

Abstract
Developing amorphous solid dispersions of water-insoluble molecules using polymeric materials is a well-defined approach to improve the dissolution rate and bioavailability. While the selected polymer plays a vital role in stabilizing the amorphous solid dispersion physically, it is equally important to improve the dissolution profile by inhibiting crystallization from the supersaturated solution generated by dissolution of the amorphous material. Furthermore, understanding the mechanism of dissolution rate enhancement is of vital importance. In this work, wetting kinetics was taken up as an alternative approach for understanding the enhanced dissolution rate for amorphous solid dispersion of a poorly soluble drug. While cilostazol (CIL) was selected as the model drug, povidone (PVP), copovidone, and hypromellose (HPMC) were the polymers of choice. The concentrations against time profiles were evaluated for the supersaturated solutions of CIL in the presence and absence of the selected polymers. The degree of supersaturation increased significantly with increase in polymer content within the solid dispersion. While povidone was found to maintain the highest level of supersaturation for the greatest length of time both in dissolution and solution crystallization experiments, copovidone and hypromellose were found to be the less effective as crystallization inhibitor. The ability of polymers to generate and maintain supersaturated drug solutions was assessed by dissolution studies. The wetting kinetics was compared against the solid dispersion composition to establish a correlation with enhanced dissolution rate.
KEY WORDS: Cilostazol, Crystallization inhibition, Solid dispersions, Supersaturated solutions, Wetting kinetics
INTRODUCTION
Developing oral formulations for poorly water-soluble drugs has always been a challenging job as the efficacy of the drugs can be severely impacted because of the poor aqueous solubility. The formulation that enters the mouth and travels down the gastrointestinal tract must make the drug available in solution form to achieve good drug bioavailability. It is apparent that the low bioavailability, large inter- and intra-subject variations, along with high variations in blood drug concentrations under fed versus fasted conditions are attributed to poor aqueous solubility (1,2). Hence, over the years in order to enhance the apparent solubility and dissolution rate, numerous drug development strategies have been explored. In this direction, amorphous drug forms have received remarkable attention because of augmented solubility they exhibit relative to their crystalline counterparts (3,4). In particular, the generation of amorphous drug forms using solid dispersions is highly attractive for enhancing the dissolution rate of poorly soluble drugs (5–7). Although a high quality of work has been done in the area of solid dispersions, still only few drug products based on solid dispersions have reached the market. Therefore, it is not surprising that it is still one of the most promising approach to explore for enhancing the dissolution rate of water-insoluble drug candidates (8,9).
Exploitation of solid dispersions as a strategy to improve the drug absorption dates back to 1961, when Sekiguchi and co-workers (9,10) reported the use of eutectic mixtures (formed by fusion) capable of enhancing dissolution and absorption rates of certain drugs. The term “solid dispersion” applies to systems in which the drug molecules are homogenously mixed with the carrier molecule throughout the binary solid matrix. Reduction of particle size of drug to nearly molecular level and transformation of crystalline drug to amorphous state are the key outcomes of solid dispersion which dictates the increased kinetic solubility or enhanced rate of dissolution and augmented oral bioavailability of a water-insoluble drug. In comparison to solid dispersions, solid solutions which represent single-phase system are more desirable because of enhanced dissolution rates. In solid solutions, the drug is completely dispersed in the carrier at molecular level and no crystalline structure can be detected (6). Generally, solid dispersions are prepared by three methods: melting, solvent evaporation, and a combination of melting and solvent evaporation (11–14). Characterization of solid dispersions is another typical area which requires multiple techniques to assess the amorphous versus crystalline state of the drugs in solid dispersion. Such techniques or methodologies include but not limited to different analytical techniques such as differential scanning calorimetry (DSC) and modulated differential scanning calorimetry (MDSC), powder X-ray diffraction (PXRD), confocal Raman spectroscopy, Fourier transform infrared spectroscopy (FTIR), solid-state nuclear magnetic resonance (ssNMR), thermal gravimetric analysis (TGA), optical, scanning (SEM), transmission electron (TEM), and atomic force (AFM) microscopy have been utilized to characterize the various properties of solid dispersions (15,16).
In general, the advantages of amorphous form in dosage form are lost because of their inadequate thermodynamic stability and their penchant to recrystallize in the solid state or during dissolution. Therefore, to achieve the full advantage of amorphous solid dispersions, it is obligatory not only to stabilize the drug during storage but also to maintain supersaturated concentration of drug during dissolution (4,5). Use of polymeric carriers for solid dispersions have previously been shown to function as stabilizing agents and thus maintain supersaturated concentrations achieved during dissolution of amorphous drugs (17). Therefore, albeit a daunting task, amorphous solid dispersions can be stabilized by rationally selecting the suitable polymeric carriers. The right polymer either by forming secondary interactions with the drug (18,19) or/and by their anti-plasticization effect, helps to stabilise the amorphous from by reducing their molecular mobility (20). Hence, the role of right polymers in developing acceptable solid dispersions is of extreme importance in stabilizing amorphous drugs. Furthermore, the rate at which a solid oral drug delivery system dissolves depends on many parameters, and occurs in a series of steps: wetting, solvent penetration, disintegration, swelling (if applicable), and dissolution of components. Particularly for composites like solid dispersions, each of these areas is complex, and their respective roles in the dissolution process need to be considered separately. In literature, the dissolution properties of individual materials have been correlated with the process of wetting and wetting kinetics. Also, in polymer science, the polymer composites have been worked out on the basis of the surface properties and wetting kinetics of individual material and how these properties changes with alteration in composite composition (21,22). However, similar understanding needs to be developed to establish a relationship between enhanced dissolution rates and wetting kinetics as per the changes in compositions of amorphous solid dispersion containing drugs. This is imperative as wetting is a reflection of the functional groups that are present at the surface of the solid; however, solubility relates to the structure of the entire molecules. It is quite possible that the various processes used to prepare solid dispersion can alter the structure of a material, and hence the surface properties and wettability and solubility behavior. Moreover, for solid dispersions where the drug is in amorphous state, wettability and their interaction strength with dissolution media are important characteristics as they can influence the supersaturation levels achieved during dissolution process and its physical stability (23).
Cilostazol (CIL) is a neutral molecule having an aqueous solubility of about 3 μg/ml at 25°C, and its octanol–water distribution coefficients (log Poct) range from 2.72 (pH 2.0) to 2.76 (pH 11.0). The apparent permeability of CIL estimated by Caco-2 cell methodology is 1.92 × 10−5 cm/s. Based on the poor solubility and good permeability, CIL is classified as BCS Class II drug, which means that its absorption is dissolution dependent (24–26). CIL is approved as immediate release tablets (only formulation available) in the USA and several European countries under the brand name Pletal® tablets (50 and 100 mg) for the treatment of intermittent claudication (7,27), and it is not surprising that its therapeutic applications are limited due to its poor aqueous solubility and bioavailability (24,25,28). For improving the solubility and dissolution of CIL, the only approach explored so far appears to be particle size reduction (26,29–31). Three crystalline forms of CIL have been reported (A, B, and C) where A is the most stable and the other two are metastable (32,33). Therefore, CIL appears a good model drug to investigate in order to develop pharmaceutically acceptable solid dispersions.
