

Abstract
For the first-aid treatment of anaphylaxis, epinephrine (Epi) 0.3 mg intramuscular (IM) injection in the thigh is the drug of choice. Epi auto-injectors are widely recommended for anaphylaxis treatment in community settings but not necessarily carried or used as prescribed when anaphylaxis occurs. We therefore developed rapidly disintegrating sublingual tablets (RDSTs) as an alternative noninvasive dosage form. Our objective in this study was to evaluate the effect of reducing Epi particle size on its in vitro and ex vivo diffusion, with the goal of enhancing Epi sublingual absorption from Epi RDSTs. Epi particle size was reduced by top-bottom technique using a microfluidizer for one pass at 30,000 Psi. The micronized Epi crystals (Epi-MC) were characterized using Zetasizer, Fourier transform infrared (FT-IR), differential scanning calorimetry (DSC), and scanning electron microscopy (SEM). Epi RDSTs were formulated and manufactured using our previously developed method. In vitro and ex vivo diffusion of Epi 10, 20, and 40 mg RDSTs and Epi-MC 10 and 20 mg RDSTs (n = 4) were evaluated using Franz cells. Epi 10 mg solution was used as a control. Mean (±standard deviation (SD)) Epi particle size was successfully reduced from 131.8 ± 10.5 to 2.5 ± 0.4 μm. Cumulative Epi diffused and influx from 40 mg Epi RDSTs and 20 mg Epi-MC RDSTs were not significantly different from each other in vitro and ex vivo (p > 0.05). Also, Epi permeability from 20 mg Epi-MC RDSTs was significantly higher than from the rest (p < 0.05). Epi-MC RDSTs improved Epi diffusion twofold and might have the potential to reduce the Epi dose needed in RDSTs by 50%.
KEY WORDS: adrenaline, anaphylaxis, diffusion, epinephrine, sublingual
INTRODUCTION
For the emergency treatment of anaphylaxis, prompt intramuscular injection of epinephrine (Epi) in the thigh is the treatment of choice (1–4). Epi auto-injectors such as EpiPen® (Mylan Inc, Basking Ridge, NJ) and Auvi-Q™ (Sanofi-Aventis, Bridgewater, NJ) are widely recommended for the treatment of anaphylaxis in community settings; however, they are not necessarily carried or used by patients at risk for anaphylaxis in the community (1–4). An alternative Epi formulation, rapidly disintegrating sublingual tablets (RDSTs), is being developed for the potential use in community settings (5–8) and might offer the advantages of a wider dose range of Epi beyond the 0.15- and the 0.3-mg fixed doses currently available in EpiPen® and Auvi-Q™ auto-injectors, and enhanced stability of Epi in a solid dosage form that might reduce the need for annual replacement associated with the use of auto-injectors containing Epi in aqueous solution (9).
The sublingual route is a promising alternative route for Epi administration (5–8). Drugs absorbed sublingually bypass metabolic conversion in the gastrointestinal tract and hepatic first-pass metabolism and reach the systemic circulation in a pharmacologically active form (5–8,10). This advantage is highly relevant to epinephrine, which is extensively metabolized after oral administration by the catechol-O-methyltransferase in the gastrointestinal tract and by monoamine oxidase in the gastrointestinal tract and in the liver (11).
The high vascularity of the sublingual mucosa and the low molecular weight of Epi, 183.2 mg, facilitate its rapid absorption directly into the venous circulation through the sublingual veins. Previously, we developed Epi RDSTs that retain sufficient hardness to withstand shipping and handling but disintegrate rapidly (≤30 s) to release Epi (12–16). In a validated rabbit model, we demonstrated that the rate and extent of absorption after administration of an Epi 40 mg RDSTs was similar to the rate and extent of absorption after administration of Epi 0.3 mg intramuscular (IM) through an EpiPen® (5,7,8). In our previous studies, the high epinephrine dose in the RDSTs was essential for establishing the concentration gradient needed to facilitate epinephrine absorption across the sublingual mucosa and achieve the required plasma epinephrine levels.
Particle size reduction to the micro- or nanosize is one of the common approaches to enhance the rate and extent of drug dissolution and absorption. Fabrication of drug microcrystals and nanocrystals requires few excipients and results in the production of virtually 100% pure drug (17). Moreover, the resulting drug crystals can be formulated into various dosage forms and a wide range of doses.
We hypothesized that significant reduction in Epi particle size might significantly increase Epi dissolution and diffusion rates and extents and enhance the Epi absorption rate and extent.
Our objective in this in vitro and ex vivo study was to evaluate the effect of the reduction in Epi particle size in the RDSTs on the rate and extent of Epi diffusion. We compared the rate and extent of in vitro and ex vivo diffusion of several doses of Epi crystals (Epi), before processing, and of our Epi microcrystals (Epi-MC), after processing, in RDSTs using Franz cell diffusion system.
MATERIALS AND METHODS
Materials
(−)-Epinephrine (+) bitartrate (EpiBit) was purchased from Sigma-Aldrich (St. Louis, MO). Ceolus® PH-301 (microcrystalline cellulose) with a mean particle size of 50 μm was supplied by Asahi Kasei Chemicals Corp. (Tokyo, Japan), and low-substituted hydroxypropyl cellulose (LH11) with a mean particle size of 50 μm was supplied by Shin-Etsu Chemical Co (Tokyo, Japan). Magnesium stearate was purchased from Mallinckrodt Baker (Phillipsburg, NJ). Isopropyl alcohol, 99.5%, was purchased from BDH (VWR, West Chester, PA). Spectra/Por® 7 dialysis membranes with 1,000 Da molecular weight cutoff (MWCO) were purchased from Spectrum Laboratories, Inc. (Rancho Dominguez, CA). Potassium phosphate monobasic was purchased from Sigma-Aldrich (St. Louis, MO), and sodium hydroxide was purchased from J.T. Baker (Philipsburg, NJ).
