapeutic approaches.
B Cell Lymphomas
B cell lymphomas (BCL) comprise a heterogeneous group of malignancies with a combined incidence of nearly 90,000 cases and 25,000 deaths in the U.S. each year (www.seer.cancer.gov). Not surprisingly, the molecular pathogenesis of BCL is also heterogeneous; however, one common feature is the deregulation of gene expression programs that promote the survival and expansion of the transformed B cells [1–6]. As we review here, recent genomic and epigenomic studies have begun to shed light on the underlying mechanisms that drive these pathogenic changes in gene expression and, importantly, implicate new therapeutic targets.
Cell of Origin Model for Lymphoma
B lymphocytes diversify genes for immunoglobulin (Ig) antigen receptors during their development from precursor cells in the bone marrow [7]. Upon expression of its unique B cell receptor (BCR), each clone exits the bone marrow as a naïve B cell and migrates to the periphery, circulating through secondary lymphoid organs until it encounters a cognate antigen. Upon BCR activation in lymph nodes, a B cell moves from the mantle zone to the T cell-rich region, aggregating into primary follicles and forming germinal centers (GCs), which are sites of sustained B cell proliferation and differentiation. GCs are also the anatomic location in which Ig genes are further modified to tailor the humoral immune response for elimination of the invading pathogen. Specifically, GC-B cells undergo somatic hypermutation (SHM), which is mediated by activation-induced cytosine deaminase (AID) and facilitates affinity maturation of BCRs [8]. The cycles of SHM, proliferation, and clonal selection of B lymphocytes in the GC are potentially oncogenic. Thus, it is not surprising that the most common BCLs are thought to derive from GC-B cells. Gene expression programs also are altered dramatically during the GC reaction, in large part, due to changes in the levels of key transcription factors (TFs), including BCL6, IRF4, IRF8, POU2AF1(OCA-B), and SPI-B [9–11].
Gene expression signatures of B cell differentiation are also a fundamental component of the “cell of origin” theory, which links individual B cell malignancies to normal counterparts in B cell differentiation stages using phenotypic and molecular similarities (Figure 1) [12]. Application of this theory segregates the most common B cell lymphoma, Diffuse Large B-Cell Lymphoma (DLBCL), into two subtypes, Germinal Center B-cell-like (GCB) and Activated B-cell-like (ABC), which also have distinct mutation profiles and prognoses [1,13]. More recently, epigenome profiling also revealed two subtypes of Follicular Lymphoma (FL), each with distinct patterns of enhancer activity, which alters the expression of target genes and suggests divergent modes of pathogenesis [14]. It is likely that similar distinctions in regulatory signatures underlie the segregation of gene expression profiles in GCB- versus ABC-DLBCL.
Figure 1. Types of B cell lymphoma and their normal cell counterparts.
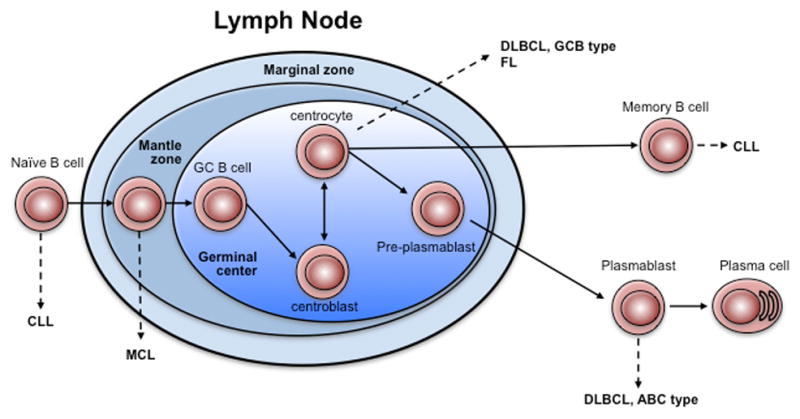
The schematic depicts B cell maturation and the relationship of normal counterpart cells to different subtypes of B cell lymphoma. While some controversies exist with regard to ABC-DLBCL, the preponderance of evidence suggests that the plasmablast is the closest normal counterpart B cell.
Based on such molecular profiling, other types of BCL are thought to originate from non-GC cells. For example, mutation patterns and gene expression profiling divide Chronic Lymphocytic Leukemia/Lymphoma (CLL) into two clinically and pathologically distinct groups postulated to derive from antigen-experienced naïve or post-GC B cells [15]. Although Mantle Cell Lymphomas (MCL) are universally aggressive and characterized by constitutive over-expression of cyclin D1, they are also subdivided by distinct mutational and gene expression profiles corresponding with naïve or post-GC B cell origins [16]. Other mature B cell malignancies, such as Hodgkin Lymphoma and Myeloma have been reviewed elsewhere [17,18].
Pathogenic Gene Regulatory Circuits in Lymphoma
The changes in gene expression that drive cellular differentiation are coordinated by a complex network of cis-regulatory elements (REs, see Glossary), including transcriptional promoters and enhancers. Connections between these two types of REs can occur over large distances, forming transcriptional circuits that control gene expression (Figure 2). Importantly, certain promoter-enhancer activities and their connections are fluid, enabling gene expression changes in response to developmental or environmental cues. The ENCODE Consortium, Roadmap Epigenome project, and others have employed chromatin profiling to identify and catalogue REs in dozens of different cell types and tissues (Box 1) [19–21]. Strikingly, the most variable component of transcriptional programs appears to be the patterns of associated enhancer activity, which regulate cell type- and context-specific gene expression [20–22]. In this regard, the expression of many cell fate genes is controlled by a specialized subset of enhancers, termed Super-Enhancers (SEs). SEs encompass hyperacetylated clusters of traditional enhancer elements that bind the master TFs of a given cell lineage or differentiation state [23].
Figure 2. Enhancers and target genes comprise transcriptional circuits.

Much like electrical circuits, transcriptional circuits regulate the direction and amplitude of their input (epigenetic modifications, chromatin organization, TF and transcriptional activator/repressor binding, RNA polymerase binding) to control their output (gene expression). In the top panel, the diagram depicts boundary elements, including CTCF sites, as electrical resistors, which set the boundaries of regions with similar transcriptional activity by blocking the spread of chromatin marks and preventing physical interactions between different transcriptional domains. The relative activation status of enhancers tunes the expression level of target genes, creating gradations in transcriptional output, and thus are depicted as amplifiers. In a normal B cell, the circuit is depicted linearly, to indicate a lack of physical interaction (i.e. chromatin looping) between each enhancer and its target gene. In a lymphoma B cell, aberrant enhancer activity changes the 3D genomic conformation, inducing interactions between the enhancer and target gene promoters within the transcriptional domain, potentiating their expression level. The bottom panel shows UCSC Genome Browser views of FAIRE, which detects nucleosome depleted regions of open chromatin; H3K27ac ChIP-seq data, which detects histone marks that are enriched in active enhancers and expressed gene bodies; and RNA-seq, which measures gene expression, for normal and lymphoma B cells. In normal B cells, the example enhancers and gene targets exhibit low levels of FAIRE and H3K27ac signal, and corresponding low expression. In lymphoma B cells, increased activity of the left enhancer and upregulated expression of Genes 1 and 2 is evident by elevated FAIRE, H3K27ac, and RNA-seq signals.