The objective of this study was to assess the process of wetting and wetting kinetics as an alternative approach to understand and correlate the enhanced dissolution rates of solid dispersions of a poorly water-soluble drug with their compositions. For this, CIL was chosen as the model drug and povidone (PVP), copovidone, and hypromellose (HPMC) were the polymers of choice to prepare the solid dispersions using spray-drying technology. The concentrations versus time profiles were evaluated for the supersaturated solutions of CIL in the presence and absence of the polymers. The ability of polymers to generate and maintain supersaturated drug solutions was assessed by dissolution studies. To correlate this enhanced dissolution rate, wetting kinetics were investigated using contact angle studies and its impact of processes of adhesion, immersion, and spreading.
MATERIALS AND METHODS
Materials
CIL was a generous gift from Daewoong Pharmaceuticals Co. (Hyderabad, India). Povidone (Plasdone K-29/32) and Copovidone (Plasdone S-630) were purchased from ISP Corporation (Hyderabad, India), while hypromellose (Methocel E5) was purchased from Colorcon (Verna, Goa India). Acetonitrile, methanol, and water (all HPLC grade) were procured from Merck. All other chemicals were of laboratory reagent grade.
Preparation of Drug/Polymer Binary Physical Mixtures
The binary physical mixtures of CIL and polymer were prepared by gently grinding accurately weighed quantities in a mortar and pestle for 5 min. The physical mixtures were prepared with 80% polymer content, which is the highest polymer concentration in study to be used as a control.
Preparation of Solid Dispersions
The binary (drug and polymer) solid dispersions with 20%, 50%, 65%, and 80% polymer content were prepared by spray-drying technology using a laboratory-scale instrument (LU-228 Advanced spray drier, Labultima, Mumbai, India). The drug and polymer were dissolved in dichloromethane (10%, w/w). The spray dryer was equipped with a spray nozzle of 0.7 mm diameter and a peristaltic pump to feed the substrate. The solutions were sprayed using a feed rate of 2–5 g/min, at an atomization pressure of 0.8 bars, an inlet temperature of 130°C, and an outlet temperature of 80°C. These process parameters were maintained for all solid dispersion compositions. The prepared solid dispersions were vacuum dried at 40–50°C for 2 h in order to remove the residual solvents. The dried solid dispersions were evaluated for residual solvent content, as the significant amount of residual solvent(s) can affect the properties of solid dispersions based on its crystalline or amorphous state at initial or in stability studies. Furthermore, during this study, dried solid dispersions were stored in desiccators with P2O5.
Effect of Polymer Type on the Solubility of CIL
The equilibrium solubility of crystalline CIL was measured at 37 ± 0.5°C using purified water in the presence or absence of the polymer. Fifty milligrams of crystalline CIL was dispersed in 500 ml of purified water, in which 500 and 1000 mg of polymer (povidone, copovidone, or hypromellose) had been previously dissolved, leading to a final polymer concentration of 1.0 and 2.0 mg/ml. The solution was stirred with a rotating paddle at 50 rpm for 24 h. Samples of 5 ml were withdrawn and filtered through 0.45 μm nylon membrane filter (Axiva, SF1510613, India). The concentrations were determined using double-beam ultraviolet–visible (UV–VIS) spectrophotometer (Shimadzu, UV-2450) at a wavelength of 257 nm. No interference from any of the polymer investigated on the CIL assay was observed. All the measurements were done in triplicate, and the average drug solubility was reported in milligrams per milliliter along with standard deviation.
X-ray Powder Diffraction
The pXRD solid-state pattern of CIL, polymers, and solid dispersions was measured with D8 Advance (Bruker, USA) using an online recorder (PM 8203A). Radiations were generated from CuKα source and filtered through Ni filters with a wavelength of 0.154 nm at a generator current of 20 mA and voltage of 35 kV, while LynxEye being the detector. The instrument was operated over the 2θ range of 2–50° at a step size of 0.015°.
Inhibition of CIL Recrystallization from Supersaturated Solutions
A concentrated solution of CIL in ethanol was prepared by dissolving 50 mg of crystalline CIL in 20 ml ethanol, which was subsequently added to 500 ml of purified water at a temperature of 37 ± 0.5°C. This generated an initial drug solution concentration of 100 μg/ml in purified water, into which 200, 50, or 12.5 mg of polymer (povidone, copovidone, or hypromellose) had been previously dissolved, leading to a final polymer concentration of 400, 100, or 25 μg/ml, respectively. The solution was stirred with a rotating paddle at 50 rpm. Samples of 5 ml each were withdrawn from each vessel at predetermined time intervals (5, 10, 15, 30, 45, 60, 120, 180, and 240 min) and filtered through 0.45 μm nylon membrane filter (Axiva, SF1510613, India). At each time point, the same volume of fresh medium was replaced. The concentrations were determined using double-beam UV–VIS spectrophotometer (Shimadzu, UV-2450) at a wavelength of 257 nm and a standard calibration curve. The percent of CIL dissolved (n = 3) was plotted as a function of time. The same experiments were performed in purified water in the absence of any polymer.
In Vitro Dissolution Studies
Dissolution studies were conducted using the US Pharmacopoeia (USP) apparatus II, paddle stirring method at speed of 50 rpm (Electrolab, TDT-08L, Mumbai, India). Dissolution media (degassed and maintained at 37 ± 0.5°C) included 500 ml of purified water. Dissolution samples were analyzed at 257 nm using double-beam UV–VIS spectrophotometer (Shimadzu, UV-2450). Dissolution samples were collected at 5, 10, 15, 30, 60, 120, 180, and 240 min through 0.45 μm nylon membrane filter (Axiva, SF1510613, India) and suitably diluted. The volume of dissolution medium was adjusted to 500 ml by replacing each 5 ml aliquot withdrawn with 5 ml of dissolution media. Stock solution was prepared by dissolving 45 mg of CIL into 100 ml volumetric flask and added 5 ml of methanol and sonicated for about 3 min and made the volume with diluent (water and methanol in 4:1). Standard was prepared by diluting 2 ml of stock solution and 2 ml of dissolution media in 50 ml volumetric flask and making the volume with diluents. For all the dissolution studies, solid dispersions equivalent to 50 mg of CIL were used and the percent of CIL dissolved for each composition (n = 3) was plotted as a function of dissolution time.
Contact Angle
Contact angle of compacted powder samples were measured by sessile drop method using Drop Shape Analyzer instrument (FTA 1000, First Ten Angstrom, Virginia, USA). The powder compacts of 12 mm diameter and a weight of 300 mg were prepared using Rimek mini PRESS-IISF (Karnavati Engineering, India). The compression force was 1000 kg for 5 s. Water contained in a syringe from which a predefined volume was dispensed onto the sample surface and video images were captured by the FTA image analyzer. Contact angle was calculated by the instrument by fitting mathematical expression to the shape of the drop and then calculating the slope of the tangent to the drop at the liquid–solid–vapor interface line. The surface tension of water was measured to be 72.8 ± 0.5 mN/m at 25°C. All measurements were performed in air under ambient conditions, and the reported values are an average of three measurements.