Preparation of Epinephrine Bitartrate Microcrystals
EpiBit microcrystals (EpiBit-MC) were prepared by a top-bottom technique using LV-1 high shear fluid processor “microfluidizer” (Microfluidics, Newton, MA) equipped with a G10Z-type reaction chamber (Fig. 1) and a maximum capacity of 6 mL. This reaction chamber has a “Z” shape to optimize the shear process of 12.25 million s−1 at 30,000 Psi. Briefly, EpiBit 16.8 mg was suspended in 6 mL isopropyl alcohol, sonicated for 30 s, and injected into the system at 30,000 Psi for one pass, based on our previous unpublished data. The microfluidizer receiving coil was immersed in ice to reduce the heat produced during the process. The fine suspension obtained was centrifuged using Avanti J-25 centrifuge (Beckman Coulter, Inc, Miami, FL) at 27,200×g and 15°C for 30 min. The upper clear isopropyl alcohol solvent layer was removed by aspiration, and the remaining particles at the bottom of the tube were dried overnight under vacuum in a dark room at ambient room temperature.
Fig. 1.
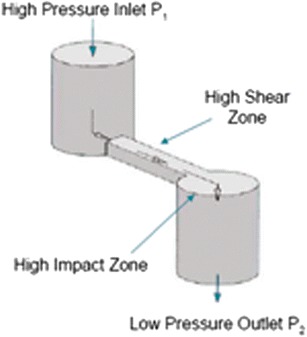
“Z” shape of G10Z-type reaction chamber of the low volume (LV-1 type) high shear fluid processor “microfluidizer” (Microfluidics, Newton, MA)
Characterization of Epinephrine Bitartrate Microcrystals
Particle Size and Zeta Potential Measurement
The average particle size distribution (by volume) of EpiBit was measured by suspending EpiBit raw crystals in isopropyl alcohol and using a laser diffraction technique using the Mastersizer (Malvern Instruments Inc, Westborough, MA), which measures particles within the range of 50 nm–900 μm. The Z-average particle size distribution (by volume) and the average zeta potential of dried EpiBit-MC were measured by suspending the processed (size-reduced) crystals in isopropyl alcohol and using a light scattering technique using the Zetasizer ZS90 (Malvern Instruments Inc, Westborough, MA), which measures particles within the range of 3.8 nm–100 μm and requires only small sample volume. The average of three measurements was reported for each sample.
Fourier Transform Infrared (FT-IR)
EpiBit-MC were tested for stability and absence of isopropyl alcohol using FT-IR spectrometer, spectrum 100 (PerkinElmer, Waltham, MA) scanned from 4,000 to 650 cm−1 after appropriate subtraction of background.
Differential Scanning Calorimetry (DSC)
EpiBit-MC were tested for purity and crystallinity changes using DSC 4000 (PerkinElmer, Waltham, MA), calibrated using an indium standard, and heated from 30°C to 300°C at a rate of 10°C/min with a nitrogen purge of 20 mL/min.
Scanning Electron Microscopy (SEM)
The morphologies of EpiBit and EpiBit-MC were examined using Quanta 200 Environmental scanning electron microscope (FEI, Hillsboro, OR) operated at an accelerating voltage of 20 kV. Fresh EpiBit-MC suspension and dispersion of EpiBit were deposited on an aluminum stub following the evaporation of isopropyl alcohol and sputter coated with gold using Cressington 108 sputter coater (Cressington Scientific Instruments Ltd, Watford, England).
Manufacturing and Quality Control of Rapidly Disintegrating Sublingual Tablets
Five RDST formulations containing EpiBit equivalent to Epi 10, 20, and 40 mg, based on our previous preclinical dose range study (7), and EpiBit-MC equivalent to Epi 10 and 20 mg, based on results obtained during this diffusion study, were manufactured by direct compression. These 150-mg tablets were formulated using microcrystalline cellulose, low-substituted hydroxypropyl cellulose, and magnesium stearate as shown in Table I and previously published (15,16). All excipients were used as supplied and stored under low humidity before mixing. The mixing process was performed in a nitrogen-pre-flushed opaque glass container using three-dimensional manual mixer (Inversina, Bioengineering AG, Wald, Switzerland) for 4 min and an additional 30 s after adding a mixture of the lubricant and one third of the superdisintegrant, which were premixed manually, as a running powder to achieve external positioning in addition to the internal positioning of the superdisintegrant to ensure rapid disintegration and to reduce the negative effect of the lubricant on disintegration. Immediately after mixing, the powder mixture was compressed using a Colton rotary press (Key Industries, Englishtown, NJ) at a pre-selected compression force for each tablet formulation to ensure sufficient hardness for shipping and handling while maintaining rapid tablet disintegration (16).
Table I.
