

Abstract
AIM: To determine the therapeutic potential of sphingosine kinase 1 (Sphk1) inhibition and its underlying mechanism in a well-characterized mouse model of D-galactosamine (D-GalN)/lipopolysaccharide (LPS)-induced acute liver failure (ALF).
METHODS: Balb/c mice were randomly assigned to different groups, with ALF induced by intraperitoneal injection of D-GaIN (600 mg/kg) and LPS (10 μg/kg). The Kaplan-Meier method was used for survival analysis. Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels at different time points within one week were determined using a multi-parametric analyzer. Serum high-mobility group box 1 (HMGB1), tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, IL-6, IL-10, and sphingosine-1-phosphate were detected by enzyme-linked immunosorbent assay. Hepatic morphological changes at 36 h after acute liver injury induction were assessed by hematoxylin and eosin staining. HMGB1 expression in hepatocytes and cytoplasmic translocation were detected by immunohistochemistry. Expression of Sphk1 in liver tissue and peripheral blood mononuclear cells (PBMCs) was analyzed by Western blot.
RESULTS: The expression of Sphk1 in liver tissue and PBMCs was upregulated in GalN/LPS-induced ALF. Upregulated Sphk1 expression in liver tissue was mainly caused by Kupffer cells, the resident macrophages of the liver. The survival rates of mice in the N,N-dimethylsphingosine (DMS, a specific inhibitor of SphK1) treatment group were significantly higher than that of the control group (P < 0.001). DMS treatment significantly decreased the levels of serum ALT and AST at 6, 12, and 24 h compared with that of the control group (P < 0.01 for all). Serum HMGB1 levels at 6, 12, and 24 h, as well as serum TNF-α, IL-6, and IL-1β levels at 12 h, were significantly lower in the DMS treatment group than in the control group (P < 0.01 for all). Furthermore, hepatic inflammation, necrosis, and HMGB1 cytoplasm translocation in liver cells were significantly decreased in the DMS treatment group compared to the control group (43.72% ± 5.51% vs 3.57% ± 0.83%, χ2 = 12.81, P < 0.01).
CONCLUSION: Inhibition of SphK1 ameliorates ALF by reducing HMGB1 cytoplasmic translocation in liver cells, and so might be a potential therapeutic strategy for this disease.
Keywords: Acute liver failure, Sphingosine kinase 1, High-mobility group box 1, Cytoplasmic translocation, Inflammatory cytokine
Core tip: Recent studies demonstrated that sphingosine kinase 1 (Sphk1) plays a critical role in sepsis-induced inflammatory responses and high-mobility group box 1 (HMGB1) cytoplasmic translocation has an important role in acute liver failure (ALF). The finding that SphK1 is able to mediate the secretion of proinflammatory mediators prompted us to investigate its role in systemic inflammatory response caused by ALF. In the present study, we demonstrated that SphK1 was critical in D-galactosamine/lipopolysaccharide-induced ALF in mice and that inhibition of SphK1 ameliorated ALF by reducing HMGB1 cytoplasmic translocation in liver cells in this animal model. Our findings suggest that inhibition of SphK1 might be a potential therapeutic strategy for ALF.
INTRODUCTION
Acute liver failure (ALF) is characterized by sudden and massive death of liver cells and remains a disease with high mortality and limited therapeutic options, often demanding liver transplantation[1]. The injured hepatocytes may themselves aggravate and exacerbate liver injury via the activation of immune cells, often leading to systemic inflammatory response syndrome, which is the most common cause of death for the disease[2,3]. Recent clinical trials and animal studies have suggested that ALF can trigger systemic inflammation. Patients with ALF have higher circulating concentrations of pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and IL-6[4,5].
Sphingosine kinases (SphKs) are intracellular signaling enzymes that catalyze the formation of the lipid mediator sphingosine-1-phosphate (S1P)[6]. Several pro-inflammatory stimuli, including TNF-α and immune complexes, activate SphK1 on human neutrophils and macrophages, and blockade of SphK1 inhibits pro-inflammatory responses triggered by these stimuli[7-9]. A recent study demonstrated that SphK1 plays a critical role in endotoxin signaling and sepsis-induced inflammatory responses[10]. The finding that SphK1 is able to mediate the secretion of proinflammatory mediators prompted us to investigate its role in the systemic inflammatory response caused by ALF.
High-mobility group box 1 (HMGB1) is a late mediator of lethal systemic inflammation. Recent studies have demonstrated that hepatocytes can actively release HMGB1 after being challenged with lipopolysaccharide (LPS) and HMGB1 cytoplasmic translocation was observed in liver cells in an animal model of ALF induced by D-galactosamine (D-GalN) and LPS, as well as in patients with ALF[11].
In the present study, we aimed to determine the therapeutic potential of Sphk1 inhibition and its underlying mechanism in a well-characterized mouse model of GalN/LPS-induced ALF. We demonstrated that SphK1 was upregulated in the liver tissue and peripheral blood mononuclear cells (PBMCs) of mice with D-GalN/LPS-induced ALF. We also found that inhibition of SphK1 with N,N-dimethylsphingosine (DMS), a specific inhibitor of SphK1, ameliorated ALF and reduced HMGB1 cytoplasmic translocation in liver cells in this animal model. Our findings suggest that inhibition of SphK1 might be a potential therapeutic strategy for ALF.