Emerging data from genome and epigenome studies now point to transcriptional REs as major drivers of pathogenic gene expression in human disease (Figure 2). Importantly, alterations in the epigenome in general, and in enhancer activity specifically, are observed in nearly all types of cancer, including BCL [14,24–27]. As we will review here, malignant transformation disrupts exquisitely controlled transcriptional programs, deregulating genes in key pathways to promote proliferation and survival of lymphoma cells. The reprogramming of gene expression in BCLs has numerous sources, including (1) altered epigenetic modification of REs (DNA methylation and histone modifications), (2) variations in the expression of TFs and chromatin modifiers, (3) inherited and acquired sequence variants in REs, and (4) changes in non-coding RNA expression. We will consider each of these in turn.
Mechanisms of Gene Deregulation in Lymphoma: Altered Epigenomic Landscapes
DNA Methylation
Epigenomic modifications can be separated into two general categories: DNA methylation and histone marks. Methylation of cytosines in CpG dinucleotides near promoters is associated with silencing, whereas CpG methylation in exons is linked to transcriptional activation [28]. Prior studies have shown that polymorphic DNA methylation patterns exist in human populations; however, their relevance to BCL remains to be established [29]. Moreover, mutations in enzymes that methylate or demethylate DNA are common in some hematopoietic malignancies [30,31], but are rare in BCL [32–36]. Nevertheless, DNA methylation profiles are deranged in all BCL subtypes profiled [12,37–40]. As with gene expression profiling, the ABC and GCB subtypes of DLBCL can be separated by discrete DNA methylation patterns [40]. More recently, genome-wide profiling revealed that DLBCL samples could be segregated into six clusters with differential methylation patterns that associate aberrant methylation with key lymphoma pathways, such as cytokine signaling or cell cycle and apoptosis [41]. Similarly, Queiros et al. identified five differentially methylated regions that segregate CLL samples into three groups based on their resemblance to normal B cells: naïve-like, intermediate, and memory-like. In addition to these phenotypic differences, epigenetically-defined subgroups manifest significant differences in disease progression and overall survival [42]. DNA methylation profiles also revealed intratumoral heterogeneity within DLBCL or CLL samples. Indeed, a higher degree of variability in DNA methylation patterns at diagnosis was associated with more aggressive disease and adverse outcome. These findings raise the intriguing possibility that, like genomic instability, epigenomic instability may provide an “evolutionary” advantage in the progression of BCL [43,44].
Histone Modifications
In contrast to DNA methylation, the language of chromatin modifications is considerably more complex, with the potential for many combinations of covalent marks on a single histone protein (Figure 3). The combination of histone marks in a genomic region, rather than a single modification, determines its “chromatin state”, which impacts relative levels of transcriptional activity or repression [20,22].
Figure 3. Post-translational modification of chromatin proteins regulates genome organization and gene expression.
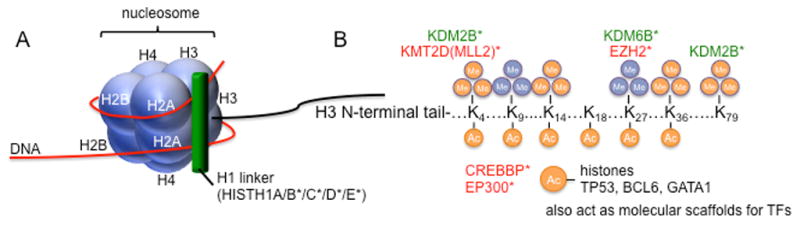
(A) The basic component of chromatin is the nucleosome, which is an octamer comprised of 2 copies each of histones H2A, H2B, H3 and H4, around which is wrapped 146 nucleotides of DNA. A fifth histone, H1, binds to DNA as it exits the nucleosome and interacts with linker DNA, which separates adjacent nucleosomes by 20–60 nucleotides. (B) Histone proteins are covalently modified on lysine and arginine residues in their N-terminal tails. Selected modifications are shown, as well as the modifier proteins mutated in BCL that target each specific residue. Orange indicates activating modifications; blue indicates repressive marks. Green indicates enzymes that remove histone marks, red indicates those that add marks. KMT2D (MLL2) and EZH2 add methyl groups, while KDM2B and KDM6B are demethylases. The HATs CREBBP and EP300 do not have specificity for particular histone lysine residues and, in fact, also acetylate non-histone proteins. * mutated in BCL
Recent BCL sequencing studies have revealed a striking pattern of recurrent mutations in chromatin modifying enzymes, particularly in GC-derived lymphomas (Figure 2B, Table 1) [36,45–48]. Intriguingly, the predicted impact of the most common mutations is a general repressive effect on chromatin states. Indeed, a recurrent mutation in the polycomb component EZH2 (Y641), which targets the histone 3 lysine 27 residue for repressive trimethylation (H3K27me3), occurs in 10–20% of FL and 22% of GCB-type DLBCL, but is absent from ABC-type tumors [32,49,50]. The Y641 mutations increase the trimethylation activity of EZH2 at the expense of monomethylation [51]. Many GC-derived lymphomas harbor a Y641 mutation or aberrantly maintain high levels of unmutated EZH2 expression, either of which generates increased global levels of the repressive H3K27me3 mark [52,53]. Recent studies indicate that mutant EZH2 represses bivalent promoters of genes involved in GC differentiation, including those mediating BCR and NF-κB signaling [52,54].
Table 1.
