

Abstract
AIM: To investigate the expression of HuR in pancreatic ductal adenocarcinoma (PDA) and to assess the effects of HuR silencing on the expression of cyclooxygenase-2 (COX-2) and heme oxygenase-1 (HO-1) and the in vitro response to gemcitabine (GEM) treatment in pancreatic cell lines.
METHODS: We compared the expression of HuR, COX-2, and HO-1 in PDA and normal pancreatic tissue using quantitative reverse transcription polymerase chain reaction (qRT-PCR) and western blot. In addition, the HuR, COX-2 and HO-1 were analyzed in four types of cancer cell lines (MiaPaca2, Su.86.86, Capan-1, and Capan-2) with and without GEM treatment. Immunocytofluorescence analysis was used to investigate HuR localization in cells. Cell viability and response to GEM after HuR silencing were determined with the 3-(4,5-dimethylthiazol-2-Yl)-2,5-diphenyltetrazolium bromide test and the crystal violet clonogenic assay, respectively. To measure apoptosis, activation of caspases 3/7 was evaluated using immunofluorescence.
RESULTS: In PDA tissue obtained from patients not treated with GEM, HuR mRNA expression was 3.2 times lower (P < 0.05) and COX-2 and HO-1 mRNA expression was 2.3-fold and 7.2-fold higher (P < 0.05), respectively, than normal pancreatic tissue (from organ donor). qRT-PCR analysis showed that HuR, COX-2, and HO-1 mRNA were overexpressed in all cancer cell lines treated with the half maximal inhibitory concentration (IC50) dose of GEM compared with control cells (P < 0.05). Western blot analysis revealed that COX-2 and HO-1 levels were significantly decreased in cancer cells after HuR silencing. Furthermore, HuR silencing increased the response to GEM treatment and decreased cell viability by 11.6%-53.7% compared to control cell lines. Caspases 3 and 7 were activated after HuR silencing and GEM treatment in all pancreatic cancer cell lines. In comparison, treatment with GEM alone did not activate caspases 3 and 7 in the same cell lines.
CONCLUSION: HuR mediated post-transcriptional upregulation of COX-2 and HO-1 expression after GEM treatment in pancreatic cancer cells. HuR silencing significantly increased the effectiveness of GEM treatment in vitro.
Keywords: HuR, Pancreatic cancer, Chemotherapy, Cyclooxygenase-2, Post-transcriptional regulation, Heme oxygenase-1
Core tip: The mRNA binding protein HuR is normally located in the nucleus and is activated by specific stressors. HuR has been reported to regulate the expression of many proteins by different post-transcriptional mechanisms, including mRNA trafficking, mRNA stabilization, and protein translation. HuR is also involved in the post-transcriptional modification of cytoprotective molecules, such as cyclooxygenase-2 and heme oxygenase-1, which may be related to the increased resistance of pancreatic cancer to chemotherapy. HuR silencing significantly increases the effectiveness of gemcitabine treatment in vitro.
INTRODUCTION
Pancreatic cancer has become the fourth leading cause of cancer related deaths worldwide[1]. The incidence of pancreatic cancer is increasing, and it threatens to become the third leading cause of cancer related deaths in Europe, if this trend persists[2]. Pancreatic cancer has a very poor prognosis, and, at present, surgical resection is the only curative treatment. Unfortunately, most patients are diagnosed at the stage of metastatic illness, and only 15%-20% of pancreatic cancer patients are eligible for curative surgery. However, even after radical surgery, many relapse locoregionally and/or with distant metastases.
The lethality of pancreatic cancer is undermined by rapid invasion of the surrounding tissue, early metastatic disease, and poor response to standard chemotherapy and radiotherapy[3]. Currently, surgical resection and chemotherapy regimens, that include gemcitabine (GEM) (2′,2′-difluorodeoxycytidine), provide the best clinical benefits. In fact, for more than 10 years, GEM has been the reference drug for the treatment of this often fatal disease[4]. A number of new anti-cancer drugs were introduced in the last decade with the hope of improving the survival of pancreatic cancer patients. Despite tenacious efforts to develop new agents, none of the currently available chemotherapeutic agents have an objective response rate higher than 10%[5]. The unchanged outcomes of pancreatic cancer treatment over the decades mandate further research to develop novel therapeutic agents.
Alterations in enzymatic activity and post-transcriptional regulation in tumor cells might be responsible for the exceptional resistance of pancreatic cancer to conventional treatment. Normally, post-transcriptional regulation plays a critical role in the process of cell proliferation and apoptosis, but in cancer cells, this control might be impaired by changes in the expression of RNA binding proteins. HuR (ELAV1) is an RNA binding protein that has been shown to regulate the expression of multiple genes by different post-transcriptional mechanisms, including messenger RNA (mRNA) trafficking, mRNA decay, and protein translation[6]. The expression of HuR and other RNA binding proteins has been shown to be altered in several pathological conditions, including cancer. Increasing evidence support HuR as the first RNA-binding protein shown to play a critical role in both carcinogenesis and cancer progression by functioning as either an oncogene or a tumor suppressor that regulates the expression of various target genes[6]. Many HuR targeted mRNAs encode stress-response, immune-response, cell cycle regulatory proteins, oncogenes, and tumor suppressor genes, including such therapeutic targets as cyclooxygenase-2 (COX-2) and heme oxygenase-1 (HO-1)[7,8]. HuR modulates these transcripts in response to stimuli, such as therapeutic agents (i.e., tamoxifen and prostaglandin), nutrient depletion (polyamines, amino acid starvation), heat shock, immune stimuli, short-wavelength ultraviolet (UV) irradiation, oxidants, and transcriptional inhibitors (actinomycin D)[9-11].
COX-2 (also known as prostaglandin endoperoxide synthase) is a key enzyme in the biochemical pathway leading to the synthesis of prostaglandins. In pancreatic tumors COX-2 is commonly overexpressed, and preclinical evidence indicates that selective COX-2 inhibition enhances both chemotherapy and radiotherapy response, without damaging normal tissue[12].
HO-1 is believed to be the key enzyme involved in protecting cells against stress. Its overexpression in different types of human cancers supports the notion that HO-1 provides a growth advantage and contributes to cellular resistance against chemotherapy and radiotherapy[13]. A study by Berberat et al[13] showed that in human pancreatic cancer there was a 6-fold and 3.5-fold upregulation of HO-1 mRNA and protein levels, respectively, when compared to normal pancreatic tissue. Treatment of the pancreatic cancer cell lines with GEM or radiation further induced HO-1 expression. In contrast, a targeted knockdown of HO-1 expression led to pronounced growth inhibition of pancreatic cancer cells and made tumor cells significantly more sensitive to radiotherapy and chemotherapy[13].
The role of HuR in regulating the cytoprotective effects of COX-2 and HO-1 in pancreatic ductal adenocarcinoma (PDA) is not clear. Previous studies have demonstrated that HuR expression in PDA specimens after chemotherapy treatments can be both cytoplasmic and nuclear[14-16]. The aim of our study was to investigate the expression of the mRNA binding protein HuR in PDA. In addition, we assessed the effects of HuR silencing on the expression of COX-2 and HO-1 and its role in chemoresistance of pancreatic cancer.
MATERIALS AND METHODS
Human pancreatic cancer tissues collection
Pancreatic carcinoma tissues were obtained from 20 patients undergoing a partial duodenopancreatectomy (Whipple resection) for pancreatic carcinoma. Normal pancreatic tissue samples were obtained, through an organ donor program, from six individuals who were free of pancreatic disease. All normal tissue samples were obtained from the head of the pancreas to ensure comparability with the tumor samples. In all experiments, tissue sections of normal and cancerous pancreas samples were processed simultaneously to ensure comparability of the results. Freshly removed tissue samples were placed in RNALater (Ambion; Huntingdon, United Kingdom), whereas tissues for protein extraction were snap frozen in liquid nitrogen in the operating room upon surgical removal and maintained at -80 °C until use. Ethical approval was issued by the Ethics Committee of the Lithuanian University of Health Sciences (Nr. BE-2-10 and BE-2-17). Consent for the use of routine blood samples, biopsies, and surgical tissue specimens for research purposes was obtained from all the patients or their representatives.