MATERIALS AND METHODS
Animal model of ALF and treatments
Male Balb/c mice aged 6-7 wk and weighing 20 ± 0.5 g were obtained from the Experimental Animal Center of Nanchang University, Nanchang, China. The mice were handled and treated in accordance with the strict guiding principles of the National Institution of Health for experimental care and use of animals. ALF was induced in mice by intraperitoneal injection of D-GalN (600 mg/kg) (Sigma-Aldrich, St. Louis, MO, United States) and LPS (10 μg/kg) (Sigma-Aldrich) as previously described. At 6 and 36 h following the onset of ALF, mice were sacrificed to harvest liver tissue for immunohistochemistry and hematoxylin and eosin (HE) staining. For deletion of Kupffer cells (KCs), mice were intraperitoneally injected with GdCL3 (20 mg/kg) (Sigma-Aldrich) 24 h before the induction of ALF[12]. DMS (Sigma-Aldrich), a specific chemical inhibitor of SphK1, was intraperitoneally injected 0.5 h prior to the onset of ALF to inhibit SphK1 activity in vivo.
Measurement of serum aminotransferase activities and cytokine levels
Serum samples were stored at -80 °C until analysis. Serum ALT and AST levels were measured using a multi-parametric analyzer (AU 5400, Olympus, Japan). Serum levels of TNF-α, IL-1β, IL-6, IL-10, and HMGB1 were determined using enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s instructions (RD Systems, Minneapolis, MN, United States).
Determination of S1P concentration
S1P concentrations were determined by ELISA using a commercial kit (Echelon, Salt Lake City, UT, United States). A 96-well microtiter plate was coated with S1P and blocked to reduce non-specific binding. The S1P standard and samples were then mixed with the anti-S1P antibody before adding the mixture to the S1P-coated plate. The antibody competes for binding to S1P bound to the plate or in the sample. Following incubation and plate wash, streptavidin-horseradish peroxidase (HRP) was added to the plate and bound to anti-S1P antibody (labeled with biotin) bound to the plate. After additional incubation and plate wash, tetramethylbenzidine substrate was added to the plate, and the reaction was stopped by the addition of sulfuric acid. The absorbance at 450 nm was measured and the concentrations of S1P in the samples were determined by comparison to the standard curve[13].
Western blot analysis
Proteins (40 μg) from total tissue or cell lysates/samples were resolved on 10% polyacrylamide gels under denaturing conditions and then transferred to 0.45 μm nitrocellulose membranes. The blots were probed using polyclonal anti-SphK1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, United States); anti-β-actin (Santa Cruz Biotechnology) was used as a loading control. Bands were visualized using HRP-conjugated anti-IgG secondary antibody and the ECL Western Blotting Detection System (GE Healthcare, United Kingdom).
Histological and immunohistochemistry assays
Liver tissue samples were fixed in 10% phosphate-buffered formalin, embedded in paraffin, and processed for immunological and histological assays. Tissue sections of 5 μm were cut and stained with hematoxylin and eosin (HE) or antibody against HMGB1. To reduce non-specific signal, slides were incubated with goat serum blocking buffer (Boster, Wuhan, China) at room temperature for 1 h. The slides were subsequently incubated with primary anti-HMGB1 antibody (1:500, Cell Signaling Technology, Danvers, MA, United States). After washing thrice with PBS, slides were incubated with HRP-conjugated goat anti-rabbit IgG (1:1000, Abcam, HK, China) at room temperature for 1 h. Histological assessment was performed by a blinded observer, who scored the liver sections using the following criteria: normal histology, “0”; minor hepatocellular death and inflammation, “1”; widely distributed patchy necrosis and inflammation, “2”; complete disruption with panlobular necrosis and inflammation, “3”; and mortality, “4”. Hepatocytes with brown staining in the nucleus area represent the normal location of HMGB1, while hepatocytes with brown staining in both cytoplasmic and nucleus areas were defined as cytoplasmic translocation of HMGB1. At least ten high-power fields were chosen randomly and > 1000 cells were counted for each section. The percentage of hepatocytes with HMGB1 cytoplasmic translocation in all hepatocytes counted in each group was calculated.
Statistical analysis
Data are expressed as the mean ± SE. Statistical significance was determined by a two-tailed Student’s t-test or one way analysis of variance (ANOVA). A log-rank test was used for survival analysis. A P value < 0.05 was considered statistically significant. Statistical image analysis was performed after determining that the data could fit with a normal distribution. A two-tailed Student’s t-test was employed after the exclusion of outliers that were less or greater than two standard deviations away from the median. All statistical analyses were performed using SPSS 13.0 for Windows.