Statistical Analysis
The effect of formulation on drug solubility/dissolution properties were statistically analyzed using a repeated measures one-way ANOVA. Individual differences in drug dissolution between formulations were statistically identified using Fisher’s PLSD test at 95% confidence interval. All the statistical calculations were performed using Minitab® 16.0 software and in all cases p < 0.05 denoted significance.
RESULTS AND DISCUSSION
Characterization CIL Received
The sample of CIL was characterized for thermal (DSC), FTIR, pXRD, and SEM for assessing the amorphous or crystalline behavior. As per these evaluations, it was concluded that the sample of CIL received was crystalline, polymorphic form A (30,31,34). The purity for CIL was 99.3%.
Assessing the Amorphous Character of Solid Dispersions
The XRD diffraction patterns of the solid dispersions prepared by using three different polymers are represented in Fig. 1a–c. Also represented are the diffraction pattern of the CIL and individual respective polymer. The XRD pattern of CIL exhibited intense and sharp characteristic peaks indicating its crystalline nature, while the XRD pattern of all the three polymers exhibited a hollow pattern due to their amorphous nature. Amorphous materials does not have long-range atomic order and therefore produces only broad scattering peaks which appears as a bump or what is referred to as “hollow pattern.”
Fig. 1.
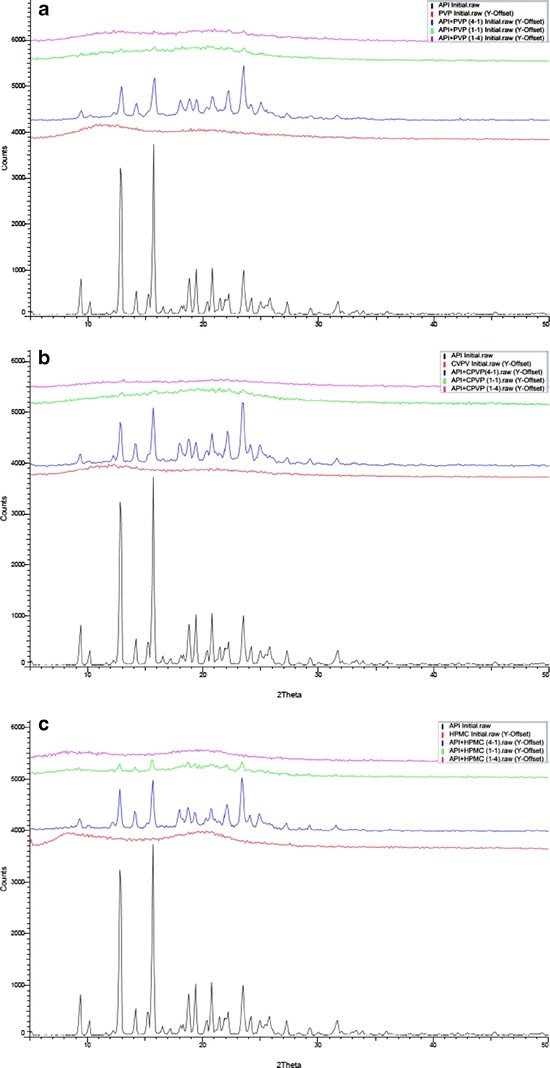
a XRD patterns of cilostazol, PVP, and solid dispersion with PVP. b XRD patterns of cilostazol, CPVP, and solid dispersion with CPVP. c XRD patterns of cilostazol, HPMC, and solid dispersion with HPMC
While, in case of solid dispersions having 20% polymer content characteristic, peaks of CIL with low intensity were observed, indicating that CIL still has some crystalline phase, the solid dispersions with 50% polymer content exhibited broad featureless peaks of CIL indicating a partial amorphous or partially dissolved form of CIL (35). However, the sample with 80% polymer content exhibited a hallow pattern, indicating formation of amorphous system of CIL (36). Even though the solid dispersion preparatory process has two drying steps: primary (spray drying) and secondary (vacuum oven drying); to ensure the critical role of residual solvent, all the solid dispersion samples were evaluated for residual solvents by gas chromatography. The level of dichloromethane in all the samples was less than 50 ppm, indicating that the samples were dried enough to rule out the development of more crystallinity in samples due to storage period. Therefore, it appears that the amount of polymeric carrier is not sufficient in solid dispersions with 20% or 50% polymer content to render them as an amorphous solid dispersion (single phase).
Estimation of Apparent Solubility
As already reported, due to recrystallization (physical instability) during dissolution, it is challenging to quantify the practical solubility of amorphous materials experimentally (37), therefore, a model developed by Parks et al. (38,39) may be used to predict the relative solubility of the amorphous and crystalline forms of material based on the difference in their free energy (ΔG) as per the below equation.
Where R is the gas constant, T is the temperature, and S is the solubility. Furthermore, the Hoffman equation can be used to estimate the free energy difference between the amorphous and crystalline forms if the melting temperature (Tm) and the heat of fusion (Hf) of the crystalline form are known, as per the below equation.
As per the Hoffman equation, at conditions where Tm ≫ T, it can be deduced that the higher the melting point and heat of fusion of a compound, the greater is the solubility enhancement that may be achieved by designing amorphous system for drug delivery. While the above approach is approximate, it does provide a useful guide to predict the extent of solubility enhancement that may be achieved through generation of amorphous drug. Using the above two equations, the solubility ratio of the amorphous and crystalline forms of CIL in purified water at 37°C was predicted to be approximately 32, which means that the theoretical solubility of amorphous CIL is about 32 times that of crystalline CIL. The equilibrium solubility of crystalline CIL was 2.63 μg/ml (Table I), so the apparent solubility of amorphous CIL was assessed to be 86.11 μg/ml.
Table I.
Equilibrium Solubility of Cilostazol (CIL) in Purified Water with or Without Dissolved Polymeric Carrier
| Polymer type | Polymer quantity (mg/ml) | Equilibrium solubility (μg/ml) |
|---|---|---|
| Absent | – | 2.63 (0.06)a |
| Povidone | 1.0 | 2.67 (0.08) |
| Povidone | 2.0 | 2.72 (0.05) |
| Copovidone | 1.0 | 2.64 (0.04) |
| Copovidone | 2.0 | 2.67 (0.07) |
| Hypromellose | 1.0 | 2.61 (0.09) |
| Hypromellose | 2.0 | 2.65 (0.07) |
aValues in parentheses represent the standard deviation, n = 3
Equilibrium Solubility in the Presence of Polymeric Carriers
The concentrated solutions of a polymeric carrier can help to assess the mechanism of dissolution of a drug from a solid dispersion (8). To investigate the solubilizing power of povidone, copovidone, and hypromellose, the equilibrium solubility of crystalline CIL in purified water containing 1.0 and 2.0 mg/ml of each of the respective polymers was determined and compared with the equilibrium solubility in the absence of polymer in purified water. The concentrations of 1.0 and 2.0 mg/ml were selected as they represented conditions wherein it would be most likely (best case scenario) to enhance solubility (high polymer to drug ratio). Furthermore, a polymer concentration of 1.0 mg/ml represents a much higher concentration of polymer that would be achieved in solution following dissolution of a drug/polymer matrix at a ratio of 1:4 (80% polymer load). All solutions reached equilibrium after 24 h. Previous studies have reported that the solubility of poorly soluble drugs was enhanced significantly in concentrated aqueous solutions of polymers as a result of the formation of weakly soluble complexes. For example, the solubility of Rofecoxib and Ibuproxam was enhanced in the presence of aqueous povidone and polyethylene glycol solutions (40,41). However, as referred in Table I, the equilibrium solubility of CIL did not change significantly with any of the polymer investigated at 1.0 and 2.0 mg/ml in purified water. Based on these solubility measurements, it can be concluded that the polymers had no influence on the solubility of crystalline CIL.