Cell lines and growing conditions
Human pancreatic cancer cell lines Capan-1, Capan-2, MiaPaca2, and Su.86.86 were used for analysis. Cells were grown in monolayers in sterile 25 cm2 capacity flasks with 5 mL RPMI medium (Gibco/Invitrogen, Carlsbad, CA, United States). Flasks with cells were cultured in an incubator and maintained at a moist temperature of 37 °C, 5% CO2 enriched environment. Capan-1, MiaPaca2, and Su.86.86 cells were grown in RPMI medium (Gibco/Invitrogen) with 10% fetal bovine serum (FBS) (Gibco/Invitrogen) and 1% penicillin/streptomycin solution (Gibco/Invitrogen). Capan-2 cells were grown in RPMI medium with 20% FBS and 1% penicillin/streptomycin solution.
GEM treatment of cells and IC50 measures
Cells were seeded in 96-well cell culture plates (3 × 103 cells/well) and were treated with GEM (Eli Lilly, Indianapolis, IN, United States) to each well at separate concentrations of 0, 1, 5, 10, 50, 100, 500, 1000, 2000, and 5000 ng/mL to test the half maximal inhibitory concentration (IC50) of GEM. Treated cells were maintained at 37 °C in 5% CO2 for 72 h. To measure the proportion of living cells, the 3-(4,5-dimethylthiazol-2-Yl)-2,5-diphenyltetrazolium bromide (MTT) method was used (see “MTT viability test”).
The IC50 was 50 ng/mL for MiaPaca2, 23 ng/mL for Su.86.86, and 5 μg/mL for Capan-1. Capan-2 failed the IC50 measurement because of stable viability at the highest GEM concentration. Therefore, we used IC40, which was 5 μg/mL GEM, for experiments in the Capan-2 cell line.
MTT viability test
Cell viability was assessed by MTT (Invitrogen). The culture medium was removed, and the cells were incubated with MTT (5 mg/mL) for 4 h at 37 °C. After 4 h incubation, the MTT solution was removed, and the formazan product was extracted and diluted with DMSO (Carl Roth GmbH, Karlsruhe, Germany) by gentle agitation for 15 min. The absorbance was measured with a Sunrise spectrophotometer (Tecan, Austria) at a wavelength of 550 nm and reference 620 nm.
Transfection
HuR (sense sequence 5’-GCGUUAUCCGGUUUGACATT-3’; antisense sequence 5’-UGUCAAACCGGAUAAACGCAA-3’) and negative control small interfering RNAs (siRNA) were purchased from Ambion (Foster City, CA, United States). Transfection was performed when cell cultures had reached 70%-80% confluence in 96 or 6-well plates or 25 cm2 flasks. Lipofectamine 2000 (Gibco/Invitrogen) was used according to the manufacturer’s instructions for all transfections with Optimem medium (Gibco/Invitrogen). Some experiments included two groups of control cells - untreated control and a control treated with a siRNA negative control. Transfection efficiency was assessed using Block-iT alexa fluor red reagents (Invitrogen). Silencing efficiency was evaluated by western blot analysis (Figure 1). Transfection of HuR siRNA were performed 24 h before the GEM treatment. All assays were performed 72 h after GEM treatment.
Figure 1.
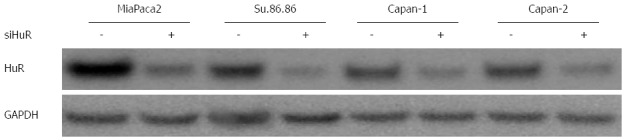
siHuR silencing efficiency in pancreatic cancer cells. siHuR silencing efficiency was evaluated by western blot analysis 72 h after transfection. HuR: Human antigen R.
RNA extraction and real-time polymerase chain reaction
Total RNA extraction was performed from tissues and cultured cells using PureLink RNA easy kit (Ambion) and TRI reagents (Zymo, Irvine, CA, United States), according to the manufacturer’s protocol. Purified RNA was quantified and assessed for purity by UV spectrophotometry (NanoDrop, Wilmington, DE, United States). Complementary DNA (cDNA) was generated from 1 μg of RNA with Super Script Vilo Master Mix (Invitrogen). The amplification of specific RNA was performed in a 20 μL reaction mixture containing 2 μL of cDNA template, 1 × PCR master mix, and the primers. The PCR primers used for detection of HuR, HO-1, and Cox-2 were from Invitrogen: COX-2: Forward, GAATCATTCACCAGGCAAATTG; Reverse, TCTGTACTGCGGGTGGAACA; HuR: Forward, GTGAACTACGTGACCGCGAA; Reverse, GACTGGAGCCTCAAGCCG; HO-1: Forward, TGCTCAACATCCAGCTCTTTGAGGA; Reverse, CAGGCAGAGAATGCTGAGTTC. The PCR products were loaded onto 1.5% agarose gels and visualized with ethidium bromide under UV light.
Western blot analysis
Whole cells were lysed using RIPA lysis buffer with protease inhibitors (Roche, Basel, Switzerland) and centrifuged at 10000 g for 10 min. The supernatants were assayed for protein concentration with bicinchoninic acid (BCA) protein assay kit (Thermo Scientific, Boston, MA, United States). Protein samples were heated at 97 °C for 5 min, and 50 μg of the samples were subjected to 4%-12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to poly-vinylidene fluoride (PVDF) membranes at 30 V for 50 min. Next, membranes were blocked with blocking buffer (Invitrogen) for 30 min at room temperature. Membranes were then incubated overnight at 4 °C with primary antibodies. The following primary antibodies were used: 1:1000 mouse monoclonal anti-HuR (Invitrogen), rabbit monoclonal anti-Cox-2 (Abcam, Cambridge, MA, United States), rabbit monoclonal anti-HO-1 (Abcam) and mouse monoclonal anti β-actin (Ambion). The membranes were washed with antibody washing buffer (Invitrogen) and incubated in the appropriate peroxidase-conjugated secondary antibody solution (Invitrogen) for 30 min. Subsequently, membranes were washed again with antibody washing buffer (Invitrogen) and incubated with chemiluminescence substrate (Invitrogen). Results were analyzed with a UVP documenting system (UVP, Upland, Canada).
Immunofluorescence
Cells were cultivated on chamber slides for 72 h with or without treatment. A mixture of 96% ethanol with 5% glacial acetic acid was used for fixation and 0.5% Triton X-100 for permeabilization. Cells were subsequently incubated with the primary mouse monoclonal anti-HuR antibody (Invitrogen) and secondary Alexa Fluor 488 goat anti-mouse immunoglobulin (IgG) (H + L) antibody and processed. Cell nuclei were stained with DAPI (Life Technologies, Carlsbad, CA, United States) and chamber slides were mounted for analysis with Olympus IX71 fluorescent confocal microscope (Olympus Corporation, Tokyo, Japan).
For caspases 3 and 7 activation analysis, CellEvent™ Caspase-3/7 Green ReadyProbes Reagent (Life Technologies) was used. Cells were prepared according to the manufacturer’s instructions and analyzed with Olympus IX71 fluorescent confocal microscope.
Crystal violet staining
The colony formation of pancreatic cancer cells was evaluated using a crystal violet (CV) stain assay. The cells were cultivated in 24-well culture plates, and after 20 min, the stain was removed, and the wells were rinsed in water. Plates were dried at room temperature, and morphology of cells was observed under an Olympus IX71 phase-contrast. Stains from cells were diluted in 0.5 mL of 50% ethanol diluent for 30 min, and absorption was measured at 550 nm for quantitative CV analysis.