Composition of the RDST Formulations of Epinephrine
| Tablet formulationsa | |||||
|---|---|---|---|---|---|
| Ingredient (mg) | 10 mg Epi | 10 mg Epi-MC | 20 mg Epi | 20 mg Epi-MC | 40 mg Epi |
| Epinephrine bitartrate | 18.2 | 0 | 36.4 | 0 | 72.8 |
| Epinephrine bitartrate (micronized) | 0 | 18.2 | 0 | 36.4 | 0 |
| Microcrystalline cellulose (Ceolus® PH-301) | 115.9 | 115.9 | 99.5 | 99.5 | 66.8 |
| Low-substituted hydroxypropyl cellulose (LH11) | 12.9 | 12.9 | 11.1 | 11.1 | 7.4 |
| Magnesium stearate | 3 | 3 | 3 | 3 | 3 |
aTablet weight was 150 mg
All tablet formulations were tested for tablet weight variation (WV), drug content uniformity (CU), and friability using the harmonized United States Pharmacopeia (USP) methods and criteria (18,19). Tablets containing ≥25 mg and ≥25% API dose require performing the WV test only (18). However, we performed both the WV and CU tests for all tablets for further investigation to confirm the uniformity of the dosage units. The acceptance value (AV) of ≤15 was considered acceptable according to the USP L1 limit.
Drug content was analyzed using a high-performance liquid chromatography (HPLC) system with ultra violet (UV) detection (PerkinElmer, Waltham, MA) as described below. Epi was extracted from the tablet by dissolving the tablet in a solution containing 0.1 N perchloric acid to maintain the stability of Epi in solution according to the USP official monograph of EpiBit (20). The tablet friability was measured using the USP Friability Tester LIC-1 (Vanguard, Spring, TX) with an acceptance value of ≤1% weight loss according to the USP criteria (19).
Tablet dimension was measured using a digital caliper (Harbor Freight Tools, Camarillo, CA). Tablet hardness was measured using the hardness tester LIH-3 (Vanguard, Spring, TX). We discriminated between the disintegration times of RDSTs using previously published method (15,16). A tablet disintegration time of 30–60 s using our previously described method is considered acceptable for the sublingual administration. Six tablets were randomly selected from each formulation for each test and the mean ± standard deviation (SD) were calculated.
In Vitro and Ex Vivo Diffusion Studies
The in vitro and ex vivo diffusion of EpiBit and EpiBit-MC formulated into RDST were evaluated using static vertical jacketed Franz cells with an OD of 20 mm and reservoir volume of 20 ± 1 mL (PermeGear Inc., Hellertown, PA). For in vitro diffusion studies, seven Spectra/Por® dialysis membranes with 1,000 Da MWCO were used as the diffusion membranes. For ex vivo diffusion studies, the center of porcine sublingual mucosa (obtained from the floor of the oral cavity) was used as the diffusion membrane. Heads from recently slaughtered pigs were obtained from a local abattoir. Porcine mucosa was excised using an established surgical technique by dissecting the sublingual mucosa and removing the underlying connective tissue using a scalpel and fine tweezers. The excised mucosa was inspected visually for integrity and then frozen on aluminum foil at −20°C until used within 4 weeks. It was defrosted at room temperature before each experiment.
The diffusion of Epi from RDSTs (n = 4) containing EpiBit equivalent to Epi 10, 20, and 40 mg and EpiBit-MC equivalent to Epi 10, and 20 mg was tested in vitro and ex vivo. EpiBit equivalent to Epi 10 mg was dissolved in 1 mL of the diffusion medium and used as a control (n = 4).
A receptor chamber with a magnetic stirrer was filled with phosphate buffer, pH 5.8 (the average pH of saliva), as the diffusion medium. The water bath was set at 37°C, and water was circulated in the jacketed Franz cells. Before beginning each experiment, air bubbles were removed after mounting the membrane between the donor and receptor chambers. The mounted membranes were equilibrated with the diffusion medium from both sides for 30 min and were checked for leaks.
Each tablet was placed at the center of the donor chamber on the membrane at T0, and 2 mL of the diffusion medium was added to facilitate tablet disintegration and dissolution. Aliquots of 200 μL were withdrawn from the receptor chamber using 6-in. needles (Popper & Sons, Inc, New Hyde Park, NY) and 1 mL syringes at 5, 10, 15, 20, 30, 45, 60, 75, and 90 min. The volumes withdrawn were replenished with fresh medium. Samples were transferred to HPLC vials for HPLC analysis using a UV detector as described below.
Epinephrine HPLC Analysis
Samples from tablets for the CU tests and from the diffusion studies were analyzed for Epi content according to the USP method for Epi injection analysis (21) using an HPLC system with UV detection (PerkinElmer, Waltham, MA). The calibration curve was linear over the range of 6.25 to 200.0 μg/mL with a correlation of coefficient (R2) of >0.99 (n = 5). The coefficient of variation (RSD%) of the system reproducibility at the lowest and highest concentrations, 6.25 and 200 μg/mL, was 1.07% and 0.40% (n = 5 each), respectively. The intra- and inter-assay RSD% at the lowest and highest concentrations were 0.40% and 0.70% (n = 2) and 2.8% and 1.5% (n = 3), respectively.
Data Analysis
The mean (±SD) cumulative diffused Epi per area (μg/cm2) and its percentage for each RDST formulation were calculated. The mean Epi influx, J (μg/cm2/min), was calculated from the slope of each graph (n = 4). Also, Epi permeability, P (cm/min), was calculated by dividing J by Epi concentration in the donor chamber at T0. Data were statistically compared by one-way ANOVA and Tukey-Kramer tests using NCSS statistical software (NCSS, Kaysville, UT). Differences were considered to be statistically significant at p < 0.05.
RESULTS
Particle Size and Zeta Potential Measurement
Mean (±SD) particle size distribution (by volume) of EpiBit crystals before processing was 131.8 ± 10.5 μm (n = 6). The 10th percentile (Dv0.1), median (Dv0.5), and 90th percentile (Dv0.9) were 39.8 ± 3.0, 113.6 ± 9.1, and 254.8 ± 20.1 μm, respectively.