Inhibition of Recrystallization from Supersaturated Solutions
A supersaturated solution of CIL in ethanol was used to assess the ability of povidone, copovidone, and hypromellose to inhibit crystallization of CIL by adding the concentrated solution of CIL to concentrated aqueous solutions of polymeric carriers while monitoring the solution concentration as a function of time. Figure 2a–c shows results obtained at different polymer solution concentrations, 400, 100, or 25 μg/ml, which correspond to the polymer solution concentrations that would be produced by total dissolution of solid dispersions containing 80%, 50%, and 20% polymer, respectively. Also, the results obtained for purified water without any polymer dissolved are shown for reference. The supersaturated solution of CIL in ethanol provided solution concentration of 100 μg/ml. As can be referred from Fig. 2, in the absence of polymer, the CIL concentration decreased rapidly, reaching about 63 μg/ml after 5 min, and then continued to decrease until achieving a plateau after 30 min with a concentration close to the equilibrium solubility of crystalline CIL. This indicated that the CIL recrystallized rapidly from a supersaturated concentration in the absence of polymer.
Fig. 2.
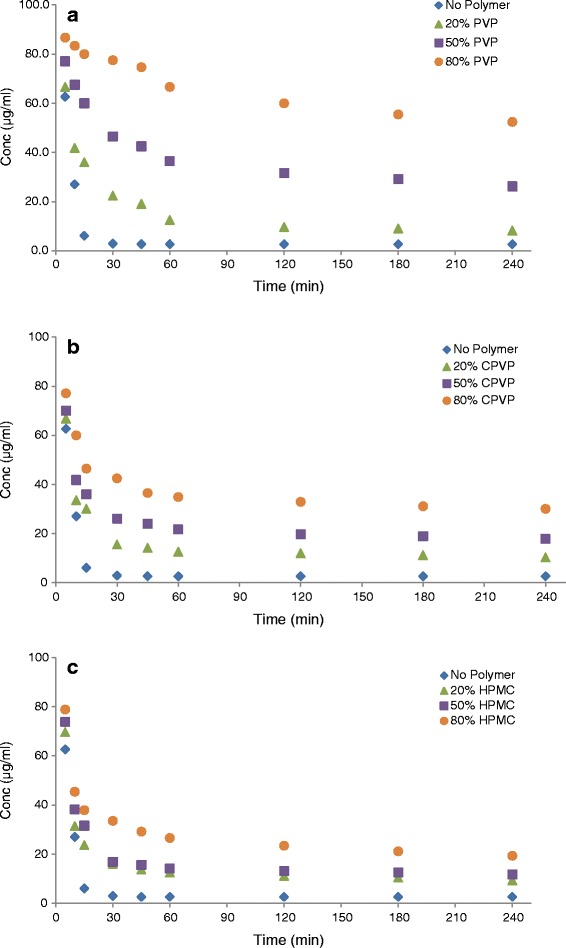
a Inhibitory effects of PVP on the recrystallization of a supersaturated solution of cilostazol in purified water. b Inhibitory effects of CPVP on the recrystallization of a supersaturated solution of cilostazol in purified water. c Inhibitory effects of HPMC on the recrystallization of a supersaturated solution of cilostazol in purified water
However, in the presence of PVP (Fig. 2a), the behavior of supersaturated solution (CIL in ethanol) changed significantly. The initial drug concentration, measured 5 min after addition of a concentrated solution of CIL, was significantly higher in purified water containing 400 and 100 μg/ml of PVP as compared with initial drug concentration in purified water containing 25 μg/ml of PVP (p < 0.0001). On the contrary, there was no significant difference between the drug concentrations achieved in purified water containing PVP concentrations of 25 μg/ml and absence of PVP (p = 0.219). At 5-min time point, CIL concentrations measured were about 86, 77, 67, and 63 μg/ml for 400, 100, 25, and 0 μg/ml PVP concentrations in purified water, respectively. These results indicate that there was a highly significant recrystallization inhibition due to the presence of PVP at concentrations of 400 and 100 μg/ml (p < 0.0001) insisting on the role and quantity of PVP to inhibit recrystallization of CIL from supersaturated solution. Furthermore, CIL touched concentrations of about 52, 26, and 8 μg/ml in purified water after 240 min in which PVP was present at concentrations of 400, 100, and 50 μg/ml, respectively. The difference in concentrations observed after 240 min indicates the role of PVP concentration to prevent the precipitation and recrystallization of CIL as a function of PVP concentration. Still, the CIL concentrations maintained even after 240 min are significantly higher than the equilibrium solubility of crystalline CIL (2.63 μg/ml; p < 0.0001 in all cases), proving the ability of PVP to maintain supersaturated levels of CIL. Similar observations were reported with HPMC, HPC, and PVP polymers in phosphate buffer to prevent the recrystallization of RS-8359 from its supersaturated solution (42).
Furthermore, both CPVP and HPMC (Fig. 2b, c) were able to prevent the recrystallization of CIL from its supersaturated solution in ethanol. The initial drug concentrations (measured 5 min) were significantly higher in purified water containing 400 and 100 μg/ml of CPVP/HPMC as compared with the initial drug concentration in purified water containing 25 μg/ml of CPVP/HPMC (p < 0.0001). On the contrary, there was no significant difference between the drug concentrations achieved in purified water containing 25 μg/ml of CPVP/HPMC and absence of any polymer (p = 0.272; 0.053). At 5-min time point, CIL concentration measured were about 77, 70, and 67 μg/ml for 400, 100, and 25 μg/ml CPVP concentrations, respectively; while 79, 74, and 70 μg/ml of concentrations were measured for 400, 100, and 25 μg/ml HPMC concentrations in purified water. These results indicate that there was a highly significant recrystallization inhibition at 5 min time point due to the presence of CPVP/HPMC at concentration of 400 and 100 μg/ml (p < 0.0001) insisting the role and quantity of these polymer to inhibit recrystallization of CIL from its supersaturated solution. However, the property of crystallization inhibition of both these polymers was not as effective as observed for PVP. After 240 min, in case of CPVP, CIL concentrations of about 30, 18, and 10 μg/ml were observed in purified water containing 400, 100, and 25 μg/ml of polymer, respectively. In case of HPMC, CIL concentrations of about 19, 12, and 9 μg/ml were observed in purified water containing 400, 100, and 25 μg/ml of polymer, respectively. The difference in concentrations observed after 240 min indicates the concentration-dependant role of CPVP/HPMC to prevent the precipitation and recrystallization of CIL. Even though the concentrations observed at 240 min are not as high as those observed for PVP, still these were significantly higher than the equilibrium solubility of crystalline CIL (2.63 μg/ml; p < 0.0001 in all cases). These findings indicate that both CPVP and HPMC play a role to prevent the precipitation and recrystallization of CIL from its supersaturated solution; however, the impact is less efficient and frail as compared with PVP, since the CIL concentrations started rapidly dropping after 10 min and reached near to a plateau in about 60 min.