Statistical analysis
Statistical analysis was performed using SPSS 18.0 software (SPSS Company, Chicago, IL, United States). The data are presented as mean ± SE or median and range. As the hypothesis of “normal distribution of data” was rejected by the Shapiro-Wilkstest, nonparametric statistical tests were used. The Mann-Whitney test was used for comparison of mRNA and protein expression levels between groups. Statistical significance was defined as P < 0.05
RESULTS
HuR, HO-1, and COX-2 expression in human PDA tissue
RT-PCR analysis (Figure 2) revealed that HuR mRNA expression was 3.2-fold lower (P < 0.05) in PDA than normal pancreatic tissue. The expression of COX-2 and HO-1 mRNA levels in pancreatic cancer tissue were 2.3 and 6.9-fold higher (P < 0.05 for both), respectively, compared to normal tissue.
Figure 2.
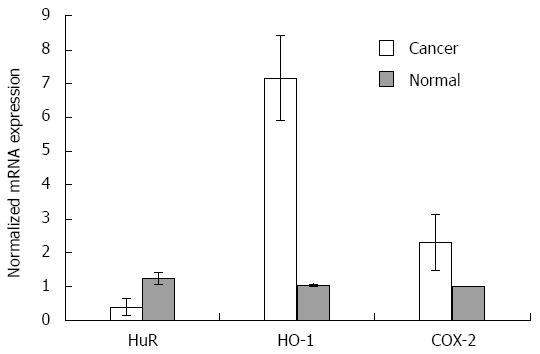
HuR, HO-1 and COX-2 mRNA expression analysis in tissues. Quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis revealed normalized HuR, HO-1, and COX-2 mRNA expression in normal tissues (n = 6) compared to pancreatic cancer tissues (n = 20) with standard deviation (P < 0.05). For normalization, β-actin housekeeping gene and ΔΔCt method was used. HO-1: Heme oxygenase-1; COX-2: Cyclooxygenase-2; HuR: Human antigen R.
Similarly, the western blot analysis (Figure 3) showed that the protein level of HuR was 1.8-fold lower in pancreatic cancer tissues compared to normal tissue (P < 0.05), while the expression of HO-1 and COX-2 proteins were, accordingly, 10.8 and 3.2-fold higher in the pancreatic cancer tissue (Figure 4).
Figure 3.
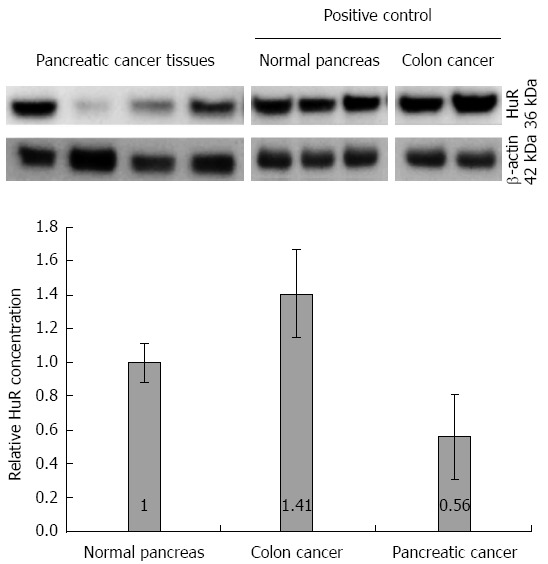
Human antigen R protein expression analysis in tissues. Quantitative western blot analysis revealed 1.8-fold higher expression of HuR in normal pancreas tissues compared to pancreatic cancer tissues. Colon cancer tissue was used as a positive control. HuR: Human antigen R.
Figure 4.
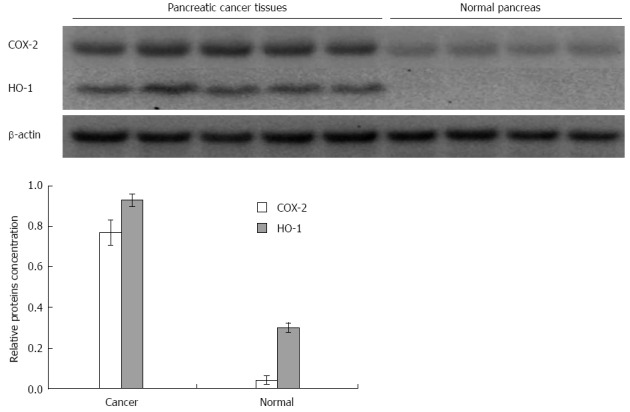
COX-2 and HO-1 protein expression analysis in tissues. Expression of COX-2 and HO-1 proteins was, accordingly, 3.2 and 10.8-fold higher in pancreatic cancer tissue compared to normal pancreatic tissue. HO-1: Heme oxygenase-1; COX-2: Cyclooxygenase-2.
Treatment with GEM induces overexpression of HuR, HO-1, and COX-2 mRNA and protein in human pancreatic cancer cells lines
RT-PCR analysis showed that HuR, COX-2, and HO-1 mRNA were overexpressed in Capan-1, Capan-2, MiaPaca2, and Su.86.86 cell lines treated with the IC50 dose of GEM compared with control cells (P < 0.05) (Figure 5). HuR mRNA expression was 1.1-1.7 fold higher in cells treated with the IC50 dose of GEM compared to untreated cell lines (P < 0.05). COX-2 mRNA overexpression was seen in all cell lines and was 2.1-6.1 fold higher in cells treated with the IC50 dose of GEM (P < 0.05). Only in MiaPaca2 control cells was COX-2 expression not observed in some of the experiments. HO-1 mRNA expression was 1.1-4.3 fold higher in cells treated with IC50 dose of GEM compared to control (P < 0.05).
Figure 5.
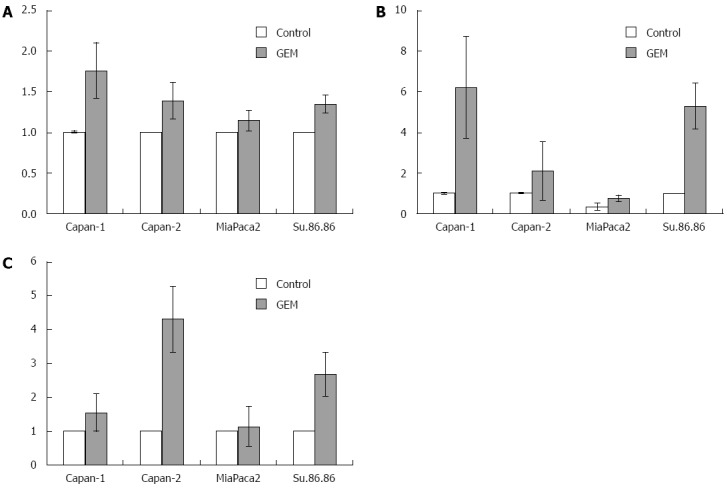
HuR, COX-2, and HO-1 mRNA expression in pancreatic cancer cells. Quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis revealed higher normalized HuR (A), COX-2 (B), and HO-1 (C) mRNA values in GEM treated cells compared to control cells (P < 0.05). Data from three independent experiments are shown. For normalization β-actin housekeeping gene and ΔΔCt method was used. HO-1: Heme oxygenase-1; COX-2: Cyclooxygenase-2; HuR: Human antigen R.
HuR silencing sensitizes cancer cells to GEM treatment by inducing apoptosis
To demonstrate that HuR silencing has an effect on COX-2 and HO-1 expression and sensitivity to chemotherapy, pancreatic cancer cell lines were transfected with either anti-HuR siRNA or control siRNA. HuR, COX-2, and HO-1 proteins were abundantly expressed in all cell lines after GEM treatment. Western blot analysis revealed that expression of these proteins was decreased in Capan-1, Capan-2, MiaPaca2, and Su.86.86 cell lines transfected with anti-HuR siRNA and treated with GEM compared to cells treated with GEM alone (Figure 6).
Figure 6.