RESULTS
Expression of Sphk1 in liver tissues and PBMCs of mice with G-GalN/LPS-induced ALF
Previous studies have indicated that SphK1 is mainly expressed in neutrophils and macrophages. To dissect the role of SphK1 in the pathogenesis of ALF, we first determined whether SphK1 expression is triggered in ALF in vivo. Liver tissues and PBMCs were harvested from mice at 0, 0.5, 1, 3, and 6 h after D-GalN/LPS injection. Compared with normal control mice, the expression of SphK1 in the liver tissues and PBMCs of mice with ALF promptly increased at 0.5 and 1 h, and then declined to the basal level after 3 h, suggesting that SphK1 expression in the liver is an early event in the development of D-GalN/LPS-induced ALF (Figure 1A and B). KCs are the resident macrophages of the liver and constitute 80%-90% of tissue macrophages in the body. During ALF, there is a remarkable increase of activated hepatic macrophages. After intraperitoneal injection of GdCL3 (20 mg/kg) for 24 h to partially delete KCs before the induction of ALF, SphK1 expression in the liver decreased at 0.5 h compared with that in normal controls. These results suggest that KCs are responsible for the increased expression of SphK1 in the liver tissue of mice with D-GalN/LPS-induced ALF (Figure 1C).
Figure 1.
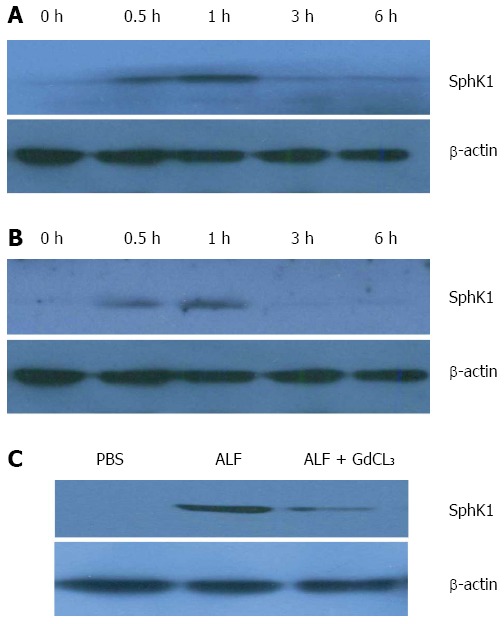
Sphk1 expression in liver tissue and peripheral blood mononuclear cells of mice with D-galactosamine/lipopolysaccharide-induced acute liver failure. Acute liver failure was induced in BALB/c mice using D-galactosamine (D-GalN; 600 mg/kg) and lipopolysaccharide (LPS; 10 μg/kg). A and B: Sphk1 expression in liver tissues and PBMCs of mice with D-GalN/LPS-induced acute liver failure; C: SphK1 expression in liver tissue of mice with acute liver failure after partial deletion of KCs with GdCL3 (20 mg/kg, i.p.) for 24 h.
Activation of SPHK1 is critical for the development of ALF
To address the functional significance of SphK1 in ALF, we treated mice with DMS, a specific chemical inhibitor of SphK1, 0.5 h prior to the onset of ALF. The inhibition of SphK1 activity in vivo was confirmed by reduced expression of sphingosine-1-phosphate (S1P), a downstream substrate of SphK1 (Figure 2A).
Figure 2.
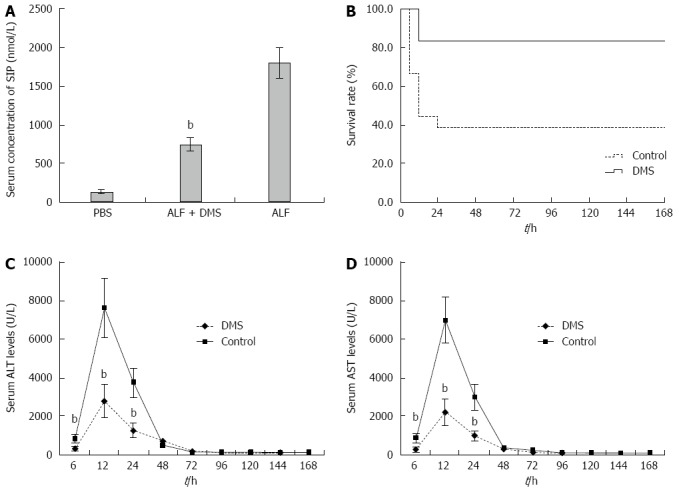
Inhibition of Sphk1 activity with DMS decreases serum S1P concentrations, improves survival, and attenuates liver enzyme release in a mouse model of acute liver failure. Animals were treated with vehicle or DMS (50 μmol/L) 30 min before the induction of acute liver failure. A: Inhibition of Sphk1 activity with DMS reduced S1P, a downstream substrate of SphK1 (735.77 nmol/L ± 87.39 nmol/L vs 1789.23 nmol/L ± 201.79 nmol/L, t = 20.39, bP < 0.01); B: Kaplan-Meier analysis of the effect of DMS on survival rate of the animals. bP = 0.004, F = 8.276, log rank test; C and D: ALT and AST levels in peripheral blood samples collected at 6, 12, 24, 48, 72, 120, 144, and 168 h after treatment. DMS treatment significantly decreased serum levels of ALT at 6, 12, and 24 h (332.8 U/L ± 124.2 U/L, 2788.2 U/L ± 839.2 U/L, 1265.1 U/L ± 371.7 U/L vs 836.6 U/L ± 221.5 U/L, 7612.8 U/L ± 1579.1 U/L, 3741.4 U/L ± 771.7 U/L, t = 6.87, 10.44, and 10.01, respectively, bP < 0.01) and AST at 6, 12, and 24 h (257.1 U/L ± 134.9 U/L, 2196.3 U/L ± 676.3 U/L, 982.3 U/L ± 255.9 U/L vs 853.7 U/L ± 202.3 U/L, 6993.6 U/L ± 1209.5 U/L, 2991.1 U/L ± 678.5 U/L, t = 8.50, 12.00, and 9.60, respectively, bP < 0.01) compared with those of the control group. The mean ± SE of three independent experiments is shown (error bar indicates standard error). DMS: N,N-dimethylsphingosine; S1P: Sphingosine-1-phosphate; ALT: Alanine aminotransferase; AST: Aspartate aminotransferase.