In Vitro Dissolution Studies
The main objective of formulating poorly water-soluble drugs in solid dispersions is to enhance their dissolution rate and saturation solubility. The dissolution studies on pure drug and solid dispersion were performed in 500 ml of purified water at 37 ± 0.5°C using paddle-stirring method (USP-II) at a speed of 50 rpm. The dissolution profiles are depicted as the concentration versus time plot in Fig. 3a–c. It is pragmatic from these figures that all the solid dispersions have significantly improved the dissolution of CIL as compared with pure drug. The concentrations of CIL observed are a function of type and amount of polymer used in the solid dispersion. Furthermore, with all the three polymers evaluated, the concentration of CIL reached a plateau after 180 min. And as the concentrations reached a plateau after 180 min, the concentrations at 240 min were used to compare the results for enhanced dissolution against pure CIL.
Fig. 3.
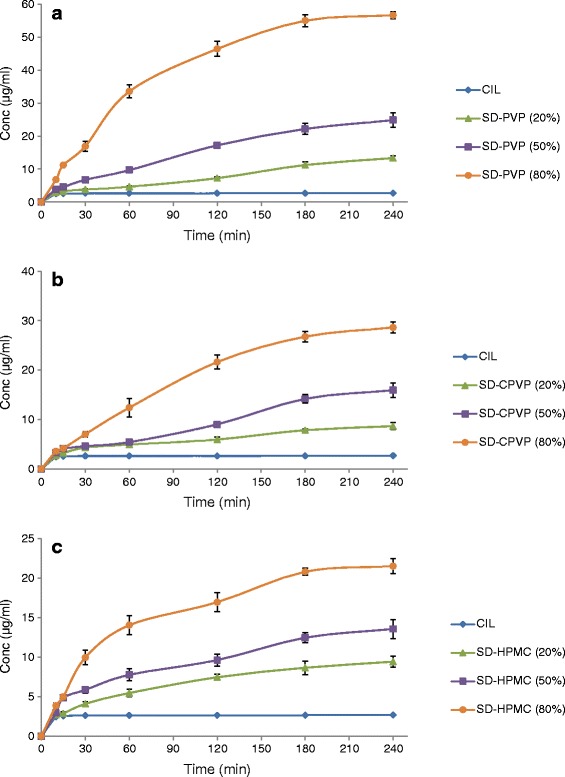
a Dissolution curve of cilostazol, along with solid dispersions containing PVP. b Dissolution curve of cilostazol, along with solid dispersions containing CPVP. c Dissolution curve of cilostazol, along with solid dispersions containing HPMC
As seen in Fig. 3, povidone provided the best results with a concentration of about 56 μg/ml as compared with 29 and 22 μg/ml observed for CPVP and HPMC, respectively, in solid dispersions having a polymer load of 80%. These concentration are exceedingly superior (p < 0.0001 in all cases) to equilibrium solubility of pure CIL (2.63 μg/ml) and indicate that supersaturated solutions were produced. Furthermore, the stabilized solution concentrations of CIL observed after 180 min are in good correlation with the equilibrium solubility observed in case of polymeric solutions of respective polymer. Also, the amount of concentration observed was significantly dependent on the polymer load in solid dispersion of the respective polymer. For example, the stabilized solution concentrations of 56 and 25 μg/ml for CIL-PVP solid dispersions matched with CIL concentration (53 and 26 μg/ml) observed in PVP polymeric solution (400 and 100 μg/ml) at 240 min, respectively, with 80% and 50% polymer load in solid dispersions. On the contrary, the PVP solid dispersion (with 20% polymer load) yielded in stabilized solution concentration of about 13 versus 8 μg/ml the equilibrium solubility observed in PVP polymeric solution (25 μg/ml) at 240 min, indicating that the lower the polymer load in solid dispersion, the lesser is the crystallization inhibition efficiency of the polymer. This suggest that PVP at a loading of 50% (w/w) or higher in solid dispersion results in dissolution of CIL closer to the amorphous solubility of the drug and prevents the precipitation and recrystallization of CIL, and the observed solubility is significantly higher than the solubility of crystalline CIL.
In comparison with the solid dispersion containing PVP, the degree of dissolution rate enhancement was much lower for solid dispersion containing CPVP or HPMC as carrier polymer. Specifically, concentrations of about 29 and 22 μg/ml observed at 240 min with solid dispersion loaded with 80% of CPVP/HPMC, respectively. Furthermore, concentrations of about 16 and 9 μg/ml were observed for solid dispersions loaded with 50% and 20% of CPVP, while concentrations of 14 and 10 μg/ml were observed for solid dispersions loaded with 50 and 20% of HPMC. Even though these concentration are far superior (p < 0.0001 in all cases) than the equilibrium solubility of CIL (2.63 μg/ml), these are comparatively much lower than the concentrations observed for PVP.
The results suggest that PVP is superior in promoting and stabilizing the dissolution of CIL as compared with CPVP/HPMC. It was observed and concluded from experiments examining the inhibitory effects of polymer (Table I) that individually the polymers as such do not have a solubilization effect. The observed dissolution enhancement of CIL when prepared in solid dispersions specifically with polymer content of 80% is related to amorphous solubility and stabilization of the drug by the respective polymer. For solid dispersions with lower amount of polymer (either 20% or 50%), the lower dissolution rate may be due to insufficient polymer content or residual crystallinity or a combination of both. In our previous work (34), we concluded that CIL has a low Tg of about 32°C and a poor glass former and to prepare the solid dispersions of CIL, a systematic approach was developed. A number of polymeric and non-polymeric materials were screened, and it was found that PVP, CPVP, and HPMC were the polymers of choice to prepare acceptable solid dispersions of CIL. Furthermore, the prepared solid dispersions with PVP, CPVP, and HPMC showed strong interactions and miscibility between CIL and the polymer as investigated by DSC, XRD, and IR spectroscopy along with combined Tg and Florey–Huggins interaction parameters. The order of interactions and miscibility observed between CIL and polymer was as follows PVP > CPVP > HPMC. Moreover, the amorphous solid dispersions (single phase) exhibited a single Tg between the Tg of CIL and respective polymer followed the same pattern of PVP > CPVP > HPMC, and it is expected to result in greater physical stability during dissolution (43). Furthermore, stronger interactions and miscibility behavior between CIL and PVP correlated well with dissolution enhancement observed for the solid dispersions loaded with more than 50% PVP. However, dissolution enhancement observed for solid dispersions containing CPVP/HPMC did not correlate to the extent for the strong interactions predicted with CPVP/HPMC. These assortments observed in dissolution/solubility data for solid dispersions containing PVP versus CPVP/HPMC represent the current status of the field and ask for further investigations to develop the mechanistic understanding driving stabilization of supersaturated drug solutions in the presence of polymeric materials and the solubilizing action of polymers (44).