Mean particle size distribution (by volume) and zeta potential of EpiBit crystals after processing using the microfluidizer were 2.5 ± 0.4 μm (pdi 0.185 ± 0.019) and −4.5 ± 1.4 mV, respectively.
FT-IR
The FT-IR spectra showed no evidence of EpiBit degradation after processing (Fig. 2c). Also, the isopropyl alcohol peaks (Fig. 2b) were missing in the FT-IR spectrum of EpiBit after processing (Fig. 2c), confirming successful removal of isopropyl alcohol.
Fig. 2.
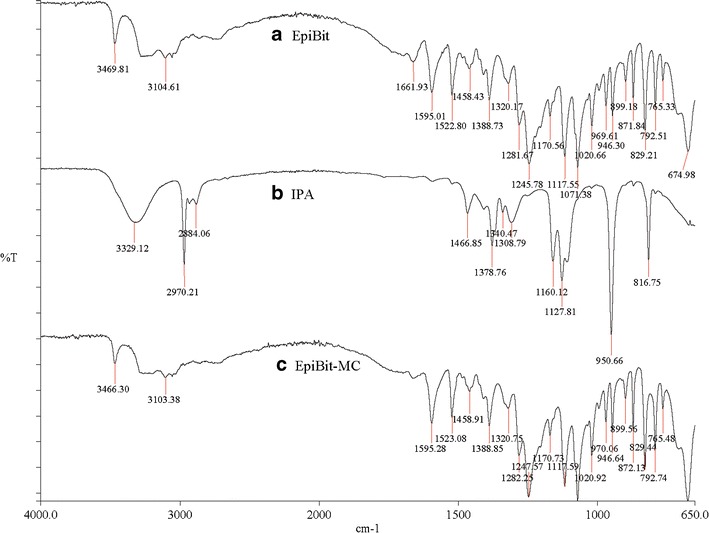
The FT-IR spectra of (a) epinephrine bitartrate (EpiBit) crystals, (b) isopropyl alcohol (IPA), and (c) processed EpiBit (EpiBit-MC) for one pass at 30,000 Psi. The spectra of EpiBit crystals were similar before and after processing and no evidence for IPA in the processed EpiBit crystals
DSC
The DSC spectra of EpiBit before (Fig. 3a) and after processing (Fig. 3b) showed no evidence for EpiBit degradation or changes in crystallinity after processing.
Fig. 3.
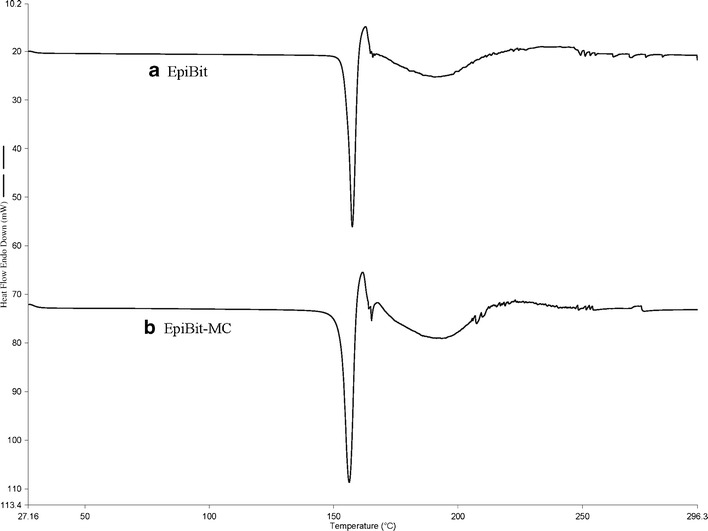
DSC spectra of epinephrine bitartrate (a) before and (b) after processing for one pass at 30,000 Psi. The spectra were similar before and after processing
SEM
The morphology of EpiBit before (Fig. 4a) and after processing (Fig. 4b) showed significant change in the size and shape of the EpiBit crystals. The large, rectangular, pin-like-shaped crystals of EpiBit were changed to much smaller spherical crystals.
Fig. 4.

SEM images of epinephrine bitartrate (a) before and (b) after processing for one pass at 30,000 Psi. There was a significant change in the size and morphology of the crystals
Quality Control of Rapidly Disintegrating Sublingual Tablets
Mean (±SD) hardness, disintegration time, WV, CU, and friability for Epi 10, 20, and 40 mg and Epi-MC 10 and 20 mg RDSTs are shown in Table II. All tablet formulations met USP criteria for WV, drug CU, and friability (18,19) and disintegrated in 30 s or less. The friability test was not performed for Epi-MC tablet formulations because Epi-MC production is a small scale, and the friability test requires a large number of tablets.
Table II.