Mutations that impact gene regulation in BCL
| Category | Genes | Diseases |
|---|---|---|
| Epigenetic | CREBBP, EP300 | DLBCL, FL, |
| EZH2, KMT2D(MLL2), KDM2B, KDM6B | DLBCL, FL, MCL | |
| HISTH1B/C/D/E | CLL, DLBCL, FL | |
| ARID1A, MEF2B, MED12 | CLL, DLBCL, FL | |
| CHD2, SMARCA4 | CLL, DLBCL | |
| Signaling | CD79B, CARD11, IRF8, GNA13, STAT3/6, MYD88, NOTCH1, PRDM1, SGK1, TNFAIP3, TNFRSF14, TRAF3, BIRC3, PIM1 | CLL, DLBCL, FL, MCL |
| Transcription Factors | FOXO1, EBF1, POU2F2 | DLBCL |
| BCL6 | DLBCL | |
| Growth, Survival, Genome Maintenance | MYC, BCL2, FAS, PIM1, CCND1, TP53, ATM, POT1, BIRC3 | CLL, DLBCL, FL, MCL |
| mRNA Processing | SF3B1, XPO1 | CLL |
Bea PNAS 2013, Zhang Blood 2014, Puente Nature 2011, Landau Cell 2013, Morin/Lohr/Pasqualucci, Okosun Nat Gen 2014, Pasqualucci Cell Reports 2014
In contrast to EZH2 mutations, deletions or sequence variants in other histone methyltransferases (HMTs) or acetyltransferases (HATs) normally reduce or cripple their enzymatic functions (Figure 3B) [32–36,48,55]. While mutations in these enzymes have not been functionally linked to specific epigenetic changes in BCL, current evidence supports an impact on transcriptional regulation. For example, the HATs CREBBP and EP300 function as transcriptional coactivators in multiple signaling and developmental pathways, modifying lysine residues on both histone and non-histone nuclear proteins. Particularly relevant to BCL, mutations in CREBBP and EP300 impair their ability to inactivate BCL6 and to activate p53 transcription factors via direct acetylation [33,56–58]. Mice with heterozygous loss of the gene encoding CREBBP develop hematopoietic defects and subsequent hematologic malignancies [59], underscoring that haploinsufficiency of this acetyltransferase is sufficient to initiate oncogenesis. Nevertheless, an outstanding question in lymphoma biology remains: what specific epigenomic changes are functionally linked to mutations in HATs and which of these changes are crucial for oncogenesis?
Outstanding questions.
Despite extensive study over the years, a number of questions remain regarding the regulatory mechanisms by which changes in gene expression contribute to lymphomagenesis.
What is the impact of HAT mutations on the epigenomic landscape of transformed B cells and how do these changes affect pathogenic gene expression?
Some of the most commonly mutated genes in BCL are HATs. However, mechanistic studies for only one such mutation, an EP300 truncation that disrupts c-Rel acetylation and function, has been described. As the field moves forward it is important to understand the mechanistic underpinnings of how various HAT mutations alter the B cell regulome.
What are the mutation profiles of regulatory elements across BCL subtypes?
Most of the available BCL mutation data arise from exome- or RNA-seq studies. However, the REs that regulate gene expression are found in non-coding DNA and are not covered in these analyses. Although RE polymorphisms have been reported for multiple BCL subtypes, mutational burdens and patterns remains less defined, but are now possible through larger scale genome-wide studies.
Can enhancer - gene target effects be more accurately predicted?
A central problem in examining the impact of mutations or epigenetic changes at REs is deciphering which nearby gene(s) are the cognate regulatory targets. Robust algorithms that incorporate concordant changes in multiple platforms of epigenetic analysis are needed (e.g. DNA methylation, chromatin interaction).
What are the origins of altered transcription factor (TF) expression in lymphoma?
Changes in enhancer chromatin state have been linked to correlative variation in the expression of TFs that bind within the enhancers. In select cases, TF expression change is due to translocation or mutation (e.g. BCL6, BCL2); however, for most TFs the mechanisms that perturb expression are unknown. Focused experiments that manipulate the regulatory components of individual TFs are needed to elucidate these mechanisms.
What role do long non-coding RNAs play in deregulating gene expression in BCL?
To date, the best characterized BCL-associated non-coding RNAs have been microRNAs. However, long non-coding RNAs (lncRNAs) have emerged as oncogenic biomolecules in a variety of cancers. Similar studies of lncRNAs in BCLs are warranted.
Is targeted epigenetic therapy a viable treatment option for BCLs?
High throughput sequencing technologies have enabled a deeper understanding of diseases at a more mechanistic level and have contributed to the concept of precision medicine, which proposes customized treatment plans for individual patients. Many of the reported mechanisms for altered gene expression programs have an epigenetic component. However, a great deal remains to be learned about whether targeted epigenetic therapies could serve as a viable option in treating BCLs.
Cis-Regulatory Elements
Although it is tempting to ascribe all changes in chromatin landscapes to mutations in modifier genes, these genetic lesions are not present in all tumors [32,33,35,36]. Nevertheless, alterations to the chromatin landscape are consistently observed in BCL [27,52,60,61]. Indeed, when compared to normal GC B cells, approximately 25% of enhancers exhibit altered activity in FL samples, whether known chromatin modifier mutations are present or absent [14]. One such mechanism that is likely to perturb enhancer activity is altered TF expression. In primary FL samples, changes in enhancer chromatin status were linked to correlative variation in the expression of TFs that bind within the enhancers. For example, compared to normal GC-B cells, loss of enhancer activity in a set of REs is associated with decreased expression of the cognate TF, SPIB, in FL samples. Moreover, when expression of SPIB was knocked down by shRNA in untransformed B cells, H3K27ac active marks were reduced in that set of enhancers and expression of their gene targets was significantly diminished [14]. Altered expression of BCL6 has also been shown to impact the chromatin landscape of normal GC-B cells and DLBCL [60]. Thus, in addition to mutations in chromatin modifiers, altered expression levels of TFs via mechanisms that remain to be identified, must also contribute to pathogenic changes in BCL epigenomes.
A foundational mechanism for the rewiring of gene expression in lymphoma is the alteration of enhancer activities, either through revisions in the epigenome or changes in TF levels. A critical step in deciphering how altered enhancer activity drives BCL is to define the affected regulatory circuits, linking enhancers to perturbations in the expression of their target genes (Figure 2). We recently defined the pathogenic regulatory circuitry of FL by comparing epigenomes and transcriptomes in primary tumors with their normal GC-B counterparts [14]. Integrative analysis of these data revealed a multi-tiered circuitry, in which GC-B enhancers are activated or decommissioned and other REs are commandeered from non-B cell lineages. The deregulated genes targeted by FL-altered enhancers include several that play crucial roles in B cell differentiation, including POU2AF1, SPIB, and TCF3, as well as genes not previously linked to FL, such as HOXA10 [14].
The pattern and activities of SEs in BCL are consistent with their cell types of origin in the germinal center [14,27]. This observation likely reflects the role of SEs in controlling expression of B lineage-specifying genes, which lymphoma cells require for their continued growth and survival, as well as staving off their terminal differentiation. Indeed, disruption of SE function reduced the expression of POU2AF1, BCL6, and MYC and caused apoptosis of DLBCL cells in vitro and in xenotransplant mouse models [27]. Collectively, current data indicate that perturbation of enhancer function resets or chronically engages regulatory circuits in B cells, altering the expression of key oncogenes to drive lymphomagenesis.