Contact Angle and Wetting Kinetics
Generally, contact angle is measured by sessile drop method using compacted powder disc or powder layer adhered to an inert support (45,46). Both the techniques are well reported; however, in the present work, compacted powder disc approach was employed as the results have better reproducibility. The limits of a contact angle are 0° for complete wetting and 180° for no wetting. Although a contact angle can be altered in different solutions, water was regularly adopted to be the wetting solution in the test (47). The contact angles were measured over a period of 0.2 to 5 s as there was no significant change in the contact angle. The contact angle at 0.2 s was considered as the initial contact angle; CIL (crystalline), PVP, CPVP, and HPMC exhibited initial contact angles of 68 ± 0.6°, 27 ± 0.5°, 33 ± 0.7°, and 52 ± 0.9°, respectively. While higher contact angle for CIL indicates the poor wetting attribute as well as hydrophobic character of the molecule, both PVP and CPVP exhibited lower contact angles, indicating good wetting properties and hydrophilic character. In comparison, even though HPMC is also water soluble (like PVP and CPVP), the contact angle of 52 indicates a mixture of hydrophilic as well as hydrophobic character and struggles to dissolve in water; the rate of dissolving in water is very slow. Furthermore, for solid dispersion of CIL with these polymers at different levels of polymer content (20%, 50%, 65%, or 80%), the contact angle reduced as the level of polymer content increased in solid dispersion proving that the introduction of a right polymer with insoluble drug improves the wettability of the composite and results in better dissolution rates. However, the change in contact angle was insignificant when the polymer content increased from 65% to 80%, indicating a plateau stage (Fig. 4). Among the three polymers studied, the reduction in contact angle of CIL was most favored by PVP followed by CPVP; however, the contribution from HMPC was insignificant.
Fig. 4.
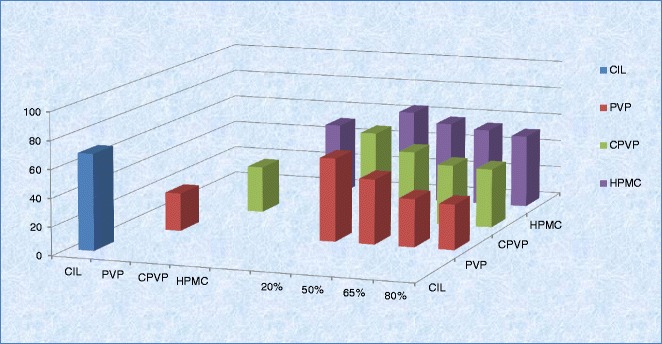
Variation of contact angle of cilostazol with water as per polymer content in solid dispersion
An extension to the contact angle, in order to understand further the enhanced dissolution rate of solid dispersions, the wetting process with water was quantified from the work of adhesion (Wa), immersion (Wi), and spreading (Ws) as per the below equations (48).
where θ is contact angle in degrees, γ is interfacial tension, and the subscripts SL, SV, and LV represent the solid–liquid, solid–vapor, and liquid–vapor interfaces, respectively. For determining the wetting process for individual components and solid dispersions, the contact angle at 5 s and surface tension of water was used. The surface tension and polarity index of water were 72.8 mJ/m2 and 0.701, respectively. In the wetting process, the thermodynamic driving force for each process, i.e., adhesion, immersion, and spreading is denoted by the work value. The work value can be positive or negative, where a negative value indicates spontaneity of the process (49).
The work of adhesion is actually the work required to separate the solid and liquid and solely depends on the contact angle and surface tension of the liquid. The negative Wa values (Fig. 5a) obtained for individual component and solid dispersions indicate that the adhesion process is spontaneous for every material studied in this study. However, the magnitude of difference is very different for pure CIL (crystalline) and the polymer studied, where both PVP and CPVP has very high work of adhesion. Furthermore, solid dispersions of CIL with PVP and CPVP exhibited the improved spontaneity over the pure CIL, indicating that the introduction of the specific polymer in solid dispersion had improved the work of adhesion (more negative); however, it was not significantly improved with HPMC. Also, the amount of polymer in solid dispersion is important because the solid dispersion with 20% or 50% polymer content exhibited less Wa as compared with solid dispersions with 80% polymer content. As observed in comparative dissolution rate in the previous section, the less significant Wa values for solid dispersion with 20% polymer content may be due to the residual crystallinity in the samples.
Fig. 5.
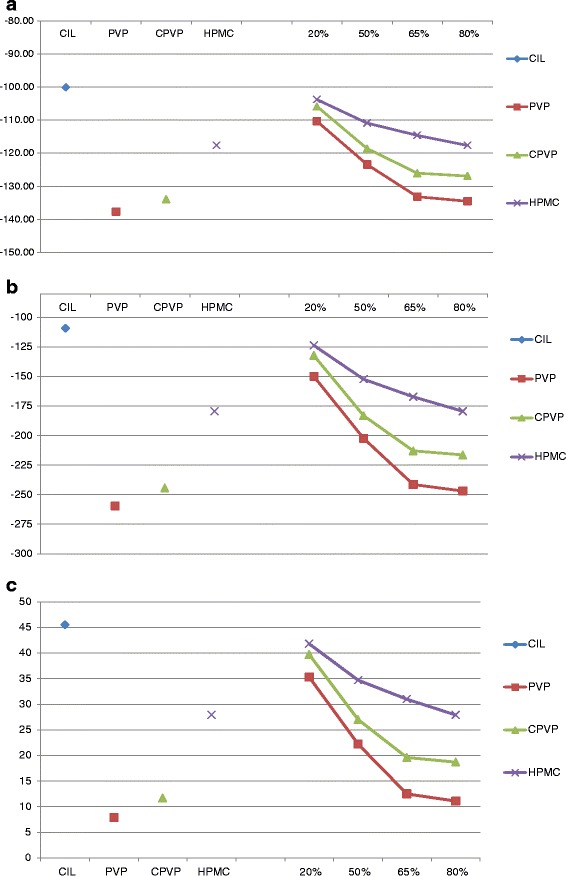
a Variation of work of adhesion (W a) with water as per polymer content in solid dispersion. b Variation of work of immersion (W i) with water as per polymer content in solid dispersion. c Variation of work of spreading (W s) with water as per polymer content in solid dispersion
The process of immersion involves a change of solid–vapor interface to solid–liquid interface, and the driving for force Wi indicated that here too all the individual components and solid dispersion had negative values proving that the process is spontaneous. However, the magnitude of difference varied prominently for CIL against PVP and CPVP (Fig. 5b). Furthermore, the work values for immersion stage became more negative as the content of polymer increased, particularly PVP and CPVP increased from 20% to 80%, indicating the improvement in spontaneity of the immersion process correlating the initial faster dissolution rate observed in case of solid dispersions. However, the role of residual crystallinity cannot be ruled out for insignificant improvement in immersion process for solid dispersions with 20% polymer content.
Work of spreading (Ws) needs an energy equivalent to the difference between the work of cohesion and adhesion; and a negative value of Ws favors the process of dispersion. In the present study, the Ws values for individual component as well as solid dispersion had positive values, indicating spreading as an unfavorable process (Fig. 5c). However, the magnitude of spreading with water was higher for CIL as compared with the polymers. Furthermore, solid dispersions exhibited the lower values showing that making solid dispersions improved the spreading process and leads to superior dissolution rates. When we increase the loading of the specific polymer in solid dispersion, the work of spreading also decreases, and a plateau stage was observed at 65% to 80% polymer content in solid dispersions. Furthermore, the decrease in the work of spreading was significant in cases of PVP and CPVP as against HPMC, and this correlates well with the dissolution rate enhancements observed in the respective solid dispersions.