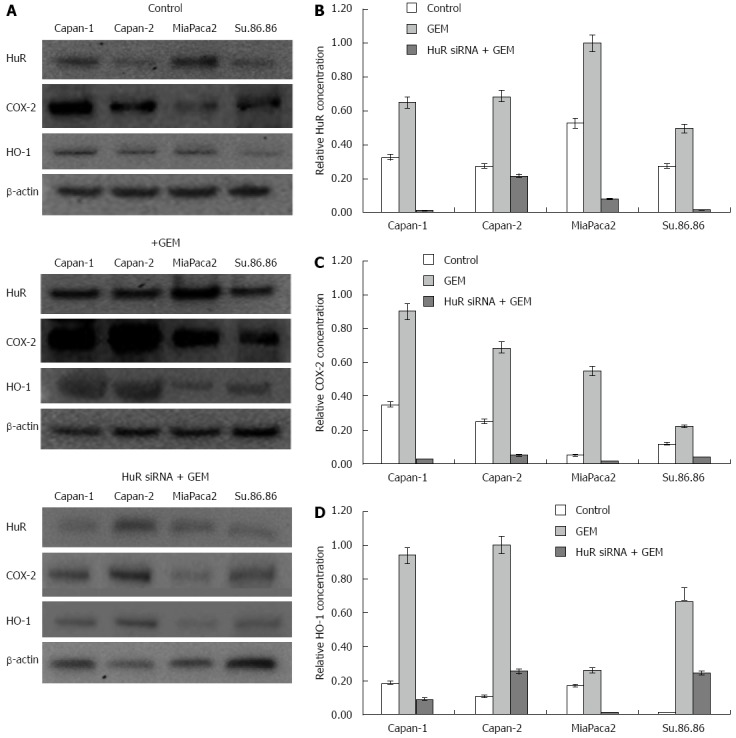
HuR, HO-1, and COX-2 protein expression analysis in pancreatic cancer cells. The western blot analysis revealed differences in protein level between control cells and cells treated with gemcitabine (GEM) and cells treated with GEM after HuR silencing (A). The quantitative Western blot analysis revealed the relative concentration of COX-2 (B), HuR (C), and HO-1 (D) in each of the experimental groups. HO-1: Heme oxygenase-1; COX-2: Cyclooxygenase-2; HuR: Human antigen R.
Treatment with GEM IC50 dose decreased the viability of all the studied pancreatic cancer cell lines. Moreover, HuR siRNA transfection further decreased cell viability in all cancer cell lines: MiaPaca2 - 53.7%, Su.86.86 - 49.4%, Capan-1 - 11.6%, and Capan-2 - 15% (Figure 7). In contrast, transfection with control siRNA did not affect cell viability or the response to GEM treatment. Although HuR silencing sensitized pancreatic cancer cells to GEM, HuR siRNA alone did not affect cell viability. CV assay also confirmed that cell viability and adhesion significantly decreased in cells transfected with anti-HuR siRNA and treated with GEM when compared to cells that were treated with GEM alone: MiaPaca2 - 44.2%, Su.86.86 - 62.9%, Capan-1 - 19.6%, and Capan-2 - 25.9% (Figure 8).
Figure 7.
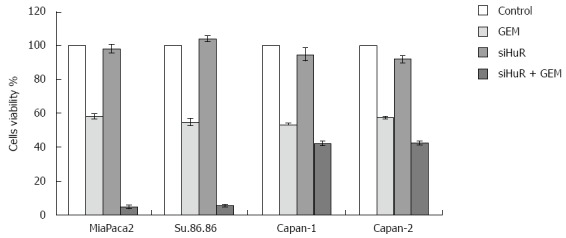
Cell viability analysis (MTT). Viability was analyzed in control cells, cells treated with gemcitabine (GEM) IC50, siHUR alone, and GEM IC50 treatment after HuR siRNA. Transfection with HuR siRNA had little or no effect on cell viability, while HuR silencing dramatically increased response of pancreatic cancer cells to the GEM treatment. HuR: Human antigen R.
Figure 8.
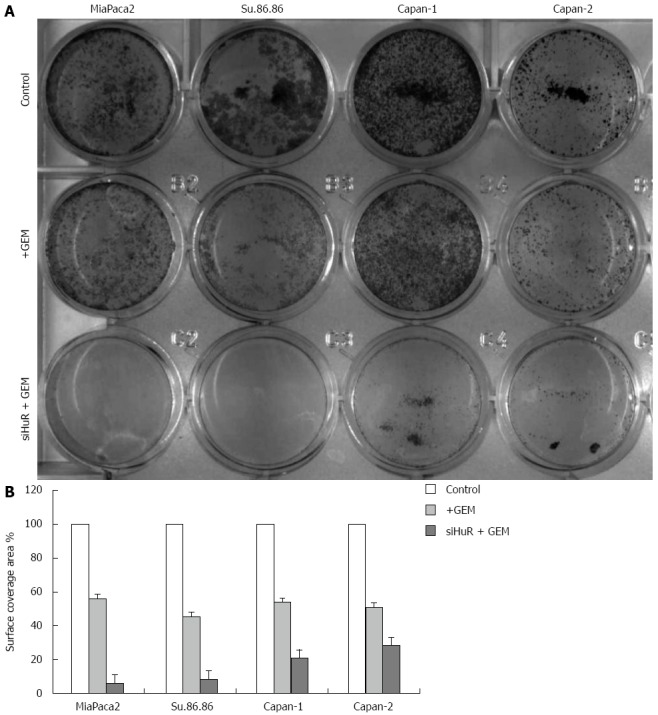
Colony formation of pancreatic cancer cells (crystal violet). A: The colony formation (CV analysis) in 12-well plates; B: Quantitative CV analysis showed that cell surface coverage area was decreased in cells treated with GEM IC50 after HuR siRNA transfection compared with cells treated only with GEM IC50. HuR: Human antigen R; CV: Crystal violet.
The microscopic image analysis revealed morphological changes of pancreatic cancer cells after adjuvant treatment (Figure 9). While GEM treatment increased cell size 2-5 times, on average, a combination of anti-HuR siRNA and GEM treatment resulted in decreased cell size and loss of typical morphology (many cells became shapeless, spherical, and/or fragmented). Based on previous findings of other authors and aforementioned observations, we hypothesized that HuR silencing induced cell cycle arrest and/or apoptosis. To verify the induction of apoptosis, the activity of cleaved caspases 3 and 7 was evaluated. Fluorescence microscopy analysis showed the activation of caspases 3 and 7 after HuR silencing and GEM treatment in all pancreatic cancer cell lines (Figure 10). In comparison, treatment with GEM alone did not activate caspases 3 and 7 in the same cell lines.
Figure 9.
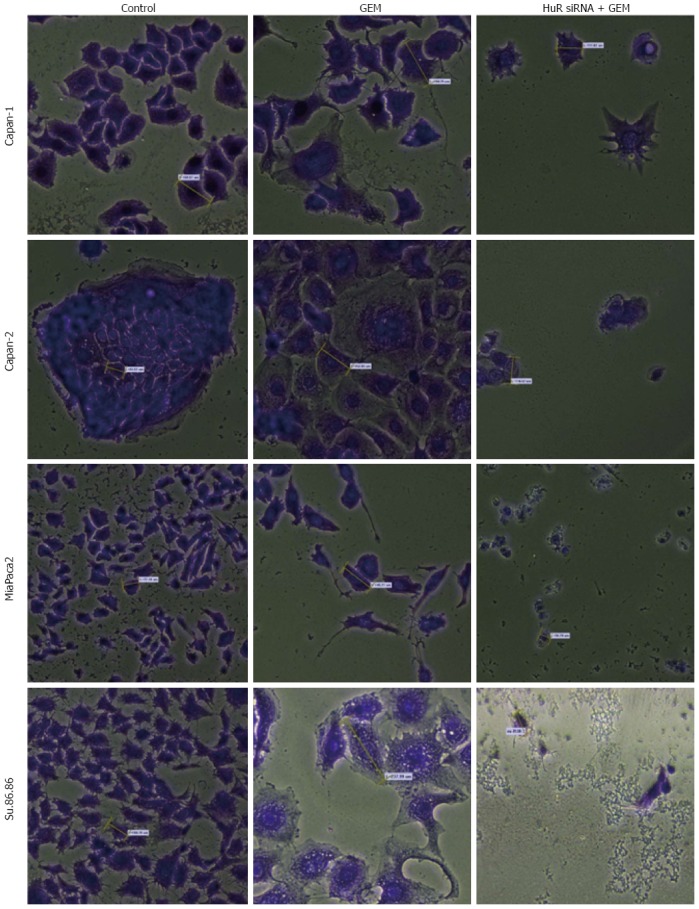
Morphological changes in the microscopic image of pancreatic cancer cells (magnification × 20). Cells became enlarged after gemcitabine (GEM) treatment but became smaller, shapeless or spherical, and fragmented after combined HuR siRNA transfection and GEM treatment. HuR: Human antigen R. Average cell size is labeled by yellow dashes.