For mortality analysis, five groups of mice, all challenged with D-GalN/LPS, were examined. The mice in group I (control) received vehicle only, the mice in groups II to IV were pre-treated with three doses (10, 25, and 50 μmol/L) of DMS before D-GalN/LPS challenge, and the mice in group V were administered DMS (50 μmol/L) 0.5 h after the onset of D-GalN/LPS-induced ALF. In the control group, the mice began to die 6 h after D-GalN/LPS injection, leading to only 38.9% (3/18) surviving at 24 h. However, pre-treatment with DMS increased the survival rate in a dose-dependent manner; at 24 h after D-GalN/LPS administration, the survival rates were 50% (9/18), 61.1% (11/18), and 83.3% (15/18) in the mice pre-treated with 10, 25, and 50 μmol/L DMS, respectively. The survival rate of mice pre-treated with 50 μmol/L DMS was significantly higher than that of the control group (P = 0.004) (Figure 2B), suggesting that DMS (50 μmol/L) provided significant protection from ALF. However, the survival rate of mice treated with DMS (50 μmol/L) at 0.5 h after onset of ALF (group V) was only 44.4% (8/18). Therefore, we selected pre-treatment with 50 μmol/L DMS for the following study.
Liver enzyme release levels measured in the peripheral blood provide a good estimate of ongoing liver injury. To address the effect of inhibiting SphK1 activity on hepatic injury, we investigated whether DMS regulates serum ALT and AST levels in mice with D-GalN/LPS-induced ALF. The peak liver enzyme levels were observed at 12 h after systemic treatment, both in the control and SphK1 inhibitor treatment groups. The peak ALT and AST levels were reduced significantly in the SphK1 inhibitor treatment group compared with the control group (Figure 2C and D). The peak ALT and AST levels were reduced by 63.4% (P < 0.001) and 68.6% (P < 0.001), respectively, in the DMS-treated animals (Figure 2A and B), and the ALT and AST levels at 6 and 24 h were also decreased significantly in the DMS-treated group compared with the control group (P < 0.001). However, no significant differences were observed at any of the other time points after 24 h.
D-GalN/LPS treatment is known to induce hepatocyte necrosis and inflammatory responses. In the present study, liver histology was normal in the vehicle-treated normal mice, but GalN/LPS treatment caused significant hepatic injury at 36 h, where the necrosis areas were more than 50% of almost all lobules examined, together with panlobular mononuclear leukocyte infiltration, cytoplasmic vacuolization, and severe distortion of tissue architecture (Figure 3B). However, DMS-treated mice showed only small areas of necrosis and inflammatory cell infiltration (Figure 3C).
Figure 3.