Mean ± SD (n = 6) Hardness, Disintegration Time, Weight Variation, Content Uniformity, Tablet Diameter, Tablet Thickness, and Friability for RDST Formulations
| Tablet characteristics | |||||||
|---|---|---|---|---|---|---|---|
| Formulations | H | DT | WV (AV) | CU (AV) | D | T | F |
| 10 mg Epi tablets | 1.7 ± 0.3 | 16.3 ± 0.3 | 100.0 ± 0.0 (0.0) | 100.6 ± 4.0 (9.6) | 7.9 ± 0.0 | 3.5 ± 0.0 | 0.4 |
| 20 mg Epi tablets | 1.6 ± 0.1 | 15.8 ± 0.4 | 99.9 ± 0.7 (1.68) | 97.7 ± 2.7 (6.48) | 7.9 ± 0.0 | 3.9 ± 0.0 | 0.5 |
| 40 mg Epi tablets | 1.7 ± 0.2 | 31.3 ± 0.4 | 100.0 ± 0.6 (1.44) | 95.6 ± 2.4 (5.76) | 7.9 ± 0.0 | 3.4 ± 0.0 | 0.6 |
| 10 mg Epi-MC tablets | 2.5 ± 0.0 | 5.5 ± 0.7 | 99.7 ± 1.2 (2.88) | 92.9 ± 0.3 (0.72) | 8.0 ± 0.1 | 3.7 ± 0.0 | NA |
| 20 mg Epi-MC tablets | 2.5 ± 0.1 | 8.7 ± 0.3 | 98.3 ± 1.7 (4.08) | 92.2 ± 4.2 (10.08) | 8.0 ± 0.1 | 3.7 ± 0.0 | NA |
H tablet hardness (kgf), DT disintegration time (values ≤60 s were considered acceptable for sublingual administration), WV weight variation (%), CU content uniformity (%), AV USP acceptance value (acceptance values ≤15.00 were considered acceptable according to the USP L1 limit), D tablet diameter (mm), T tablet thickness (mm), F friability (values ≤1% weight loss were considered acceptable according to the USP limit)
In Vitro and Ex Vivo Diffusion Studies
The mean (±SD) cumulative of diffused Epi in vitro and ex vivo versus time for each RDST formulation is shown in Figs. 5 and 6, respectively. The mean cumulative of diffused Epi and J from Epi-MC 20 mg and Epi 40 mg RDSTs were not significantly different (p > 0.05) from each other but were significantly higher (p < 0.05) than from the rest of the formulations in both in vitro and ex vivo studies (Tables III and IV, respectively). The mean percentage of cumulative Epi diffused and P from Epi-MC 20 mg were significantly higher (p < 0.05) than from the rest of the formulations, including Epi 40 mg RDSTs, in both in vitro and ex vivo studies (Tables III and IV, respectively). The in vitro/ex vivo cumulative Epi diffusion correlation was significant (p < 0.05), and the linear correlation coefficient (R2) ranged from 0.87 to 0.99 for all tested formulations (Fig. 7).
Fig. 5.
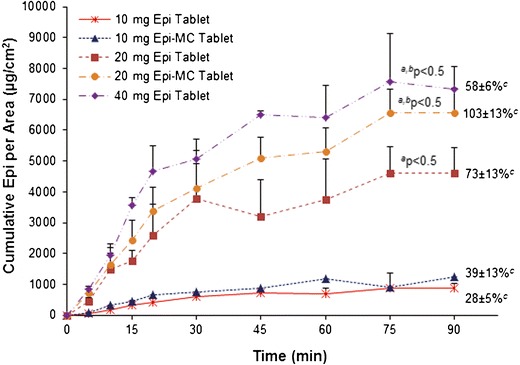
Mean ± SD (n = 4) cumulative of diffused epinephrine per area versus time using dialysis membranes. Superscript a: P < 0.05 compared to 10 mg Epi tablets and Epi-MC tablets; superscript b: P < 0.05 compared to 20 mg Epi tablets; superscript c: cumulative percentage of diffused epinephrine per area at 90 min
Fig. 6.
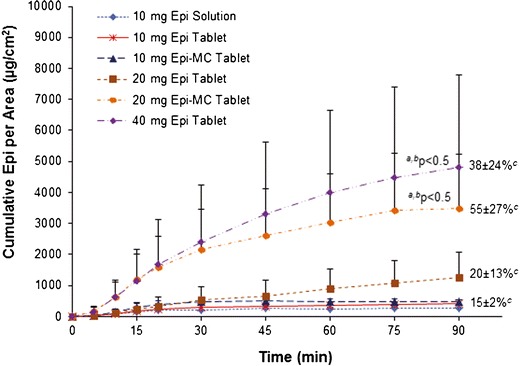
Mean ± SD (n = 4) cumulative of diffused epinephrine per area versus time using excised sublingual mucosal membranes. Superscript a: P < 0.05 compared to 10 mg Epi tablets, Epi-MC tablets, and Epi solution; superscript b: P < 0.05 compared to 20 mg Epi tablets; superscript c: cumulative percentage of diffused epinephrine per area at 90 min
Table III.
Mean ± SD (n = 4) of Cumulative Diffused Epinephrine, J, and P Through Dialysis Membranes (In Vitro) for Each RDST Formulation
| 10 mg Epi tablet | 10 mg Epi-MC tablet | 20 mg Epi tablet | 20 mg Epi-MC tablet | 40 mg Epi tablet | |
|---|---|---|---|---|---|
| Cumulative Epi per area (μg/cm2) | 533 ± 99 | 724 ± 219 | 2,919 ± 584a | 3,982 ± 558a,b | 4,833 ± 308a,b |
| Cumulative Epi (%) | 17 ± 3 | 23 ± 7 | 46 ± 9a | 63 ± 9a,b,c | 38 ± 2a |
| J (μg/cm2/min) | 25 ± 5 | 37 ± 14 | 87 ± 9 | 155 ± 28a,b | 180 ± 41a,b |
| P (cm/min) | 5 ± 1 | 7 ± 3 | 9 ± 1 | 16 ± 3a,b,c | 9 ± 2 |
Cumulative Epi mean cumulative Epi concentration per area over 90 min, J mean Epi influx over 90 min. P mean Epi permeability
a P < 0.05 compared to 10 mg Epi tablets and Epi-MC tablets
b P < 0.05 compared to 20 mg Epi tablets
c P < 0.05 compared to 40 mg Epi tablets
Table IV.