Mechanisms of Gene Deregulation in Lymphoma: Inherited and Acquired Sequence Variants
While most sequencing studies have focused on the coding genome, or exome, non-coding sequence variants also play a significant role in pathogenic gene regulation. The vast majority of inherited single nucleotide polymorphisms (SNPs) are located in non-coding regions, a feature that complicates SNP-disease associations. However, recent epigenome studies revealed that many of these non-coding SNPs co-localize with regulatory regions and their composite TF binding sites [62–66]. In fact, many disease-associated SNPs, including those linked to auto-immunity and lymphoma, localize preferentially in distal REs and SEs, disrupt TF binding, and correlate with changes in histone modifications and neighboring gene expression (Figure 4, Key Figure) [67–71]. Some of these correlative findings have been validated experimentally, using oligonucleotide binding assays or CRISPR-mediated removal of the SNP, either of which abrogated TF binding and reduced target gene expression [14,72]. Thus, SNPs are clearly one genetic source for perturbations in enhancer activity and, consequently, gene expression changes associated with BCL.
Figure 4. Sequence and structural variants alter enhancer function via multiple mechanisms.
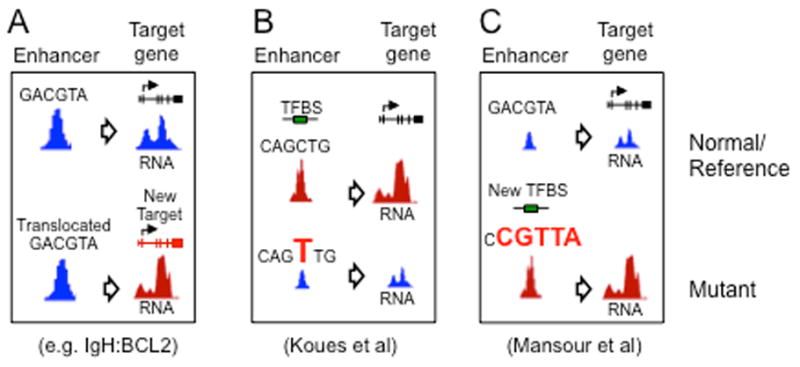
(A) In several BCL subtypes, translocation of the IgH enhancer juxtaposes it with one of several oncogenes (BCL2, MYC, BCL6, etc) causing overexpression and contributing to lymphomagenesis. (B) Koues et al demonstrated that inherited polymorphisms (SNPs) and acquired somatic mutations alter enhancer function in FL by disrupting transcription factor (TF) binding and the expression of target genes involved in oncogenesis. (C) Mansour et al showed that somatic mutations upstream of the TAL1 oncogene create a super-enhancer that drives its overexpression in T-ALL.
Acquired somatic mutations and structural genomic variants in regulatory regions also contribute to the deregulation of linked genes in lymphoma (Figure 4). An example of the latter is the pathologic hallmark of FL, the t(14;18) [IgH-BCL2] chromosomal translocation. This structural variant juxtaposes the powerful IgH enhancer with BCL2, resulting in over-expression of the anti-apoptotic BCL2 protein [73]. Similarly, an RE-based driver of Burkitt lymphoma is its hallmark translocation of the IGH enhancer into the MYC locus, leading to high level expression of this oncogene [74]. Recently, the role of acquired somatic sequence variants was revealed by integrative analyses of mutational and chromatin landscapes in B cell tumors. These studies showed that somatic variants are enriched in REs that drive key cell-identity programs associated with normal counterpart B cells [14,61,75]. Similar to inherited SNPs, the presence of some somatic mutations in FL diminished TF binding, altered enhancer chromatin patterns, and attenuated target gene expression. For example, one of these enhancer mutations diminishes binding of the B cell activation TF, TCF3, and is associated with decreased expression of the linked target gene, IRF8 [14]. In this way, the acquisition of somatic mutations in cell identity-associated REs may enable transforming B cells to alter the expression of genes in differentiation pathways, thereby preventing normal maturation and promoting lymphomagenesis.
In normal GC-B cells, this mechanism presumably is amplified by AID-mediated somatic hypermutation, which leads to lymphomagenic perturbations in enhancer activity and deregulation of linked oncogenes or tumor suppressors. Both on- and off-target mutations caused by AID are predominantly located within heavily acetylated and hyperaccessible SEs. Indeed, several independent lines of evidence indicate that AID-induced mutations are highly enriched at topologically complex SEs that are active in GC-B cells, including analyses of (i) SHM targets in normal mouse B cells, (ii) AID-induced kataegis in human DLBCL samples, and (iii) translocations in mouse embryonic fibroblasts engineered to express AID [76]. Collectively, these findings indicate that sequence variation in REs is a common mechanism for altering the activity of transcriptional circuits in BCL.
Mechanisms of Gene Deregulation in Lymphoma: Non-coding RNA
A recent addition to the menu of “epigenetic” mechanisms for gene regulation is non-coding RNAs (ncRNAs), which encompass a diverse array of transcripts lacking potential for translation into protein products. The persistence of ncRNAs as stable biomolecules permits them to function in gene regulation by a variety of mechanisms. For instance, ncRNAs can serve as ligands for proteins or complexes that modify chromatin landscapes, guiding these enzymes to their genomic targets via base pairing [77–81].
Of the ncRNAs involved in transcriptional regulation, miRs are the best characterized, with diverse roles in development, differentiation, and malignant transformation [82]. During B cell development, discrete groups of miRs regulate the expression of key transcription factors, including c-myb, Foxp1, and PU.1 [83–86]. Some miRs are deregulated in BCL, contributing to lymphomagenesis either as oncogenes (oncomiRs) or tumor suppressors [83,87]. The best example of a lymphoma oncomiR is miR-155, whose overexpression in multiple subtypes of BCL correlates with poorer prognosis [88–91]. miR-155 normally regulates the GC response via its direct targeting of transcripts encoding PU.1 and AID, attenuating their translation [84,92–96]. Recent evidence from DLBCL and knockout models suggests that miR-155 contributes to lymphomagenesis by blocking TGF-β1-mediated activation of the retinoblastoma protein [97,98]. miRs may also act as tumor suppressors, as evidenced by recurrent deletions in lymphoma. For example, a miR cluster in the 13q14 region is frequently deleted in CLL [99]. For a more extensive review of miRs involved in B lymphocyte differentiation and lymphomagenesis, see [83,87,100,101].
Long non-coding RNAs (lncRNAs, >200 bp) are also devoid of open reading frames and fold into complex secondary structures, which often mediate their interaction with specific proteins [80,81]. Importantly, lncRNA expression is even more restricted to specific cell types and differentiation stages than the expression of coding genes, suggesting that lncRNAs contribute to cell fate determination [102–104]. A specialized category of lncRNA, called eRNAs, originate bidirectionally from active enhancers. In both normal and transformed B cells, this “divergent” mode of transcription targets AID activity, leading to higher rates of somatic hypermutation and translocations involving enhancer/SE regions [76,105].