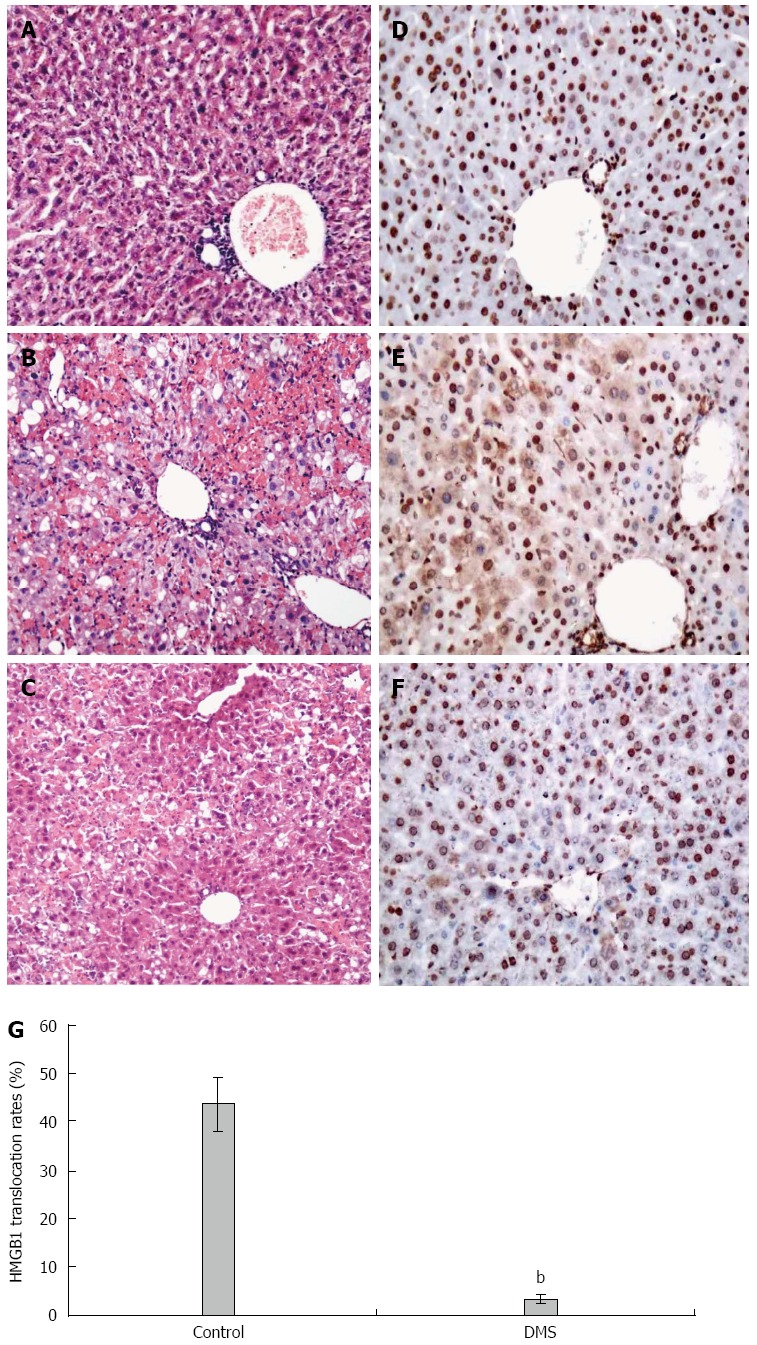
Immune cell infiltration and tissue damage and HMGB1 cytoplasmic translocation in hepatocytes of mice 36 h after D-galactosamine/lipopolysaccharide challenge. Immune cell infiltration, tissue damage, and HMGB1 cytoplasmic translocation in hepatocytes at 6 and 36 h after the onset of acute liver failure were detected by hematoxylin and eosin staining (A-C) and immunohistochemistry (D-F); magnification × 10 or × 20. A and B: Normal mice; C and D: Acute liver failure mice; E and F: DMS-treated mice; G: Percentage of hepatocytes with HMGB1 cytoplasmic translocation. χ2 = 12.81, bP < 0.01, (3.57% ± 0.83%) vs controls (43.72% ± 5.51%). The mean ± SE of three independent experiments is shown (error bar indicates standard error).
Taken together, these data demonstrated that inhibition of Sphk1 activity with DMS improved the survival rate, attenuated liver enzyme release, and reduced liver inflammation and necrosis in ALF, suggesting that Sphk1 activation is critical for the development of ALF.
Inhibition of SphK1 activation downregulates inflammatory cytokine and HMGB1 levels
Massive liver injury results in local and systemic inflammatory responses that can ultimately lead to multi-organ failure and death. Analysis of serum cytokine levels revealed a significant decrease in TNF-α, IL-1β, and IL-6 after DMS treatment (P < 0.01). However, the level of the anti-inflammatory cytokine IL-10 was not increased in the DMS treatment group (Figure 4A). These pro-inflammatory cytokines are known to be upregulated during liver injury. Extracellular HMGB1 is released by phagocytes and damaged or necrotic cells, and functions as a damage-associated molecular pattern (DAMAP) that contributes to the pathogenesis of ALF. In the control group, serum HMGB1 levels increased 6 h after ALF induction, peaked at 12 h, and then dropped rapidly. However, DMS treatment decreased serum concentrations of HMGB1 at all three time points (Figure 4B). These data demonstrate that inhibition of Sphk1 activity with DMS decreased pro-inflammatory cytokine and HMGB1 levels.
Figure 4.