Mean ± SD (n = 4) of Cumulative Diffused Epinephrine, J, and P Through Excised Sublingual Mucosal Membranes (Ex Vivo) for Each RDST Formulation
| 10 mg Epi solution | 10 mg Epi tablet | 10 Mg Epi-Mc tablet | 20 mg Epi tablet | 20 mg Epi-MC tablet | 40 mg Epi tablet | |
|---|---|---|---|---|---|---|
| Cumulative Epi per area (μg/cm2) | 150 ± 81 | 259 ± 66 | 367 ± 80 | 574 ± 424 | 2,042 ± 1138a,b | 2,510 ± 1765a,b |
| Cumulative Epi (%) | 6 ± 2 | 8 ± 2 | 12 ± 3 | 9 ± 7 | 32 ± 18a,b,c | 20 ± 14a,b |
| J (μg/cm2/min) | 12 ± 3 | 17 ± 7 | 25 ± 7 | 20 ± 16 | 112 ± 26a,b | 94 ± 66a,b |
| P (cm/min) | 2.3 ± 0.6 | 3.4 ± 1.3 | 5.0 ± 1.3 | 2.0 ± 1.6 | 11 ± 3a,b,c | 5 ± 3 |
Cumulative Epi mean cumulative Epi concentration per area over 90 min, J mean Epi influx over 90 min, P mean Epi permeability
a P < 0.05 compared to 10 mg Epi tablets, Epi-MC tablets, and Epi solution
b P < 0.05 compared to 20 mg Epi tablets
c P < 0.05 compared to 40 mg Epi tablets
Fig. 7.
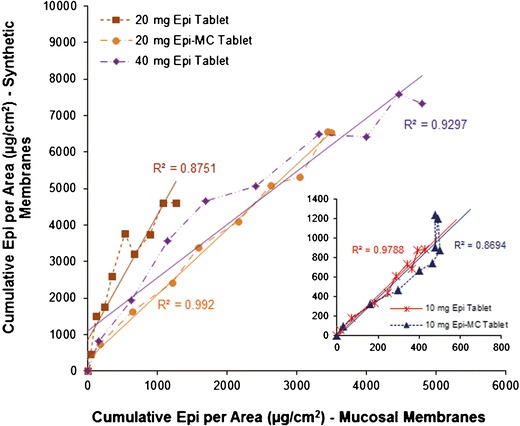
Correlation between the cumulative diffused epinephrine per area through dialysis and excised sublingual mucosal membranes
Diffusion parameters for both Epi 10 mg solution and Epi 10 mg RDST did not differ significantly (p > 0.05) from each other (Table IV).
DISCUSSION
In previous studies in our preclinical model, we confirmed that epinephrine is well-absorbed after sublingual administration using novel RDSTs (5,7,8). In these formulations, an epinephrine dose of 40 mg administered as RDSTs was bioequivalent to an epinephrine dose of 0.3 mg administered by IM injection using an EpiPen®. RDSTs of Epi 40 mg resulted in rapid and high plasma epinephrine concentrations similar to those achieved following the IM injection of Epi 0.3 mg.
For the emergency treatment of anaphylaxis, prompt Epi IM injection administration is required in order to reduce the risk and severity of anaphylactic reactions. The administration of the proposed Epi RDSTs would ultimately require the same recommendations to administer the prescribed dose as soon as the patient identifies the initial signs and symptoms of anaphylaxis.
In order to enhance the sublingual bioavailability of Epi, we reduced the particle size of EpiBit crystals up to 55-fold. Significant reduction in drug particle size increased the saturation solubility and the dissolution rate. This increased the concentration gradient across the diffusional membrane and promoted permeation, which will ultimately increase its bioavailability, leading to a significant reduction in the dose required (6,22). Particle size is extremely important in sublingual drug delivery because of the small saliva volume available for drug dissolution and the short sublingual residence time in comparison to the relatively large volumes of gastrointestinal fluid and long residence times in the gastrointestinal tract.
We aimed to reduce the particle size of EpiBit to the nanosize range (1,000 nm or less). The concentration of the EpiBit suspension, the pressure applied, and the number of cycles were optimized to obtain the smallest particle size range with the lowest possible number of cycles (data not shown). We came close to accomplishing our goal; however, the technical problems of reaching the nanosize range without using any surfactant and processing the EpiBit for only one cycle to reduce potential stress on EpiBit that might affect its stability remain to be solved (23). The zeta potential of the processed particles was low and may not have the potential for particle aggregation problems.
The FT-IR spectra of EpiBit before and after processing were similar, confirming the stability of the EpiBit during the particle size reduction process using LV-1 microfluidizer (Fig. 2a, c). Additionally, after drying the processed EpiBit to obtain the micro-sized EpiBit crystals, there was no evidence in the FT-IR spectrum for any residual isopropyl alcohol, the carrier used to process the EpiBit (Fig. 2b, c), confirming the efficiency of the drying step.
The DSC spectra of EpiBit before and after processing were also similar with a single endothermic peak around 157°C, confirming the absence of any change in the purity and crystallinity of EpiBit (Fig. 3a, b).