Emerging evidence supports a significant role for lncRNAs in normal B cell development and lymphomagenesis [106]. For example, a lncRNA that is antisense to the transcript for Fas, called FAS-AS1, prevents skipping of exon 6 in the processed mRNA, which suppresses production of the soluble isoform of Fas receptor (sFas). Because sFas inhibits apoptosis by sequestering Fas ligand, higher levels of FAS-AS1 correlate with better DLBCL prognoses, presumably because lymphoma cells are less protected from Fas-mediated death. Importantly, EZH2-mediated repression of Fas-AS1 can be reversed by treatment with the EZH2 inhibitor, DZNeP, augmenting DLBCL apoptosis [107]. The expression of another lncRNA, PEG10, which originates from an imprinted region on chr7, is upregulated in several types of cancer, including DLBCL, and correlates with poor prognosis. PEG10 depletion in DLBCL cell lines reduced their proliferation and survival [108]. However, the relevant molecular targets for PEG10 and its underlying modes of action remain unclear.
LncRNAs have also been implicated in the regulation of apoptosis in CLL. The lncRNAs NEAT1 and lincRNA-p21 are induced in response to DNA damage if the CLL cells express functional p53. Both of these lncRNAs are direct targets of p53, and their expression levels correlate with DNA damage-induced cell death [109]. Importantly, lincRNA-p21 was previously shown to serve as a repressor in p53-dependent transcriptional responses via physical association with hnRNP-K. This interaction is required for proper genomic localization of hnRNP-K at repressed genes and regulation of p53 mediated apoptosis [110]. Given the crucial role that lncRNAs play in gene expression control during normal cellular development and differentiation, we predict that future studies will uncover new oncogenic mechanisms associated with lncRNA deregulation in BCL.
Therapeutic Reversal of Transformative Expression Programs
Many therapeutic approaches aim to reverse pathogenic gene expression in BCL by targeting distinct epigenetic modifiers. Treatment with one class of therapeutics, general histone deacetylase inhibitors (HDACi), attempted to mitigate the effects of chromatin hypoacetylation stemming from HAT dysfunction, but clinical response was relatively low [111]. A new class of HDACi is more selective, inhibiting specific HDAC proteins, which may enhance the effectiveness of this epigenetic approach while minimizing its side effects [112]. Indeed, the HDAC6-selective inhibitor, tubastatin A, suppresses growth of MCL and DLBCL cells in xenograft mouse models [113]. Another class of targeted epigenetic therapy is a new generation of small molecules that inhibit EZH2 function. A subset of these compounds effectively inhibit both wild-type and Y641 mutant forms of EZH2 in DLBCL cells, suppressing H3K27 methylation and reactivating the expression of target genes, including those controlling cell cycle checkpoints and GC-B differentiation [114,115]. Importantly, these agents caused tumor regression in animal models of DLBCL [114] and are currently in clinical trials.
An entirely novel therapeutic approach has been innovated by Bradner and colleagues, who created a small molecule inhibitor, JQ1, which targets SEs. JQ1 competitively binds bromodomain and extra terminal (BET) domains, such as the transcriptional co-activator BRD4, which is highly enriched at hyperacetylated SEs [27,116]. The BRD4 enrichment appears to render SEs particularly sensitive to JQ1, which selectively attenuates the expression of linked transcriptional circuits controlling the expression of certain oncogenes and lineage-specific factors [27,116–119]. In DLBCL, treatment with JQ1 impaired cell proliferation and viability in a cMYC-dependent manner, both in cultured cells and xenograft models [27,120], and this BRD4 inhibitor is now in Phase 1 and 2 clinical trials.
Although these novel therapeutic approaches are promising, they are nevertheless non-specific in their alteration of epigenomes and, perhaps, other cellular proteins. In turn, these “off target” effects may impact the expression of many bystander genes or perturb essential cellular processes. Given the recent identification of REs that are pathogenically altered in BCL, it is tempting to envision a more targeted approach: using sequence-specific reagents (e.g., zinc fingers or CRISPR) to direct fused chromatin modifiers to REs that are linked functionally to crucial oncogenes, disabling only these transcriptional circuits while leaving others unaffected [121–124]. Such a precision medicine approach is predicted to generate fewer side effects, while effectively eradicating lymphoma cells.
Concluding Remarks
In summary, recent studies of normal and transformed B cells have revealed widespread perturbations in BCL epigenomes, many of which can be linked to altered enhancer profiles. The remodeled regulomes disrupt the normal transcriptional circuitry of B lymphocytes, rewiring gene expression programs to drive the proliferation and survival of lymphoma cells. Several mechanisms underlie these changes, including altered epigenetic modification of the enhancers themselves, variable expression of key TFs, mutation/aberrant expression of chromatin modifiers, SNPs/mutations in enhancers, and changes in non-coding RNA expression. These discoveries define the epigenetic and genetic agents that transform normal B lymphocytes. Importantly, the reversible nature of epigenetic modifications has enabled the development of new therapies that target certain types of altered REs, including the super-enhancers that regulate key lineage-specifying genes. Looking forward, a truly personalized medicine approach may be possible by specifically targeting individual pathogenic enhancers with locus-specific chromatin modifiers. However, many open questions still remain (Outstanting Questions Box).
Trends.