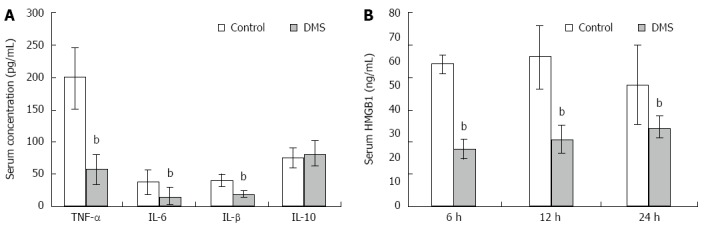
Inhibition of SPHK1 activation downregulates serum inflammatory cytokine and HMGB1 levels in mice with acute liver failure. A: DMS reduced serum levels of TNF-α, IL-1β, and IL-6 significantly (57.07 pg/mL ± 22.83 pg/mL vs 198.55 pg/mL ± 48.17 pg/mL; 15.66 pg/mL ± 13.13 pg/mL vs 37.44 pg/mL ± 18.68 pg/mL; 19.13 pg/mL ± 5.32 pg/mL vs 40.13 pg/mL ± 9.37 pg/mL, t = 27.6, 10.12 and 4.33, respectively, bP < 0.01), but did not increase the level of anti-inflammatory cytokine IL-10 at 12 h; B DMS reduced HMGB1 levels at 6, 12, and 24 h in mice (23.49 ng/mL ± 3.89 ng/mL vs 58.6 ng/mL ± 3.65 ng/mL; 61.62 ng/mL ± 13.07 ng/mL vs 27.32 ng/mL ± 5.97 ng/mL; 49.91 ng/mL ± 16.6 ng/mL vs 32.42 ng/mL ± 4.23 ng/mL, t = 11.27, 4.07, and 8.42, respectively, bP < 0.01). The mean ± SE of three independent experiments is shown (error bar indicates standard error).
SPHK1 inhibitor attenuates HMGB1 cytoplasmic translocation in liver cells
The pathological findings of the liver after exposure to D-GalN and LPS for 6 h showed minor derangement of the hepatic plate and the appearance of ballooning degeneration in several hepatocytes. Liver specimens from normal mice revealed a nuclear localization of HMGB1 in most hepatocytes (Figure 3D). In contrast, HMGB1 cytoplasmic staining was easily observed in liver tissue at 6 h after administration of D-GalN and LPS. HMGB1 staining was found in both the cytoplasm and nucleus of many hepatocytes in the lobes (Figure 3E). In some hepatocytes, HMGB1 was only found in the cytoplasm, showing different stages of HMGB1 cytoplasmic translocation. However, HMGB1 staining was found in the cytoplasm of very few hepatocytes in DMS-treated mice (Figure 3F). Consistently, the percentage of hepatocytes with HMGB1 cytoplasmic staining was significantly higher (43.42% ± 5.51%) in the D-GalN and LPS-treated groups than in the DMS-treated group (3.57% ± 0.83%) (Figure 3G).
DISCUSSION
ALF is an acute inflammatory process of the liver which may lead to systemic inflammatory response syndrome and multi-organ dysfunction syndrome[14]. SphK1 has been implicated in inflammatory response and various immune cell functions. Recent studies showed that Sphk1 regulates macrophage cytokine production, and inhibition of Sphk1 protects mice from endotoxin-induced shock. However, the potential role for SphK1 in systemic inflammatory response in ALF has not yet been explored. In a previous in vivo study, compared with vehicle-treated normal mice, the administration of LPS together with a sub-lethal dose of D-GalN induced severe hepatic damage accompanied by necrotic changes and severe inflammation in the liver, which is similar to human liver failure[15]. Therefore, it is of interest to further investigate the hepatoprotective potential of Sphk1 inhibition in D-GalN/LPS-induced ALF and the mechanism involved.
SphK1 is mainly expressed in neutrophils and macrophages[6,10]. KCs are the resident macrophages of the liver and constitute the majority of tissue macrophages in the body. During ALF, there is a remarkable increase in activated hepatic macrophages, and partial deletion of KCs with GdCL3 prevents D-GalN/LPS-induced ALF[12]. Our current study demonstrated that expression of SphK1 in liver tissue and PBMCs promptly increased at 0.5 h after D-GalN/LPS injection; however, after intraperitoneal injection of GdCL3 to partially delete KCs, SphK1 expression in the liver decreased at 0.5 h compared with that in normal controls. These results suggest that SphK1 expression in the liver is an early event in the acute phase of ALF, and that KCs are responsible for the upregulated expression of SphK1 in the liver tissue of mice with D-GalN/LPS-induced ALF.
To address the functional significance of SphK1 in ALF, we treated mice with DMS; a specific chemical inhibitor of SphK1. Pre-treatment with DMS reduced S1P level and increased the survival rate in a dose-dependent manner in animals with D-GalN/LPS-induced ALF. Peak liver enzyme levels were observed 12 h after systemic treatment, with the peak ALT and AST levels being significantly reduced in the DMS treatment group compared to that of the control group. Furthermore, DMS-treated mice showed only small areas of necrosis and inflammatory cell infiltration, although GalN/LPS treatment caused significant hepatic injury in mice at 36 h, where necrosis areas were more than 50% of almost all lobules examined, together with panlobular mononuclear leukocyte infiltration, cytoplasmic vacuolization, and severe distortion of tissue architecture. These data demonstrate that inhibition of Sphk1 activity with DMS improved the survival rate, attenuated liver enzyme release, and reduced liver inflammation and necrosis in ALF, suggesting that Sphk1 activation is critical for the development of ALF in mice.