All RSDTs were tested for quality control and had almost the same hardness, 1.5–2.5 kgf, and disintegrated within 30 s (Table II). Despite the rapid tablet disintegration, these RDST were hard enough to pass the USP friability test. The USP friability test requires tablets that are equivalent to 6.5 g in weight (19), which is about 44 RDSTs. Therefore, the friability test for RDSTs containing Epi-MC was not performed because we were limited with the number of tablets available using the small-scale Microfluidics-LV1 machine to produce limited amounts of Epi-MC. However, RDSTs containing the same dose of raw Epi have already passed the friability test at even slightly lower hardness range, 1.5–2.0 kgf (Table II).
The initial diffusion studies were conducted using dialysis membranes. Later, they were performed using excised porcine sublingual mucosa. In previous studies, sublingual mucosa of pigs and rabbits appeared to be similar to human sublingual mucosa (24,25). This was the rationale for our decision to use porcine mucosa in the ex vivo diffusion study reported here and our decision to use rabbits in our previous in vivo studies (5–8). Porcine sublingual mucosa has a large surface area and is easy to excise surgically for ex vivo studies, and rabbits are easy to house and handle for in vivo studies.
Results from both in vitro and ex vivo experiments were highly correlated (R2 ≥ 87; Fig. 7) and demonstrated that the cumulative percentage of Epi diffused from Epi-MC 20 mg RDST was significantly higher than that of the other formulations including Epi 40 mg RDST (Tables III and IV). These results were further confirmed by calculating Epi permeability using individual data from each RDST formulation. The analysis of the data showed that the mean permeability of Epi from Epi-MC 20 mg RDST was also significantly higher than that of the other formulations including Epi 40 mg RDST (Tables III and IV). This resulted in similar cumulative Epi diffusion and influx from both Epi-MC 20 mg RDST and Epi 40 mg RDST (Tables III and IV). Also, formulating EpiBit into our RDST formulation in comparison to Epi solution formulation did not decrease EpiBit diffusion, as shown in Table IV, further confirming complete and rapid tablet disintegration and Epi release (Table II).
Although that the cumulative amounts of Epi diffused and Epi influx in vitro and ex vivo (Tables III and IV) from Epi-MC 10 mg RDST were higher than those from the Epi 10 mg RDST, they were not significantly different, mainly because the Epi dose available was insufficient to yield the required concentration gradient across the membrane to result in significant drug permeability and ultimately absorption. Similar results were observed in our previous in vivo studies following the sublingual administration of an Epi 10-mg dose from RDSTs (5,7,8). An Epi 10-mg dose was insufficient to result in significant sublingual absorption when compared to placebo.
The significant reduction of the particle size of EpiBit increased its permeability twofold for an Epi 20-mg dose, confirming the potential for these micro-sized Epi RDSTs to reduce the Epi sublingual dose by 50%. In a preclinical pilot study, the plasma concentrations of these micro-sized Epi particles from the RDSTs were similar to those from Epi 40 mg RDSTs and Epi 0.3 mg IM injection (6).
CONCLUSION
In this study, we were able to demonstrate that reducing the particle size of EpiBit almost to the nanosize range improved its diffusion and permeability from RDST by twofold. These micro-sized Epi RDST tablets have the potential to reduce the bioequivalent dose of sublingually administered Epi by 50%. Preclinical and clinical studies will be further pursued to evaluate the in vivo absorption and bioavailability of Epi from these tablets.
ACKNOWLEDGMENTS
Dr. Rawas-Qalaji would like to acknowledge the financial support received from the President’s Faculty Research & Development Grant (PFRDG) at Nova Southeastern University. No financial or in-kind support for this study was provided by any corporate sponsor.
Conflict of Interest
Mutasem Rawas-Qalaji reports no declarations of interest.
Shima Werdy reports no declarations of interest.
Ousama Rachid reports no declarations of interest.
F. Estelle R. Simons reports no declarations of interest.
Keith J. Simons reports no declarations of interest.
REFERENCES
- 1.Kemp SF, Lockey RF, Simons FE. Epinephrine: the drug of choice for anaphylaxis. A statement of the World Allergy Organization. Allergy. 2008;63(8):1061–70. doi: 10.1111/j.1398-9995.2008.01733.x. [DOI] [PubMed] [Google Scholar]
- 2.Simons FER, Sheikh A. Anaphylaxis: the acute episode and beyond. BMJ. 2013;346:f602. doi: 10.1136/bmj.f602. [DOI] [PubMed] [Google Scholar]
- 3.Simons KJ, Simons FE. Epinephrine and its use in anaphylaxis: current issues. Curr Opin Allergy Clin Immunol. 2010;10(4):354–61. doi: 10.1097/ACI.0b013e32833bc670. [DOI] [PubMed] [Google Scholar]