Aberrant epigenomes and deregulated gene expression characterize B cell lymphoma
Altered transcription factor and enhancer activity modulate the output of transcriptional circuits
Mutations in chromatin modifiers and enhancers underlie some oncogenic changes in gene expression
Epigenetic changes that drive B cell transformation could provide targets for novel therapies
Glossary
- BCL
B cell lymphoma, a heterogeneous group of mature B lymphocyte neoplasms
- Bivalent promoter
promoters that carry both activating (H3K4me3) and repressive (H3K27me3) modifications, indicating that they are “poised” for transcription
- ChIP-Seq
Chromatin Immunoprecipitation Sequencing, combines traditional chromatin immunoprecipitation with NGS to characterize global TF or histone protein interactions with DNA
- CLL
Chronic Lymphocytic Leukemia/Lymphoma, the most common leukemia in the U.S. Its normal counterpart cells are naïve and memory B cells, depending on the CLL subtype
- DLBCL
Diffuse Large B cell Lymphoma, an aggressive neoplasm that is curable in 60% of cases. Molecular profiling segregates DLBCL into two subtypes, Germinal Center (GCB) and Activated B cell (ABC), which resemble GC centrocytes or plasmablasts, respectively
- DMTases
DNA methyltransferases, enzymes that catalyze the transfer of a methyl group to a cytosine forming 5-methylcytosine (5mC)
- DNase-Seq
DNase I hypersensitive sites sequencing, based on the fact that nucleosome poor regions of the genome are more sensitive to cleavage by DNase I. DNase-Seq indentifies accessible regions of DNA associated with regulatory elements (REs)
- FAIRE-Seq
Formaldehyde-Assisted Isolation of Regulatory Elements, Based on the fact that formaldehyde crosslinks nucleosome-bound DNA more efficiently that nucleosome depleted regions. FAIRE-Seq identifies the nucleosome depleted REs
- FL
Follicular Lymphoma, an indolent yet incurable BCL. The normal counterpart cell is a germinal center centrocyte
- GC
Germinal Centers, sites of B cell proliferation and differentiation within secondary lymphoid organs
- HDACs
Histone Deactylases, enzymes that remove acetyl groups from lysine residues on histones, tightening the histone-DNA interaction and blocking access to transcriptional machinery. The 11 HDAC proteins are divided into four classes
- HATs
Histone Acetyltransferases, enzymes that catalyze the transfer of an acetyl group to lysine residues of histones, establishing a histone-DNA environment permissive for active transcription. There are multiple HAT families, the members of which have overlapping substrate specificity, including an ability to acetylate non-histone proteins. HATs are often components of multi-subunit protein complexes and their interacting partners may contribute to their target profiles
- HMTases
Histone Methyltransferases, enzymes that catalyze the transfer of one, two, or three methyl groups onto lysine or arginine residues of histone proteins. Histone methylation is a principal epigenetic modification that determines gene expression
- Kataegis
a pattern of concentrated hypermutation identified in some cancer genomes, often localized with structural genomic variants or rearrangements
- MCL
Mantle Cell Lymphoma, an aggressive BCL characterized by the overexpression of CCND1 and the hallmark t(11;14)(q13;q32) translocation. The normal counterpart cell is the mantle zone B cell
- ncRNA
Non-coding RNA, RNA not translated into protein. They are categorized by size and function. For example, microRNAs (miRs) are 22 nucleotides in length, while long ncRNAs (lncRNAs) are >200 nucleotides to several kilobases. ncRNA functions include recruiting chromatin modifiers, forming core components of ribosomes (rRNA), otherwise involved in translation (tRNA), splicing (URNAs, snRNP), and genome defense (piRNAs – silence retrotransposons in germ cells)
- RE
Regulatory Element, non-coding regions of the genome that control gene expression. Common REs include promoters, enhancers and insulators
- RNA-seq
RNA Sequencing, characterization of global steady state RNA levels (transcriptomes) using high throughput sequencing
- SE
Super-Enhancer, large hyperacetylated genomic regions that contain clusters of REs
- TF
Transcription Factor, proteins that bind REs and control gene expression
- Xenograft mouse model
Implantation of human tumor tissue into an immunocompromised mouse
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alizadeh AA, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 2.Rosenwald A, et al. Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med. 2001;194:1639–47. doi: 10.1084/jem.194.11.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klein U, et al. Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. J Exp Med. 2001;194:1625–38. doi: 10.1084/jem.194.11.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenwald A, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell. 2003;3:185–97. doi: 10.1016/s1535-6108(03)00028-x. [DOI] [PubMed] [Google Scholar]
- 5.Dave SS, et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N Engl J Med. 2004;351:2159–2169. doi: 10.1056/NEJMoa041869. [DOI] [PubMed] [Google Scholar]
- 6.Staudt LM, Dave S. The biology of human lymphoid malignancies revealed by gene expression profiling. Adv Immunol. 2005;87:163–208. doi: 10.1016/S0065-2776(05)87005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miyazaki K, et al. The establishment of B versus T cell identity. Trends Immunol. 2014;35:205–10. doi: 10.1016/j.it.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murphy KM, et al. Janeway Immunologie. 2009;7 Aufl. [Google Scholar]
- 9.Victora GD, et al. Identification of human germinal center light and dark zone cells and their relationship to human B-cell lymphomas. Blood. 2012;120:2240–8. doi: 10.1182/blood-2012-03-415380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Silva NS, Klein U. Dynamics of B cells in germinal centres. Nat Rev Immunol. 2015;15:137–148. doi: 10.1038/nri3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Basso K, Dalla-Favera R. Germinal centres and B cell lymphomagenesis. Nat Rev Immunol. 2015;15:172–184. doi: 10.1038/nri3814. [DOI] [PubMed] [Google Scholar]
- 12.Shaffer AL, et al. Pathogenesis of human B cell lymphomas. Annu Rev Immunol. 2012;30:565–610. doi: 10.1146/annurev-immunol-020711-075027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Young RM, et al. B cell Receptor Signaling in Diffuse Large B cell Lymphoma. Semin Hematol. 2015;52:77–85. doi: 10.1053/j.seminhematol.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koues OI, et al. Enhancer Sequence Variants and Transcription Factor Deregulation Synergize to Construct Pathogenic Regulatory Circuits in B Cell Lymphoma. Immunity. 2015;42:186–198. doi: 10.1016/j.immuni.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lanasa MC. Novel insights into the biology of CLL. Hematology Am Soc Hematol Educ Program. 2010;2010:70–6. doi: 10.1182/asheducation-2010.1.70. [DOI] [PubMed] [Google Scholar]
- 16.Campo E, Rule S. Mantle cell lymphoma: evolving management strategies. Blood. 2015;125:48–55. doi: 10.1182/blood-2014-05-521898. [DOI] [PubMed] [Google Scholar]
- 17.Bianchi G, Munshi NC. Pathogenesis beyond the cancer clone(s) in multiple myeloma. Blood. 2015;125:3049–3058. doi: 10.1182/blood-2014-11-568881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Küppers R. New insights in the biology of Hodgkin lymphoma. Hematology Am Soc Hematol Educ Program. 2012;2012:328–34. doi: 10.1182/asheducation-2012.1.328. [DOI] [PubMed] [Google Scholar]
- 19.Bernstein BE, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Consortium RE, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andersson R, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thurman RE, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whyte WAA, et al. Master Transcription Factors and Mediator Establish Super-Enhancers at Key Cell Identity Genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herz HM, et al. Enhancer malfunction in cancer. Mol Cell. 2014;53:859–66. doi: 10.1016/j.molcel.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akhtar-Zaidi B, et al. Epigenomic enhancer profiling defines a signature of colon cancer. Science. 2012;336:736–9. doi: 10.1126/science.1217277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chapuy B, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013;24:777–90. doi: 10.1016/j.ccr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 29.Heyn H, et al. DNA methylation contributes to natural human variation. Genome Res. 2013;23:1363–1372. doi: 10.1101/gr.154187.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Solary E, et al. The Ten-Eleven Translocation-2 (TET2) gene in hematopoiesis and hematopoietic diseases. Leukemia. 2014;28:485–96. doi: 10.1038/leu.2013.337. [DOI] [PubMed] [Google Scholar]