Although etiologies of ALF vary between Western countries and the Eastern developing world, the resulting clinical manifestation is remarkably similar. This reflects common patterns of innate immune responses to various pathogenic factors, such as bacteria, toxins, cytokines, and free radicals[16]. Among many other factors, proinflammatory cytokines (e.g., TNF-α, IL-1b, and IL-6) may play a common role in the pathophysiology of ALF. In the current study, inhibition of Sphk1 decreased the levels of pro-inflammatory cytokines TNF-α, IL-1β, and IL-6, indicating that Sphk1 may positively participate in the innate immune response to liver injury.
HMGB1 is a non-histone nuclear protein ubiquitously expressed in eukaryotes that exerts distinct functions at different subcellular localizations. Within the nucleus, HMGB1 plays an important role in the regulation of gene transcription[17]. Upon release by phagocytes and damaged/necrotic cells[18-21], extracellular HMGB1 functions as a DAMAP and contributes to the pathogenesis of various inflammatory diseases[22,23]. HMGB1 exerts its effects through a number of Toll-like receptors (TLR2/4)[21], and this leads to the activation of immune cells and the consequent release of multiple pro-inflammatory cytokines[24]. In animal models of infection or local tissue injury, HMGB1 functions as a critical mediator of systemic or local inflammatory injury[25]. As a result, HMGB1 has been established as a late mediator of lethal systemic inflammatory disease. In the clinical setting, elevated serum HMGB1 levels have been described in patients with sepsis[18,26,27], pneumonia[28], acute pancreatitis[29], and ALF[30].
Recent studies demonstrated that hepatocytes can actively release HMGB1 after challenge with LPS, and that HMGB1 cytoplasmic translocation was observed in liver cells in the animal model of ALF induced by D-GalN and LPS, as well as in patients with ALF[11]. In this study, liver specimens from normal mice revealed nuclear localization of HMGB1 in most hepatocytes, and HMGB1 cytoplasmic staining was easily observed in liver tissue at 6 h after administration of D-GalN and LPS. However, HMGB1 staining was found in the cytoplasm of very few hepatocytes in DMS-treated mice. This result shows that Sphk1 inhibitor attenuated HMGB1 cytoplasmic translocation in liver cells. HMGB1 is abundantly expressed in hepatocytes and predominantly localized in the nucleus of quiescent cells. Given the huge number of hepatocytes in the liver, potential HMGB1 release by hepatocytes could contribute to the pathogenesis of liver failure.
In summary, Sphk1 inhibition represents a potent and safe strategy to ameliorate ALF. This approach may attenuate liver enzyme release, reduce liver inflammation and necrosis, and decrease pro-inflammatory cytokines levels, thus ultimately improving survival in mice with D-GalN/LPS-induced ALF. Furthermore, we demonstrated that inhibition of Sphk1 with DMS attenuated HMGB1 cytoplasmic translocation in liver cells. Further preclinical studies with chemical inhibitors of Sphk1 may pave the way for the development of a clinically-applicable therapeutic strategy against ALF and create a potential new avenue for the treatment of this devastating disorder.
COMMENTS
Background
Recent studies demonstrated that sphingosine kinase 1 (Sphk1) plays a critical role in sepsis-induced inflammatory responses and that high-mobility group box 1 (HMGB1) cytoplasmic translocation has an important role in acute liver failure. The finding that SphK1 is able to mediate the secretion of proinflammatory mediators prompted us to investigate its role in systemic inflammatory response caused by acute liver failure.
Research frontiers
Acute liver failure is an acute inflammatory process of the liver, which may lead to systemic inflammatory response syndrome. Recent studies showed that Sphk1 regulates macrophage cytokine production and that inhibition of Sphk1 protects mice from endotoxin shock. However, the potential role for SphK1 in systemic inflammatory response in acute liver failure has not yet been explored.
Innovations and breakthroughs
SphK1 was critical in acute liver failure in mice and inhibition of SphK1 attenuated acute liver failure by reducing HMGB1 cytoplasmic translocation in liver cells.
Applications
The results suggest that inhibition of SphK1 may be a potential therapeutic strategy for acute liver failure.
Terminology
Acute liver failure is an acute inflammatory process of the liver.
Peer-review
Acute liver failure is an acute inflammatory process of the liver and HMGB1 cytoplasmic translocation plays an important role in acute liver failure. Recent studies demonstrated that SphK1 plays a critical role in sepsis-induced inflammatory responses. It is the first time that the authors demonstrated that SphK1 was critical in D-galactosamine/lipopolysaccharide-induced acute liver failure and inhibition of SphK1 ameliorated acute liver failure by reducing HMGB1 cytoplasmic translocation in liver cells in this animal model. These results have potential clinical application in acute liver failure treatment.
Footnotes
Supported by the National Natural Science Foundation of China, No. 81160065.
Institutional review board statement: The study was reviewed and approved by Zhejiang hospital Institutional Review Board.
Institutional animal care and use committee statement: All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of Zhejiang Hospital (IACUC protocol number: 126).
Conflict-of-interest statement: We declare that there are no conflicts of interest to disclose.