- 4.Soar J, Pumphrey R, Cant A, Clarke S, Corbett A, Dawson P, et al. Emergency treatment of anaphylactic reactions—guidelines for healthcare providers. Resuscitation. 2008;77(2):157–69. doi: 10.1016/j.resuscitation.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 5.Rachid O, Rawas-Qalaji MM, Simons FE, Simons KJ. Epinephrine (adrenaline) absorption from new-generation, taste-masked sublingual tablets: a preclinical study. J Allergy Clin Immunol. 2013;131(1):236–8. doi: 10.1016/j.jaci.2012.10.016. [DOI] [PubMed] [Google Scholar]
- 6.Rawas-Qalaji M, Rachid O, Mendez BA, Losada A, Simons FE, Simons KJ. Adrenaline (epinephrine) microcrystal sublingual tablet formulation: enhanced absorption in a preclinical model. J Pharm Pharmacol. 2015;67(1):20–5. doi: 10.1111/jphp.12312. [DOI] [PubMed] [Google Scholar]
- 7.Rawas-Qalaji MM, Simons FE, Simons KJ. Sublingual epinephrine tablets versus intramuscular injection of epinephrine: dose equivalence for potential treatment of anaphylaxis. J Allergy Clin Immunol. 2006;117(2):398–403. doi: 10.1016/j.jaci.2005.12.1310. [DOI] [PubMed] [Google Scholar]
- 8.Rawas-Qalaji MM, Simons FE, Simons KJ. Epinephrine for the treatment of anaphylaxis: do all 40 mg sublingual epinephrine tablet formulations with similar in vitro characteristics have the same bioavailability? Biopharm Drug Dispos. 2006;27(9):427–35. doi: 10.1002/bdd.519. [DOI] [PubMed] [Google Scholar]
- 9.Rawas-Qalaji MM, Rachid O, Simons FE, Simons KJ. Long-term stability of epinephrine sublingual tablets for the potential first-aid treatment of anaphylaxis. Ann Allergy Asthma Immunol. 2013;111(6):568–70. doi: 10.1016/j.anai.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 10.Rolan P, Lim S, Sunderland V, Liu Y, Molnar V. The absolute bioavailability of racemic ketamine from a novel sublingual formulation. Br J Clin Pharmacol. 2014;77(6):1011–6. doi: 10.1111/bcp.12264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoffman BB, Taylor P. Neurotransmission: the autonomic and somatic motor nervous systems. In: Hardman JG, Limbird LE, Gilman AG, editors. Goodman & Gilman’s the pharmacological basis of therapeutics. 9. New York: McGraw-Hill Companies, Inc; 2001. pp. 115–53. [Google Scholar]
- 12.Rachid O, Rawas-Qalaji M, Simons FER, Simons KJ. Dissolution testing of sublingual tablets: a novel in vitro method. AAPS PharmSciTech. 2011;12(2):544–52. doi: 10.1208/s12249-011-9615-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rachid O, Rawas-Qalaji M, Simons FER, Simons KJ. Rapidly-disintegrating sublingual tablets of epinephrine: role of non-medicinal ingredients in formulation development. Eur J Pharm Biopharm. 2012;82(3):598–604. doi: 10.1016/j.ejpb.2012.05.020. [DOI] [PubMed] [Google Scholar]
- 14.Rachid O, Simons FE, Rawas-Qalaji M, Simons KJ. An electronic tongue: evaluation of the masking efficacy of sweetening and/or flavoring agents on the bitter taste of epinephrine. AAPS PharmSciTech. 2010;11(2):550–7. doi: 10.1208/s12249-010-9402-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rawas-Qalaji MM, Simons FE, Simons KJ. Fast-disintegrating sublingual epinephrine tablets: effect of tablet dimensions on tablet characteristics. Drug Dev Ind Pharm. 2007;33(5):523–30. doi: 10.1080/03639040600897150. [DOI] [PubMed] [Google Scholar]
- 16.Rawas-Qalaji MM, Simons FER, Simons KJ. Fast-disintegrating sublingual tablets: effect of epinephrine load on tablet characteristics. AAPS PharmSciTech. 2006;7(2):E72–8. doi: 10.1208/pt070241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muller RH, Gohla S, Keck CM. State of the art of nanocrystals—special features, production, nanotoxicology aspects and intracellular delivery. Eur J Pharm Biopharm. 2011;78(1):1–9. doi: 10.1016/j.ejpb.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 18.USP/NF . Physical tests: <905> uniformity of dosage units. United States Pharmacopeia 3. 37/32. Rockville: Pharmacopeial Convention, Inc; 2014. pp. 491–4. [Google Scholar]
- 19.USP/NF . General chapters: <1216> tablet friability. United States Pharmacopeia 6. 37/32. Rockville: Pharmacopeial Convention, Inc; 2014. pp. 1145–6. [Google Scholar]
- 20.USP/NF . Official monograph: epinephrine bitartrate. 31/26. Rockville: United States Pharmacopeial Convention, Inc; 2008. [Google Scholar]
- 21.USP/NF. Official monograph: epinephrine injection. 31/26 ed. Rockville: United States Pharmacopeial Convention, Inc; 2008.
- 22.Liu Y, Sun C, Hao Y, Jiang T, Zheng L, Wang S. Mechanism of dissolution enhancement and bioavailability of poorly water soluble celecoxib by preparing stable amorphous nanoparticles. J Pharm Pharm Sci. 2010;13(4):589–606. doi: 10.18433/j3530j. [DOI] [PubMed] [Google Scholar]
- 23.Ma Q, Sun H, Che E, Zheng X, Jiang T, Sun C, et al. Uniform nano-sized valsartan for dissolution and bioavailability enhancement: influence of particle size and crystalline state. Int J Pharm. 2013;441(1–2):75–81. doi: 10.1016/j.ijpharm.2012.12.025. [DOI] [PubMed] [Google Scholar]
- 24.Dali MM, Moench PA, Mathias NR, Stetsko PI, Heran CL, Smith RL. A rabbit model for sublingual drug delivery: comparison with human pharmacokinetic studies of propranolol, verapamil and captopril. J Pharm Sci. 2006;95(1):37–44. doi: 10.1002/jps.20312. [DOI] [PubMed] [Google Scholar]
- 25.Ong CM, Heard CM. Permeation of quinine across sublingual mucosa, in vitro. Int J Pharm. 2009;366(1–2):58–64. doi: 10.1016/j.ijpharm.2008.08.048. [DOI] [PubMed] [Google Scholar]