Figure 10.
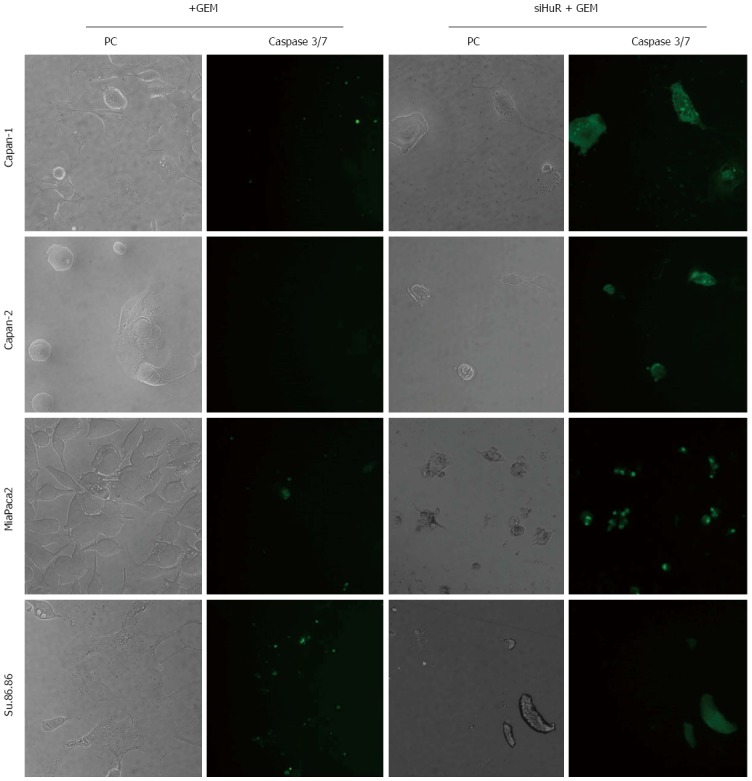
Analysis of caspases 3 and 7 activation in cells. Caspases 3 and 7 were activated in all cell lines treated with GEM after HuR silencing but not after GEM alone treatment.
HuR proteins shuttle from the nucleus to the cytoplasm in human pancreatic cancer cell lines
HuR is predominantly nuclear, but in response to various stimuli, it mobilizes to the cytoplasm, prolonging target mRNA half-life and modulating target mRNA translation[6]. In our analysis, HuR protein was mainly localized to the nucleus of non-treated pancreatic cancer cells (Figure 11). Strong cytoplasmic expression was seen only in MiaPaca2 and Su.86.86 cells after GEM treatment. Cytoplasmic HuR expression was less pronounced in Capan-1 and Capan-2 cell lines.
Figure 11.
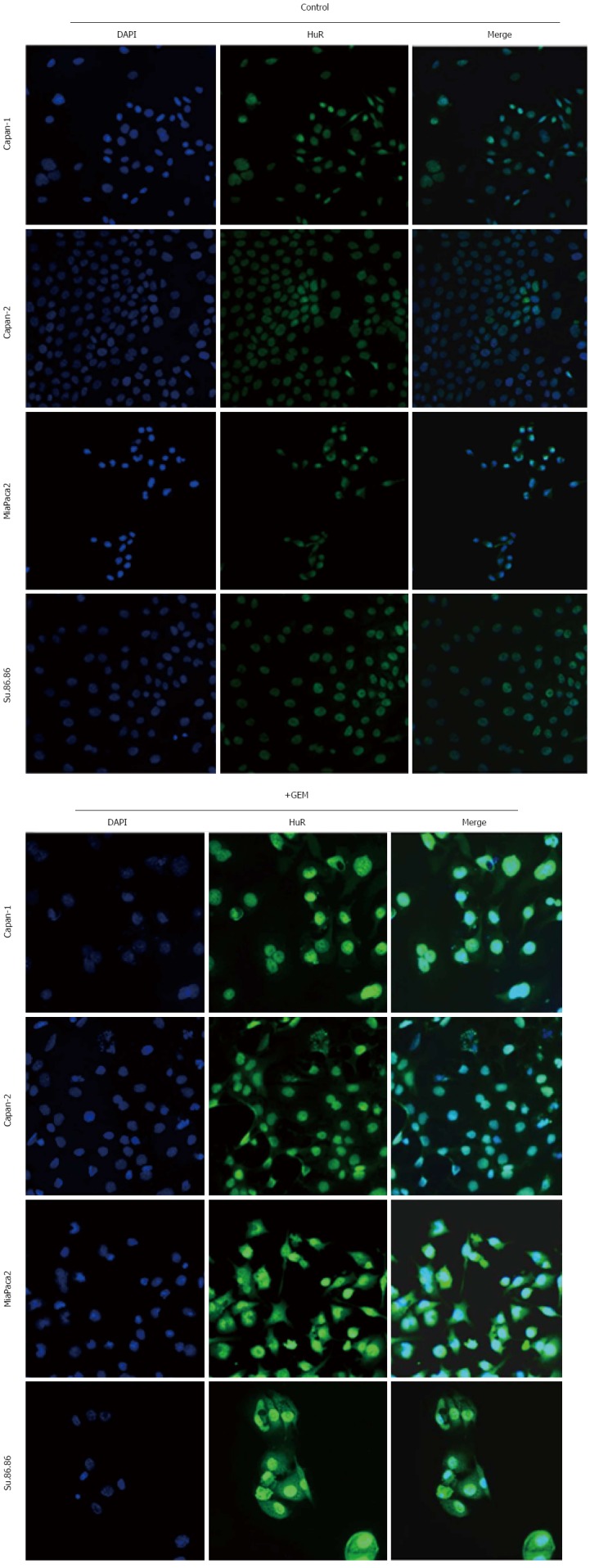
Immunofluorescence analysis of HuR protein expression. Nuclear and cytoplasmic HuR expression was analyzed in control and treated with gemcitabine (GEM) cell lines. Shuttling of HuR from the nucleus to the cytoplasm was seen in cells after GEM treatment.
DISCUSSION
Published investigations on the biological activity of HuR indicate that it is a crucial regulator of post-transcriptional gene expression and a key player in carcinogenesis and chemoresistance of panreatic cancer. Multiple functions of HuR are linked to its ability to recognize, bind, and stabilize a large subset of AU rich element (ARE)-containing mRNAs. The HuR target mRNAs encode a variety of factors required for cancer cell proliferation, survival, angiogenesis, invasion, and metastasis[6,17,18].