This study reveals that the improvement in the wetting process, step by step through wetting kinetics correlates well with the composition of respective solid dispersion. The solid dispersion with PVP as carrier exhibited faster wetting process as compared with CPVP or HMPC as polymeric carrier. Even the polymer load in the respective solid dispersion is important as it affects the levels of improvement in the wetting process. A very significant improvement in solid dispersions with 80% polymer content was observed and not so significant improvement with 20% polymer content. However, the less significant improvement for solid dispersions with lower polymer content (either 20% or 50%) could either be because of lower polymer load or the residual crystallinity in the respective solid dispersion.
CONCLUSIONS
In this work, we have investigated the role of polymer by solid dispersion approach in the dissolution enhancement of poorly soluble drug and its correlation with the wetting kinetics. In the first series of experiments, the dissolution behavior of amorphous solid dispersions of poorly water-soluble drug cilostazol was investigated. The type and content of polymer affected the supersaturated drug concentrations generated from the dissolution of amorphous solid dispersions. Solid dispersions prepared with PVP were found to result in solutions with highest level of supersaturation, whereas the solid dispersion with CPVP and HPMC were less efficient. At equivalent supersaturation, the crystallinity inhibition followed the order of PVP > CPVP > HMPC containing solid dispersions. These findings indicate the significant role of selecting the appropriate polymer and its concentration in solid dispersion for stabilizing the supersaturated solution generated following the dissolution of the amorphous solid. In the second series of experiments, the contact angle and wetting kinetics were studied to understand surface properties. The solid surface free energy of the solid dispersions decreased, thereby increasing the work of adhesion. The interaction between liquid and solid surface (immersion stage) for solid dispersions became high as compared with the pure drug. The enhanced dissolution rate as per the type and content of polymer in solid dispersion correlated well with contact angle and wetting kinetics studies. The solid dispersion with faster dissolution rate exhibited a superior wetting kinetics and vice-versa.
Acknowledgments
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Horter D, Dressman JB. Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract. Adv Drug Deliv Rev. 1997;25:3–14. doi: 10.1016/S0169-409X(96)00487-5. [DOI] [PubMed] [Google Scholar]
- 2.Kaushal AM, Gupta P, Bansal AK. Amorphous drug delivery systems: molecular aspects, design and performance. Crit Rev Ther Drug Carrier Syst. 2004;21:133–193. doi: 10.1615/CritRevTherDrugCarrierSyst.v21.i3.10. [DOI] [PubMed] [Google Scholar]
- 3.Chiou WL, Riegelman S. Pharmaceutical applications of solid dispersion systems. J Pharm Sci. 1971;60:1281–1302. doi: 10.1002/jps.2600600902. [DOI] [PubMed] [Google Scholar]
- 4.Hancock BC, Zografi G. Characteristics and significance of the amorphous state in pharmaceutical systems. J Pharm Sci. 1997;86:1–12. doi: 10.1021/js9601896. [DOI] [PubMed] [Google Scholar]
- 5.Serajuddin AM. Solid dispersion of poorly water-soluble drugs: early promises subsequent problems, and recent breakthroughs. J Pharm Sci. 1999;88(10):1058–1066. doi: 10.1021/js980403l. [DOI] [PubMed] [Google Scholar]
- 6.Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50:47–60. doi: 10.1016/S0939-6411(00)00076-X. [DOI] [PubMed] [Google Scholar]
- 7.Vo CL, Park C, Lee BJ. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur J Pharm Biopharm. 2013;85:799–813. doi: 10.1016/j.ejpb.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 8.Craig DQM. Review: the mechanism of drug release from solid dispersions in water-soluble polymers. Int J Pharm. 2002;231:131–144. doi: 10.1016/S0378-5173(01)00891-2. [DOI] [PubMed] [Google Scholar]
- 9.Sekiguchi K, Obi N. Studies on absorption of eutectic mixture. I. A comparison of the behaviour of eutectic mixture of sulfathiazole and that of ordinary sulfathiazole in man. Chem Pharm Bull. 1961;9:866–872. doi: 10.1248/cpb.9.866. [DOI] [Google Scholar]
- 10.Sekiguchi K, Obi N, Ueda Y. Studies on absorption of eutectic mixtures. II: absorption of fused conglomerates of chloramphenicol and urea in rabbits. Chem Pharm Bull. 1964;12:134–139. doi: 10.1248/cpb.12.134. [DOI] [PubMed] [Google Scholar]
- 11.Lakshman JP, et al. Application of melt extrusion in the development of a physically and chemically stable high-energy amorphous solid dispersion of a poorly water-soluble drug. Mol Pharm. 2008;5(6):994–1002. doi: 10.1021/mp8001073. [DOI] [PubMed] [Google Scholar]
- 12.Riikka Laitinen R, et al. Emerging trends in the stabilization of amorphous drugs. Int J Pharm. 2013;453:65–79. doi: 10.1016/j.ijpharm.2012.04.066. [DOI] [PubMed] [Google Scholar]
- 13.Shah S, et al. Melt extrusion with poorly soluble drugs. Int J Pharm. 2013;453:233–252. doi: 10.1016/j.ijpharm.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 14.Paudel A, et al. Manufacturing of solid dispersions of poorly water soluble drugs by spray drying: formulation and process considerations. Int J Pharm. 2013;453:253–284. doi: 10.1016/j.ijpharm.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 15.Baird JA, Taylor LS. Evaluation of amorphous solid dispersion properties using thermal analysis techniques. Adv Drug Deliv Rev. 2012;64:396–421. doi: 10.1016/j.addr.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 16.Bugay DE. Characterization of the solid-state: spectroscopic techniques. Adv Drug Deliv Rev. 2001;48:43–65. doi: 10.1016/S0169-409X(01)00101-6. [DOI] [PubMed] [Google Scholar]
- 17.Tanno F, Nishiyama Y, Kokubo H, Obara S. Evaluation of hypromellose acetate succinate (HPMCAS) as a carrier in solid dispersions. Drug Dev Ind Pharm. 2004;30:9–17. doi: 10.1081/DDC-120027506. [DOI] [PubMed] [Google Scholar]
- 18.Huang J, Wigent RJ, Schwartz JB. Drug–polymer interaction and its significance on the physical stability of nifedipine amorphous dispersion in microparticles of an ammonio methacrylate copolymer and ethylcellulose binary blend. J Pharm Sci. 2008;97(1):251–262. doi: 10.1002/jps.21072. [DOI] [PubMed] [Google Scholar]