- 31.Im AP, et al. DNMT3A and IDH mutations in acute myeloid leukemia and other myeloid malignancies: associations with prognosis and potential treatment strategies. Leukemia. 2014;28:1774–1783. doi: 10.1038/leu.2014.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morin RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pasqualucci L, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189–195. doi: 10.1038/nature09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lohr JG, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. 2012;109:3879–84. doi: 10.1073/pnas.1121343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Landau DA, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152:714–26. doi: 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beà S, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci U S A. 2013;110:18250–5. doi: 10.1073/pnas.1314608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kulis M, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet. 2012;44:1236–42. doi: 10.1038/ng.2443. [DOI] [PubMed] [Google Scholar]
- 38.Martín-Subero JI, et al. New insights into the biology and origin of mature aggressive B-cell lymphomas by combined epigenomic, genomic, and transcriptional profiling. Blood. 2009;113:2488–97. doi: 10.1182/blood-2008-04-152900. [DOI] [PubMed] [Google Scholar]
- 39.O’Riain C, et al. Array-based DNA methylation profiling in follicular lymphoma. Leukemia. 2009;23:1858–66. doi: 10.1038/leu.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shaknovich R, et al. DNA methylation signatures define molecular subtypes of diffuse large B-cell lymphoma. Blood. 2010;116:e81–9. doi: 10.1182/blood-2010-05-285320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chambwe N, et al. Variability in DNA methylation defines novel epigenetic subgroups of DLBCL associated with different clinical outcomes. Blood. 2014;123:1699–708. doi: 10.1182/blood-2013-07-509885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Queirós AC, et al. A B-cell epigenetic signature defines three biologic subgroups of chronic lymphocytic leukemia with clinical impact. Leukemia. 2014;29:598–605. doi: 10.1038/leu.2014.252. [DOI] [PubMed] [Google Scholar]
- 43.Pan H, et al. Epigenomic evolution in diffuse large B-cell lymphomas. Nat Commun. 2015;6:6921. doi: 10.1038/ncomms7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Landau DA, et al. Locally Disordered Methylation Forms the Basis of Intratumor Methylome Variation in Chronic Lymphocytic Leukemia. Cancer Cell. 2014;26:813–825. doi: 10.1016/j.ccell.2014.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gruber M, Wu CJ. Evolving understanding of the CLL genome. Semin Hematol. 2014;51:177–87. doi: 10.1053/j.seminhematol.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang Y, Melnick A. The Epigenetic basis of diffuse large B-cell lymphoma. Semin Hematol. 2015;52:86–96. doi: 10.1053/j.seminhematol.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kishimoto W, Nishikori M. Molecular pathogenesis of follicular lymphoma. J Clin Exp Hematop. 2014;54:23–30. doi: 10.3960/jslrt.54.23. [DOI] [PubMed] [Google Scholar]
- 48.Okosun J, et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet. 2013;46:176–81. doi: 10.1038/ng.2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bödör C, et al. EZH2 mutations are frequent and represent an early event in follicular lymphoma. Blood. 2013;122:3165–8. doi: 10.1182/blood-2013-04-496893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morin RD, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181–185. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yap DB, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. 2011;117:2451–9. doi: 10.1182/blood-2010-11-321208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Béguelin W, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013;23:677–92. doi: 10.1016/j.ccr.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Velichutina I, et al. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood. 2010;116:5247–55. doi: 10.1182/blood-2010-04-280149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berg T, et al. A transgenic mouse model demonstrating the oncogenic role of mutations in the polycomb-group gene EZH2 in lymphomagenesis. Blood. 2014 doi: 10.1182/blood-2012-12-473439. [DOI] [PubMed] [Google Scholar]
- 55.Pasqualucci L, et al. Genetics of follicular lymphoma transformation. Cell Rep. 2014;6:130–40. doi: 10.1016/j.celrep.2013.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bereshchenko OR, et al. Acetylation inactivates the transcriptional repressor BCL6. Nat Genet. 2002;32:606–13. doi: 10.1038/ng1018. [DOI] [PubMed] [Google Scholar]
- 57.Lill NL, et al. Binding and modulation of p53 by p300/CBP coactivators. Nature. 1997;387:823–7. doi: 10.1038/42981. [DOI] [PubMed] [Google Scholar]
- 58.Ogryzko VV, et al. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–9. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- 59.Kung AL, et al. Gene dose-dependent control of hematopoiesis and hematologic tumor suppression by CBP. Genes Dev. 2000;14:272–7. [PMC free article] [PubMed] [Google Scholar]
- 60.Hatzi K, et al. A hybrid mechanism of action for BCL6 in B cells defined by formation of functionally distinct complexes at enhancers and promoters. Cell Rep. 2013;4:578–88. doi: 10.1016/j.celrep.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang J, et al. The genomic landscape of mantle cell lymphoma is related to the epigenetically determined chromatin state of normal B cells. Blood. 2014;123:2988–96. doi: 10.1182/blood-2013-07-517177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Corradin O, et al. Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome Res. 2014;24:1–13. doi: 10.1101/gr.164079.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Khurana E, et al. Integrative annotation of variants from 1092 humans: application to cancer genomics. Science (80-) 2013;342:1235587–1235587. doi: 10.1126/science.1235587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maurano MT, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science (80-) 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou X, et al. Epigenomic annotation of genetic variants using the Roadmap Epigenome Browser. Nat Biotechnol. 2015;33:345–346. doi: 10.1038/nbt.3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Leung D, et al. Integrative analysis of haplotype-resolved epigenomes across human tissues. Nature. 2015;518:350–354. doi: 10.1038/nature14217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cerhan JR, et al. Genome-wide association study identifies multiple susceptibility loci for diffuse large B cell lymphoma. Nat Genet. 2014;46:1233–1238. doi: 10.1038/ng.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Farh KK-H, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2014 advance on. [Google Scholar]
- 69.Berndt SI, et al. Genome-wide association study identifies multiple risk loci for chronic lymphocytic leukemia. Nat Genet. 2013;45:868–76. doi: 10.1038/ng.2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cowper-Sal lari R, et al. Breast cancer risk-associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nat Genet. 2012;44:1191–1198. doi: 10.1038/ng.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang Q, et al. A prostate cancer susceptibility allele at 6q22 increases RFX6 expression by modulating HOXB13 chromatin binding. Nat Genet. 2014;46:126–135. doi: 10.1038/ng.2862. [DOI] [PubMed] [Google Scholar]
- 72.Yao L, et al. Functional annotation of colon cancer risk SNPs. Nat Commun. 2014;5:5114. doi: 10.1038/ncomms6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kridel R, et al. Pathogenesis of follicular lymphoma. J Clin Invest. 2012;122:3424–31. doi: 10.1172/JCI63186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Love C, et al. The genetic landscape of mutations in Burkitt lymphoma. Nat Genet. 2012;44:1321–5. doi: 10.1038/ng.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Polak P, et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature. 2015;518:360–364. doi: 10.1038/nature14221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Qian J, et al. B Cell Super-Enhancers and Regulatory Clusters Recruit AID Tumorigenic Activity. Cell. 2014;159:1524–37. doi: 10.1016/j.cell.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morris KV, Mattick JS. The rise of regulatory RNA. Nat Rev Genet. 2014;15:423–37. doi: 10.1038/nrg3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Holoch D, Moazed D. RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet. 2015;16:71–84. doi: 10.1038/nrg3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.StLaurent G, et al. The Landscape of long noncoding RNA classification. Trends Genet. 2015;31:239–251. doi: 10.1016/j.tig.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15:7–21. doi: 10.1038/nrg3606. [DOI] [PubMed] [Google Scholar]