Cellular expression of HuR is associated with carcinogenesis and poor clinical outcomes in lung, breast, oral, gallbladder, colon, and urinary tract cancers[6,19-28]. Recent findings established that HuR is a critical regulator of pancreatic cancer cell metabolism and survival under acute glucose deprivation[29]. Another study demonstrated that stressing PDA cells by treatment with DNA-damaging anticancer agents [mitomycin C, oxaliplatin, cisplatin, carboplatin, and a poly (ADP-ribose) polymerase (PARP) inhibitor] resulted in translocation of HuRs from the nucleus to the cytoplasm. Importantly, silencing of HuR in pancreatic cancer cells sensitized the cells to these agents, whereas overexpressing HuR caused resistance. The role of HuR in the efficacy of DNA-damaging agents in pancreatic cancer cells was, in part, attributed to the acute upregulation of WEE1 by HuR. WEE1, a mitotic inhibitor kinase, regulates the DNA damage repair pathway. Therapeutic inhibition of WEE1, in combination with chemotherapy, is currently in early phase trials for the treatment of pancreatic cancer[30].
In the present study, we hypothesised that HuR silencing would sensitize pancreatic cancer cells to GEM treatment by reducing the activity of COX-2 and HO-1 and inducing apoptosis. We speculated that the upregulation of these cytoprotective and anti-apoptotic molecules is mediated by the mRNA binding protein HuR, resulting in increased cancer cell resistance to adjuvant chemotherapy. Data from the present study showed that mRNA and protein expression of HuR was 2-3 times lower in PDA tissue samples from patients not treated with GEM relative to normal pancreas tissue samples. Many previous reports showed that HuR expression in cancer cells was higher than normal cells. We want to emphasize, however, that these studies used GEM treated pancreatic cancer tissues, whereas we used pancreatic cancer tissue not treated with GEM. Cellular analysis showed that HuR expression was increased after GEM treatment.
The expression of the HuR regulated cytoprotective molecules HO-1 and COX-2 proteins was 10.8 and 3.2-fold higher, respectively, in pancreatic cancer tissue. An in vitro study revealed that treatment with an IC50 dose of GEM significantly upregulated HuR, HO-1, and COX-2 expression, both at the mRNA and protein level, in Capan-1 Capan-2, MiaPaca2, and Su.86.86 cell lines.
It is well established that intracellular HuR is predominantly localized within the nucleus of resting cells. The translocation from the nucleus to the cytoplasm appears to be an important aspect of the stabilizing function of HuR. Many stress stimulators, including chemotherapy by GEM, have been reported to induce the shuttling of HuR from the nucleus to the cytoplasm, and most exogenous stimuli significantly increase cytoplasmic accumulation of HuR protein[6]. The present study confirmed that GEM treatment leads to the shuttling of HuR protein from the nucleus into the cytoplasm in MiaPaca2, Su.86.86, Capan-1, and Capan-2 human pancreatic cancer cell lines. These findings support the hypothesis that HuR is an important molecule that is associated with the regulation of increased cancer cell resistance to extrinsic insults, such as chemotherapy, oxidative stress, heat shock, and/or nutrient deprivation.
The need for adjuvant therapy for pancreatic cancer is based on the high risk of local and systemic disease recurrence and the overall poor prognosis after tumor resection. For more than a decade, GEM has been used as the first-line chemotherapy drug for pancreatic cancer. Treatment failure using GEM or other chemotherapy agents indicates suboptimal blockage of cancer survival signals, which may be maintained by the crosstalk of signal transduction pathways or drug resistance[31]. One of the possible molecular mechanisms determining the increased resistance to chemotherapeutic agents is post-transcriptional regulation by mRNA binding proteins. The expression and activity of RNA binding proteins could be regulated by a number of intrinsic and extrinsic pathways, and, thus, they potentially could become an important group of new therapeutic targets[32]. The data from the current study supports the notion that GEM induces HuR shuttling from the nucleus into the cytoplasm in pancreatic cancer cells. This results in upregulation of cytoprotective and/or anti-apoptotic molecules and leads to the resistance to the standard anti-cancer treatment. Moreover, HuR silencing by siRNA transfection significantly increased cancer cell susceptibility to GEM in all cancer cell lines. The fluorescent microscopy analysis showed cleaved caspases 3 and 7 activation after HuR silencing and GEM treatment in all pancreatic cancer cells lines, while treatment with GEM alone did not activate caspases 3 and 7 in the same cell lines.
These findings are in contrast with previously published data showing that patients who receive GEM with low cytoplasmic HuR levels (compared to high cytoplasmic HuR levels) have a 7-fold higher risk of mortality, after adjusting for variables, such as as age, sex, capecitabine use, and radiation therapy. That study revealed in pancreatic cancer cells that HuR associated with deoxycytidine kinase (dCK) mRNA, which encodes the enzyme that metabolizes and thereby activates GEM. Accordingly, HuR overexpression elevated dCK protein expression in pancreatic cancer cells and sensitized them to GEM treatment[33,34]. We did not analyze CK protein expression in this study, and available data does not support this concept. Possibly, post-transciptional regulation via the mRNA binding proteins is much more complex and multimodal. Therefore, these findings mandate further in vitro and clinical research in this field.
However, some findings of our group are in complete concordance with the data from other studies. In the HuR-knockdown oral cancer cells, the cytoplasmic expression of c-fos, c-myc, and COX-2 mRNAs was inhibited compared to cells transfected with control siRNA. In addition, the half-lives of these mRNAs were shorter than those of their counterparts in the control cells. The expression of cell cycle proteins, such as cyclin A, cyclin B1, cyclin D1, and cyclin-dependent kinase 1, was reduced in HuR-knockdown cancer cells. Additionally, the motile and invasive activities of the cells decreased remarkably after the HuR knockdown. Therefore, HuR knockdown changes the features of oral cancer cells, at least in part, by affecting their cell cycle and shows potential as an effective therapeutic approach[35]. Another group reported that HuR translocates from the nucleus to the cytoplasm of PDA cells upon treatment with a death receptor 5 (DR5) agonist. High doses of DR5 agonist induced cleavage of both HuR and caspase 8. This finding demonstrates a feedback mechanism elicited by HuR-mediated repression of the key apoptotic membrane protein DR5[14]. HuR translocated into the cytoplasm upon lethal stress, by a mechanism involving its association with the apoptosome activator pp32/putative HLA II-associated protein 1 (PHAP-I). Depleting the expression of pp32/PHAP-I by RNA interference reduced both HuR cytoplasmic accumulation and the efficiency of caspases 3 and 7 activation. This model, in which HuR association with pp32/PHAP-I and its caspase-mediated cleavage constitutes a regulatory step that, contributes to an amplified apoptotic response[36].
These data indicated that HuR silencing not only downregulated the activity of COX-2 and HO-1 but also increased the pancreatic cancer cell susceptibility to GEM by altering the cell cycle and inducing apoptosis. Earlier studies supported the notion that reduced levels of COX-2 and HO-1 proteins can increase the response to GEM in pancreatic and other cancers[13,37].
Previous studies have demonstrated that COX-2 was scarcely expressed in the majority of normal pancreatic tissue but increased levels of the molecule were detected in pancreatic cancer tissue[38]. A large amount of epidemiological and experimental evidence supports the role of COX-2 in human tumorigenesis, particularly in colorectal cancer. COX-2 mediates carcinogenesis through the production of 15(R)-15-methyl prostaglandin E2 (mPGE2), which inhibits apoptosis, promotes cell proliferation, stimulates angiogenesis, and downgrades the immune response[39]. These findings are fully in accordance with the results of our current study.
Our study demonstrated that HO-1 was overexpressed in the PDA tissues. Normally, HO catabolizes the free heme and its stress-responsive isoenzyme HO-1 is known to protect against apoptosis. Catabolism of free heme, which sensitizes cells to apoptosis, is believed to underlie the cytoprotective mechanism of HO-1[40].
There are some limitations of our study. It would be useful to investigate and compare the expression of HuR, COX-2, and HO-1 in PDA tissue obtained both from GEM treated and non-treated patients in order to fully understand the underlying mechanism and the role of HuR mediated post-transcriptional regulation for the exceptional resistance of pancreatic cancer to the conventional treatment. However, it is not routine practice to give neoadjuvant chemotherapy with GEM (or any other chemotherapeutic drug) prior to pancreatoduodenal resection in our hospital. Therefore, it is not possible to obtain pancreatic cancer tissue samples after GEM treatment for research purposes in our institution. Although this issue is a limitation of our study, we believe that the overall results are sufficient to demonstrate that HuR plays an important role in the chemoresistance of pancreatic cancer cells.