- 19.Andrews GP, Abu-Diak O, Jones DS. Physicochemical characterization of hot-melt extruded bicalutamide-polyvinylpyrrolidone solid dispersions. J Pharm Sci. 2010;99(3):1322–1335. doi: 10.1002/jps.21914. [DOI] [PubMed] [Google Scholar]
- 20.Van den Mooter G, et al. Physical stabilization of amorphous ketoconazole in solid dispersions with polyvinylpyrrolidone K25. Eur J Pharm Sci. 2001;12:261–269. doi: 10.1016/S0928-0987(00)00173-1. [DOI] [PubMed] [Google Scholar]
- 21.Manias E, et al. Intercalation kinetics of long polymers in 2 nm confinements. Macromolecules. 2000;33:7955–7966. doi: 10.1021/ma0009552. [DOI] [Google Scholar]
- 22.Park SJ, Jin JS. Effect of silane coupling agent on interphase and performance of glass fibers/unsaturated polyester composites. J Colloid Interface Sci. 2001;242:174–179. doi: 10.1006/jcis.2001.7788. [DOI] [Google Scholar]
- 23.Matteucci, et al. Highly supersaturated solutions of amorphous drugs approaching predictions from configurational thermodynamic properties. J Phys Chem B. 2008;112:16675–16681. doi: 10.1021/jp805991f. [DOI] [PubMed] [Google Scholar]
- 24.Shimizu T, Osumi T, Niimi K, Nakagawa K. Physico-chemical properties and stability of cilostazol. Arzneimittelforschung. 1985;35:1117–1123. [PubMed] [Google Scholar]
- 25.Toyobuku H, Tamai I, Ueno K, Tsuji A. Limited influence of P-glycoprotein on small-intestinal absorption of cilostazol, a high absorptive permeability drug. J Pharm Sci. 2003;92:2249–2259. doi: 10.1002/jps.10490. [DOI] [PubMed] [Google Scholar]
- 26.Jinno JJ, et al. Effect of particle size reduction on dissolution and oral absorption of a poorly water-soluble drug, cilostazol, in beagle dogs. J Control Release. 2006;111:56–64. doi: 10.1016/j.jconrel.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 27.Dawson DL. Comparative effects of cilostazol and other therapies for intermittent claudication. Am J Cardiol. 2001;87:19D–27D. doi: 10.1016/S0002-9149(01)01673-3. [DOI] [PubMed] [Google Scholar]
- 28.Money SR, et al. Effect of cilostazol on walking distances in patients with intermittent claudication caused by peripheral vascular disease. J Vasc Surg. 1998;27:267–275. doi: 10.1016/S0741-5214(98)70357-X. [DOI] [PubMed] [Google Scholar]
- 29.Pinnamaneni S, Das NG, Das SK. Formulation approaches for orally administered poorly soluble drugs. Pharmazie. 2002;57:291–300. [PubMed] [Google Scholar]
- 30.Jinno JJ, et al. In vitro–in vivo correlation for wet-milled tablet of poorly water-soluble cilostazol. J Control Release. 2008;130:29–37. doi: 10.1016/j.jconrel.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 31.Kim MS, Lee S, Park JS, Woo JS, Hwang JS. Micronization of cilostazol using supercritical antisolvent (SAS) process: effect of process parameters. Powder Technol. 2007;177:64–70. doi: 10.1016/j.powtec.2007.02.029. [DOI] [Google Scholar]
- 32.Whittall LB, Whittle RR, Stowell GW. Polymorphic forms of cilostazol. Acta Crystallogr. 2002;C58:525–527. doi: 10.1107/s0108270102012544. [DOI] [PubMed] [Google Scholar]
- 33.Stowell GW, et al. Thermally-prepared polymorphic forms of cilostazol. J Pharm Sci. 2002;9:2481–2488. doi: 10.1002/jps.10240. [DOI] [PubMed] [Google Scholar]
- 34.Verma S, Rudraraju VS. A systematic approach to design and prepare solid dispersion of poorly water soluble drug. AAPS Pharm Sci Tech. doi:10.1208/s12249-014-0093-z). [DOI] [PMC free article] [PubMed]
- 35.Bley H, Fussneggerb B, Roland BR. Characterization and stability of solid dispersions based on PEG/polymer blends. Int J Pharm. 2010;390:165–173. doi: 10.1016/j.ijpharm.2010.01.039. [DOI] [PubMed] [Google Scholar]
- 36.Eerdenbrugh BV, et al. Itraconazole/TPGS/Aerosil®200 solid dispersions: characterization, physical stability and in vivo performance. Eur J Pharm Sci. 2009;38:270–278. doi: 10.1016/j.ejps.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 37.Hancock BC, Parks M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm Res. 2000;17:397–404. doi: 10.1023/A:1007516718048. [DOI] [PubMed] [Google Scholar]
- 38.Parks GS, Huffman HM, Cattor FR. Studies on glass. II: the transition between the glassy and liquid states in the case of glucose. J Phys Chem. 1928;32:1366–1379. doi: 10.1021/j150291a008. [DOI] [Google Scholar]
- 39.Parks GS, Snyder LJ, Cattoir FR. Studies on glass. XI: some thermodynamic relations of glassy and alpha-crystalline glucose. J Chem Phys. 1934;2:595–598. doi: 10.1063/1.1749540. [DOI] [Google Scholar]
- 40.Cirri M, et al. Characterization of ibuproxam binary and ternary dispersions with hydrophilic carriers. Drug Dev Ind Pharm. 2004;30(1):65–74. doi: 10.1081/DDC-120027513. [DOI] [PubMed] [Google Scholar]
- 41.Ahuja N, Katare OP, Singh B. Studies on dissolution enhancement and mathematical modelling of drug release of a poorly water-soluble drug using water-soluble carriers. Eur J Pharm Biopharm. 2007;65:26–38. doi: 10.1016/j.ejpb.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 42.Usui F, et al. Inhibitory effects of water-soluble polymers on precipitation of RS-8359. Int J Pharm. 1997;154:59–66. doi: 10.1016/S0378-5173(97)00129-4. [DOI] [Google Scholar]
- 43.Gupta P, Kakumanu KV, Bansal AK. Stability and solubility of celecoxib:PVP amorphous dispersions: a molecular perspective. Pharm Res. 2004;21(10):1762–1769. doi: 10.1023/B:PHAM.0000045226.42859.b8. [DOI] [PubMed] [Google Scholar]
- 44.Osama A, et al. An investigation into the dissolution properties of celecoxib melt extrudates: understanding the role of polymer type and concentration in stabilizing supersaturated drug concentrations. Mol Pharm. 2011;8:1362–1371. doi: 10.1021/mp200157b. [DOI] [PubMed] [Google Scholar]
- 45.Buckton G. Interfacial phenomena in drug delivery and targeting. Switzerland: Harwood Academic Publishers; 1995. [Google Scholar]
- 46.Ahfat NM, et al. An exploration of interrelationships between contact angle, inverse phase gas chromatography and triboelectric charging data. Eur J Pharm Sci. 2000;9:271–276. doi: 10.1016/S0928-0987(99)00063-9. [DOI] [PubMed] [Google Scholar]
- 47.Tian, et al. Influence of polymorphic form, morphology, and excipient interactions on the dissolution of carbamazepine compacts. J Pharm Sci. 2007;96:584–594. doi: 10.1002/jps.20756. [DOI] [PubMed] [Google Scholar]
- 48.Buckton G, Beezer AE. A microcalorimetric study of powder surface energetics. Int J Pharm. 1988;41:139–145. doi: 10.1016/0378-5173(88)90146-9. [DOI] [Google Scholar]
- 49.Young SA, Buckton G. Particle growth in aqueous suspensions: the influence of surface energy and polarity. Int J Pharm. 1990;60:235–241. doi: 10.1016/0378-5173(90)90077-H. [DOI] [Google Scholar]