- 81.Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152:1298–307. doi: 10.1016/j.cell.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hausser J, Zavolan M. Identification and consequences of miRNA-target interactions - beyond repression of gene expression. Nat Rev Genet. 2014;15:599–612. doi: 10.1038/nrg3765. [DOI] [PubMed] [Google Scholar]
- 83.De Yébenes VG, et al. Regulation of B-cell development and function by microRNAs. Immunol Rev. 2013;253:25–39. doi: 10.1111/imr.12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.De Yébenes VG, et al. miR-181b negatively regulates activation-induced cytidine deaminase in B cells. J Exp Med. 2008;205:2199–206. doi: 10.1084/jem.20080579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xiao C, et al. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell. 2007;131:146–59. doi: 10.1016/j.cell.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 86.Ventura A, et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008;132:875–86. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Musilova K, Mraz M. MicroRNAs in B-cell lymphomas: how a complex biology gets more complex. Leukemia. 2015;29:1004–1017. doi: 10.1038/leu.2014.351. [DOI] [PubMed] [Google Scholar]
- 88.Eis PS, et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U S A. 2005;102:3627–32. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cui B, et al. MicroRNA-155 influences B-cell receptor signaling and associates with aggressive disease in chronic lymphocytic leukemia. Blood. 2014;124:546–54. doi: 10.1182/blood-2014-03-559690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhao JJ, et al. microRNA expression profile and identification of miR-29 as a prognostic marker and pathogenetic factor by targeting CDK6 in mantle cell lymphoma. Blood. 2010;115:2630–9. doi: 10.1182/blood-2009-09-243147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Roehle A, et al. MicroRNA signatures characterize diffuse large B-cell lymphomas and follicular lymphomas. Br J Haematol. 2008;142:732–44. doi: 10.1111/j.1365-2141.2008.07237.x. [DOI] [PubMed] [Google Scholar]
- 92.Rodriguez A, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–11. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thai TH, et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–8. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 94.Vigorito E, et al. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity. 2007;27:847–59. doi: 10.1016/j.immuni.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dorsett Y, et al. MicroRNA-155 suppresses activation-induced cytidine deaminase-mediated Myc-Igh translocation. Immunity. 2008;28:630–8. doi: 10.1016/j.immuni.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tan LP, et al. miRNA profiling of B-cell subsets: specific miRNA profile for germinal center B cells with variation between centroblasts and centrocytes. Lab Invest. 2009;89:708–16. doi: 10.1038/labinvest.2009.26. [DOI] [PubMed] [Google Scholar]
- 97.Rai D, et al. Targeting of SMAD5 links microRNA-155 to the TGF-beta pathway and lymphomagenesis. Proc Natl Acad Sci U S A. 2010;107:3111–6. doi: 10.1073/pnas.0910667107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jiang D, Aguiar RCT. MicroRNA-155 controls RB phosphorylation in normal and malignant B lymphocytes via the noncanonical TGF-β1/SMAD5 signaling module. Blood. 2014;123:86–93. doi: 10.1182/blood-2013-07-515254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Klein U, et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010;17:28–40. doi: 10.1016/j.ccr.2009.11.019. [DOI] [PubMed] [Google Scholar]
- 100.Baumjohann D, Ansel KM. MicroRNA regulation of the germinal center response. Curr Opin Immunol. 2014;28:6–11. doi: 10.1016/j.coi.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen C-Z, et al. Regulation of immune responses and tolerance: the microRNA perspective. Immunol Rev. 2013;253:112–28. doi: 10.1111/imr.12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Derrien T, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–89. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482:339–46. doi: 10.1038/nature10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cabili MN, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–27. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Meng FL, et al. Convergent Transcription at Intragenic Super-Enhancers Targets AID-Initiated Genomic Instability. Cell. 2014;159:1538–48. doi: 10.1016/j.cell.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ranzani V, et al. The long intergenic noncoding RNA landscape of human lymphocytes highlights the regulation of T cell differentiation by linc-MAF-4. Nat Immunol. 2015;16:318–325. doi: 10.1038/ni.3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sehgal L, et al. FAS-antisense 1 lncRNA and production of soluble versus membrane Fas in B-cell lymphoma. Leukemia. 2014;28:2376–87. doi: 10.1038/leu.2014.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Peng W, et al. Upregulation of long noncoding RNA PEG10 associates with poor prognosis in diffuse large B cell lymphoma with facilitating tumorigenicity. Clin Exp Med. 2015 doi: 10.1007/s10238-015-0350-9. [DOI] [PubMed] [Google Scholar]
- 109.Blume CJ, et al. p53-dependent non-coding RNA networks in Chronic Lymphocytic Leukemia. Leukemia. 2015 doi: 10.1038/leu.2015.119. [DOI] [PubMed] [Google Scholar]
- 110.Huarte M, et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142:409–19. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Crump M, et al. Phase II trial of oral vorinostat (suberoylanilide hydroxamic acid) in relapsed diffuse large-B-cell lymphoma. Ann Oncol. 2008;19:964–969. doi: 10.1093/annonc/mdn031. [DOI] [PubMed] [Google Scholar]
- 112.Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13:673–91. doi: 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- 113.Lwin T, et al. A microenvironment-mediated c-Myc/miR-548m/HDAC6 amplification loop in non-Hodgkin B cell lymphomas. J Clin Invest. 2013;123:4612–26. doi: 10.1172/JCI64210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.McCabe MT, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 115.Garapaty-Rao S, et al. Identification of EZH2 and EZH1 small molecule inhibitors with selective impact on diffuse large B cell lymphoma cell growth. Chem Biol. 2013;20:1329–39. doi: 10.1016/j.chembiol.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 116.Lovén J, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–34. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mertz JA, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A. 2011;108:16669–74. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Delmore JE, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–17. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Filippakopoulos P, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–73. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Trabucco SE, et al. Inhibition of bromodomain proteins for the treatment of human diffuse large B-cell lymphoma. Clin Cancer Res. 2015;21:113–22. doi: 10.1158/1078-0432.CCR-13-3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wilber A, et al. A zinc-finger transcriptional activator designed to interact with the gamma-globin gene promoters enhances fetal hemoglobin production in primary human adult erythroblasts. Blood. 2010;115:3033–41. doi: 10.1182/blood-2009-08-240556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gilbert LA, et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell. 2013;154:442–51. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Perez-Pinera P, et al. Synergistic and tunable human gene activation by combinations of synthetic transcription factors. Nat Methods. 2013;10:239–42. doi: 10.1038/nmeth.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hilton IB, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015 doi: 10.1038/nbt.3199. advance on. [DOI] [PMC free article] [PubMed] [Google Scholar]