In conclusion, results of the study demonstrated that the expression of the mRNA binding protein HuR is relatively low in PDA tissue (from patients without previous adjuvant chemo- and/or radiotherapy). Our in vitro study confirms that GEM treatment strongly induces HuR expression in all pancreatic cancer cells lines evaluated. The stabilization and increased translation of HuR target mRNAs results in overexpression of cytoprotective and anti-apoptotic proteins COX-2 and HO-1, which translated to increased resistance to GEM treatment. HuR silencing significantly decreased COX-2 and HO-1 expression and sensitized pancreatic cancer cells to GEM treatment. We propose that HuR is a new important therapeutic target in adjuvant treatment of human pancreatic cancer.
ACKNOWLEDGMENTS
We thank to Dovilė Gataveckaite and Augustina Gasianec (Vytautas Magnus University) for technical assistance and Dr. Rima Ramonaite (Lithuanian university of health sciences) for advice in experiment planning. We would also like to thank Lithuanian Centre for Evidence-based Medicine for editing this manuscript.
COMMENTS
Background
Post-transcriptional regulation plays a critical role in the process of cell proliferation and apoptosis, but, in cancer, this control might be impaired by changes in expression or function of RNA binding proteins. HuR is an RNA binding protein that has been reported to regulate the expression of multiple genes by different post-transcriptional mechanisms. The expression of HuR has been shown to be altered in several pathological conditions, including pancreatic cancer.
Research frontiers
Alterations in enzymatic activity and post-transcriptional regulation in tumor cells might be responsible for the exceptional resistance of pancreatic cancer to conventional treatment. The aim of this study was to assess the changes of HuR expression and function (i.e., the translocation from the nucleus to the cytoplasm) after chemotherapy and to evaluate the effects of HuR silencing in the pancreatic cancer cell lines on in vitro response to gemcitabine treatment.
Innovations and breakthroughs
This is the first study demonstrating that HuR mediated post-transcriptional changes play an important role in the regulation of heme oxygenase-1 (HO-1) expression in pancreatic cancer after chemotherapy. Previously, one study had reported HuR dependent HO-1 upregulation in human fibroblasts.
Applications
Data from this study suggest that HuR is one of the key molecules responsible for the exceptional chemoresistance of pancreatic cancer cells. HuR silencing appears to be a potential new therapeutic target in adjuvant treatment of human pancreatic cancer. This hypothesis needs to be tested further in studies using animal models of cancer.
Terminology
Post-transcriptional regulation is the control of genes expression at the RNA level. HuR (ELAV1) is an RNA that regulates the expression of multiple genes by different post-transcriptional mechanisms, including mRNA trafficking, mRNA decay, and protein translation.
Peer-review
This study demonstrates a role of HuR in chemoresistance of pancreatic cancer. The authors found that treatment with GEM induced HuR expression and its translocation from the nucleus to the cytoplasm, affecting cyclooxygenase-2 (COX-2) and HO-1 protein expression. They also revealed that knockdown of HuR sensitized pancreatic cancer cells to GEM treatment. Finally, they concluded that HuR regulated the posttranscriptional modification of cytoprotective molecules, including COX-2 and HO-1, and that HuR may be a key molecule for induction of chemoresistance in pancreatic cancer.
Footnotes
Supported by The European Social Fund under the Global Grant measure.
Institutional review board statement: Ethical approval was issued by the Ethics Committee of the Lithuanian University of Health Sciences (Nr. BE-2-10 and BE-2-17).
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: March 5, 2015
First decision: April 24, 2015
Article in press: August 31, 2015
P- Reviewer: Masuda K S- Editor: Yu J L- Editor: Filipodia E- Editor: Ma S
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Malvezzi M, Bertuccio P, Levi F, La Vecchia C, Negri E. European cancer mortality predictions for the year 2014. Ann Oncol. 2014;25:1650–1656. doi: 10.1093/annonc/mdu138. [DOI] [PubMed] [Google Scholar]
- 3.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 4.Burris HA, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 5.Kroep JR, Pinedo HM, van Groeningen CJ, Peters GJ. Experimental drugs and drug combinations in pancreatic cancer. Ann Oncol. 1999;10 Suppl 4:234–238. [PubMed] [Google Scholar]
- 6.Wang J, Guo Y, Chu H, Guan Y, Bi J, Wang B. Multiple functions of the RNA-binding protein HuR in cancer progression, treatment responses and prognosis. Int J Mol Sci. 2013;14:10015–10041. doi: 10.3390/ijms140510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuwano Y, Rabinovic A, Srikantan S, Gorospe M, Demple B. Analysis of nitric oxide-stabilized mRNAs in human fibroblasts reveals HuR-dependent heme oxygenase 1 upregulation. Mol Cell Biol. 2009;29:2622–2635. doi: 10.1128/MCB.01495-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barbisan F, Mazzucchelli R, Santinelli A, Lopez-Beltran A, Cheng L, Scarpelli M, Montorsi F, Montironi R. Overexpression of ELAV-like protein HuR is associated with increased COX-2 expression in atrophy, high-grade prostatic intraepithelial neoplasia, and incidental prostate cancer in cystoprostatectomies. Eur Urol. 2009;56:105–112. doi: 10.1016/j.eururo.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 9.Hinman MN, Lou H. Diverse molecular functions of Hu proteins. Cell Mol Life Sci. 2008;65:3168–3181. doi: 10.1007/s00018-008-8252-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hostetter C, Licata LA, Witkiewicz A, Costantino CL, Yeo CJ, Brody JR, Keen JC. Cytoplasmic accumulation of the RNA binding protein HuR is central to tamoxifen resistance in estrogen receptor positive breast cancer cells. Cancer Biol Ther. 2008;7:1496–1506. doi: 10.4161/cbt.7.9.6490. [DOI] [PubMed] [Google Scholar]
- 11.Kuwano Y, Kim HH, Abdelmohsen K, Pullmann R, Martindale JL, Yang X, Gorospe M. MKP-1 mRNA stabilization and translational control by RNA-binding proteins HuR and NF90. Mol Cell Biol. 2008;28:4562–4575. doi: 10.1128/MCB.00165-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crane CH, Mason K, Janjan NA, Milas L. Initial experience combining cyclooxygenase-2 inhibition with chemoradiation for locally advanced pancreatic cancer. Am J Clin Oncol. 2003;26:S81–S84. doi: 10.1097/00000421-200308002-00009. [DOI] [PubMed] [Google Scholar]
- 13.Berberat PO, Dambrauskas Z, Gulbinas A, Giese T, Giese N, Künzli B, Autschbach F, Meuer S, Büchler MW, Friess H. Inhibition of heme oxygenase-1 increases responsiveness of pancreatic cancer cells to anticancer treatment. Clin Cancer Res. 2005;11:3790–3798. doi: 10.1158/1078-0432.CCR-04-2159. [DOI] [PubMed] [Google Scholar]
- 14.Pineda DM, Rittenhouse DW, Valley CC, Cozzitorto JA, Burkhart RA, Leiby B, Winter JM, Weber MC, Londin ER, Rigoutsos I, et al. HuR’s post-transcriptional regulation of Death Receptor 5 in pancreatic cancer cells. Cancer Biol Ther. 2012;13:946–955. doi: 10.4161/cbt.20952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brody J, Dasgupta A, Costantino CL, Kennedy E, Yeo CJ, Witkiewicz AK. Correlation of HuR cytoplasmic expression in pancreatic cancer and overall patient survival when treated with gemcitabine in the adjuvant setting [Abstracts] J Clin Oncol. 2009;27:S11097. [Google Scholar]
- 16.Williams TK, Costantino CL, Bildzukewicz NA, Richards NG, Rittenhouse DW, Einstein L, Cozzitorto JA, Keen JC, Dasgupta A, Gorospe M, et al. pp32 (ANP32A) expression inhibits pancreatic cancer cell growth and induces gemcitabine resistance by disrupting HuR binding to mRNAs. PLoS One. 2010;5:e15455. doi: 10.1371/journal.pone.0015455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.López de Silanes I, Zhan M, Lal A, Yang X, Gorospe M. Identification of a target RNA motif for RNA-binding protein HuR. Proc Natl Acad Sci USA. 2004;101:2987–2992. doi: 10.1073/pnas.0306453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.López de Silanes I, Lal A, Gorospe M. HuR: post-transcriptional paths to malignancy. RNA Biol. 2005;2:11–13. doi: 10.4161/rna.2.1.1552. [DOI] [PubMed] [Google Scholar]
- 19.Lauriola L, Granone P, Ramella S, Lanza P, Ranelletti FO. Expression of the RNA-binding protein HuR and its clinical significance in human stage I and II lung adenocarcinoma. Histol Histopathol. 2012;27:617–626. doi: 10.14670/HH-27.617. [DOI] [PubMed] [Google Scholar]
- 20.Kim I, Kwak H, Lee HK, Hyun S, Jeong S. β-Catenin recognizes a specific RNA motif in the cyclooxygenase-2 mRNA 3’-UTR and interacts with HuR in colon cancer cells. Nucleic Acids Res. 2012;40:6863–6872. doi: 10.1093/nar/gks331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyata Y, Watanabe S, Sagara Y, Mitsunari K, Matsuo T, Ohba K, Sakai H. High expression of HuR in cytoplasm, but not nuclei, is associated with malignant aggressiveness and prognosis in bladder cancer. PLoS One. 2013;8:e59095. doi: 10.1371/journal.pone.0059095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang PI, Li WM, Wang YH, Wu TF, Wu WR, Liao AC, Shen KH, Wei YC, Hsing CH, Shiue YL, et al. HuR cytoplasmic expression is associated with increased cyclin A expression and poor outcome with upper urinary tract urothelial carcinoma. BMC Cancer. 2012;12:611. doi: 10.1186/1471-2407-12-611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J, Li D, Wang B, Wu Y. Predictive and prognostic significance of cytoplasmic expression of ELAV-like protein HuR in invasive breast cancer treated with neoadjuvant chemotherapy. Breast Cancer Res Treat. 2013;141:213–224. doi: 10.1007/s10549-013-2679-7. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Yu J, DU D, Fu S, Chen Y, Yu F, Gao P. Involvement of post-transcriptional regulation of FOXO1 by HuR in 5-FU-induced apoptosis in breast cancer cells. Oncol Lett. 2013;6:156–160. doi: 10.3892/ol.2013.1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun DP, Lin CY, Tian YF, Chen LT, Lin LC, Lee SW, Hsing CH, Lee HH, Shiue YL, Huang HY, et al. Clinicopathological significance of HuR expression in gallbladder carcinoma: with special emphasis on the implications of its nuclear and cytoplasmic expression. Tumour Biol. 2013;34:3059–3069. doi: 10.1007/s13277-013-0872-2. [DOI] [PubMed] [Google Scholar]
- 26.Zhu Z, Wang B, Bi J, Zhang C, Guo Y, Chu H, Liang X, Zhong C, Wang J. Cytoplasmic HuR expression correlates with P-gp, HER-2 positivity, and poor outcome in breast cancer. Tumour Biol. 2013;34:2299–2308. doi: 10.1007/s13277-013-0774-3. [DOI] [PubMed] [Google Scholar]
- 27.Habiba U, Kitamura T, Yanagawa-Matsuda A, Hida K, Higashino F, Ohiro Y, Totsuka Y, Shindoh M. Cytoplasmic expression of HuR may be a valuable diagnostic tool for determining the potential for malignant transformation of oral verrucous borderline lesions. Oncol Rep. 2014;31:1547–1554. doi: 10.3892/or.2014.3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Govindaraju S, Lee BS. Adaptive and maladaptive expression of the mRNA regulatory protein HuR. World J Biol Chem. 2013;4:111–118. doi: 10.4331/wjbc.v4.i4.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burkhart RA, Pineda DM, Chand SN, Romeo C, Londin ER, Karoly ED, Cozzitorto JA, Rigoutsos I, Yeo CJ, Brody JR, et al. HuR is a post-transcriptional regulator of core metabolic enzymes in pancreatic cancer. RNA Biol. 2013;10:1312–1323. doi: 10.4161/rna.25274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lal S, Burkhart RA, Beeharry N, Bhattacharjee V, Londin ER, Cozzitorto JA, Romeo C, Jimbo M, Norris ZA, Yeo CJ, et al. HuR posttranscriptionally regulates WEE1: implications for the DNA damage response in pancreatic cancer cells. Cancer Res. 2014;74:1128–1140. doi: 10.1158/0008-5472.CAN-13-1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang ZQ, Saluja AK, Dudeja V, Vickers SM, Buchsbaum DJ. Molecular targeted approaches for treatment of pancreatic cancer. Curr Pharm Des. 2011;17:2221–2238. doi: 10.2174/138161211796957427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abdelmohsen K, Gorospe M. Posttranscriptional regulation of cancer traits by HuR. Wiley Interdiscip Rev RNA. 2010;1:214–229. doi: 10.1002/wrna.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Costantino CL, Witkiewicz AK, Kuwano Y, Cozzitorto JA, Kennedy EP, Dasgupta A, Keen JC, Yeo CJ, Gorospe M, Brody JR. The role of HuR in gemcitabine efficacy in pancreatic cancer: HuR Up-regulates the expression of the gemcitabine metabolizing enzyme deoxycytidine kinase. Cancer Res. 2009;69:4567–4572. doi: 10.1158/0008-5472.CAN-09-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong A, Soo RA, Yong WP, Innocenti F. Clinical pharmacology and pharmacogenetics of gemcitabine. Drug Metab Rev. 2009;41:77–88. doi: 10.1080/03602530902741828. [DOI] [PubMed] [Google Scholar]
- 35.Kakuguchi W, Kitamura T, Kuroshima T, Ishikawa M, Kitagawa Y, Totsuka Y, Shindoh M, Higashino F. HuR knockdown changes the oncogenic potential of oral cancer cells. Mol Cancer Res. 2010;8:520–528. doi: 10.1158/1541-7786.MCR-09-0367. [DOI] [PubMed] [Google Scholar]
- 36.Mazroui R, Di Marco S, Clair E, von Roretz C, Tenenbaum SA, Keene JD, Saleh M, Gallouzi IE. Caspase-mediated cleavage of HuR in the cytoplasm contributes to pp32/PHAP-I regulation of apoptosis. J Cell Biol. 2008;180:113–127. doi: 10.1083/jcb.200709030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eibl G, Reber HA, Wente MN, Hines OJ. The selective cyclooxygenase-2 inhibitor nimesulide induces apoptosis in pancreatic cancer cells independent of COX-2. Pancreas. 2003;26:33–41. doi: 10.1097/00006676-200301000-00007. [DOI] [PubMed] [Google Scholar]
- 38.Hill R, Li Y, Tran LM, Dry S, Calvopina JH, Garcia A, Kim C, Wang Y, Donahue TR, Herschman HR, et al. Cell intrinsic role of COX-2 in pancreatic cancer development. Mol Cancer Ther. 2012;11:2127–2137. doi: 10.1158/1535-7163.MCT-12-0342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doré M. Cyclooxygenase-2 expression in animal cancers. Vet Pathol. 2011;48:254–265. doi: 10.1177/0300985810379434. [DOI] [PubMed] [Google Scholar]
- 40.Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol. 2010;50:323–354. doi: 10.1146/annurev.pharmtox.010909.105600. [DOI] [PubMed] [Google Scholar]