Data sharing statement: No additional data are available.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: May 29, 2015
First decision: June 25, 2015
Article in press: October 13, 2015
P- Reviewer: Cignarelli M S- Editor: Qi Y L- Editor: Rutherford A E- Editor: Wang CH
References
- 1.Stravitz RT, Kramer DJ. Management of acute liver failure. Nat Rev Gastroenterol Hepatol. 2009;6:542–553. doi: 10.1038/nrgastro.2009.127. [DOI] [PubMed] [Google Scholar]
- 2.Malhi H, Gores GJ. Cellular and molecular mechanisms of liver injury. Gastroenterology. 2008;134:1641–1654. doi: 10.1053/j.gastro.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atillasoy E, Berk PD. Fulminant hepatic failure: pathophysiology, treatment, and survival. Annu Rev Med. 1995;46:181–191. doi: 10.1146/annurev.med.46.1.181. [DOI] [PubMed] [Google Scholar]
- 4.Tavares Fortuna JF, Lourdes Pratas M. Coarctation of the umbilical cord: a cause of intrauterine fetal death. Int J Gynaecol Obstet. 1978;15:469–473. doi: 10.1002/j.1879-3479.1977.tb00735.x. [DOI] [PubMed] [Google Scholar]
- 5.Wu Z, Han M, Chen T, Yan W, Ning Q. Acute liver failure: mechanisms of immune-mediated liver injury. Liver Int. 2010;30:782–794. doi: 10.1111/j.1478-3231.2010.02262.x. [DOI] [PubMed] [Google Scholar]
- 6.Melendez AJ. Sphingosine kinase signalling in immune cells: potential as novel therapeutic targets. Biochim Biophys Acta. 2008;1784:66–75. doi: 10.1016/j.bbapap.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 7.Abdin AA. Targeting sphingosine kinase 1 (SphK1) and apoptosis by colon-specific delivery formula of resveratrol in treatment of experimental ulcerative colitis in rats. Eur J Pharmacol. 2013;718:145–153. doi: 10.1016/j.ejphar.2013.08.040. [DOI] [PubMed] [Google Scholar]
- 8.Zhang W, Mottillo EP, Zhao J, Gartung A, VanHecke GC, Lee JF, Maddipati KR, Xu H, Ahn YH, Proia RL, et al. Adipocyte lipolysis-stimulated interleukin-6 production requires sphingosine kinase 1 activity. J Biol Chem. 2014;289:32178–32185. doi: 10.1074/jbc.M114.601096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Melendez AJ, Harnett MM, Pushparaj PN, Wong WS, Tay HK, McSharry CP, Harnett W. Inhibition of Fc epsilon RI-mediated mast cell responses by ES-62, a product of parasitic filarial nematodes. Nat Med. 2007;13:1375–1381. doi: 10.1038/nm1654. [DOI] [PubMed] [Google Scholar]
- 10.Lufrano M, Jacob A, Zhou M, Wang P. Sphingosine kinase-1 mediates endotoxemia-induced hyperinflammation in aged animals. Mol Med Rep. 2013;8:645–649. doi: 10.3892/mmr.2013.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou RR, Zhao SS, Zou MX, Zhang P, Zhang BX, Dai XH, Li N, Liu HB, Wang H, Fan XG. HMGB1 cytoplasmic translocation in patients with acute liver failure. BMC Gastroenterol. 2011;11:21. doi: 10.1186/1471-230X-11-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang M, Xu S, Han Y, Cao X. Apoptotic cells attenuate fulminant hepatitis by priming Kupffer cells to produce interleukin-10 through membrane-bound TGF-β. Hepatology. 2011;53:306–316. doi: 10.1002/hep.24029. [DOI] [PubMed] [Google Scholar]
- 13.Hao J, Huang YM, Zhao MH, Chen M. The interaction between C5a and sphingosine-1-phosphate in neutrophils for antineutrophil cytoplasmic antibody mediated activation. Arthritis Res Ther. 2014;16:R142. doi: 10.1186/ar4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology. 2000;32:734–739. doi: 10.1053/jhep.2000.17687. [DOI] [PubMed] [Google Scholar]
- 15.Chen L, Ren F, Zhang H, Wen T, Piao Z, Zhou L, Zheng S, Zhang J, Chen Y, Han Y, et al. Inhibition of glycogen synthase kinase 3β ameliorates D-GalN/LPS-induced liver injury by reducing endoplasmic reticulum stress-triggered apoptosis. PLoS One. 2012;7:e45202. doi: 10.1371/journal.pone.0045202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leifeld L, Dumoulin FL, Purr I, Janberg K, Trautwein C, Wolff M, Manns MP, Sauerbruch T, Spengler U. Early up-regulation of chemokine expression in fulminant hepatic failure. J Pathol. 2003;199:335–344. doi: 10.1002/path.1298. [DOI] [PubMed] [Google Scholar]
- 17.Bustin M. Regulation of DNA-dependent activities by the functional motifs of the high-mobility-group chromosomal proteins. Mol Cell Biol. 1999;19:5237–5246. doi: 10.1128/mcb.19.8.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 19.Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ. HMG-1 as a mediator of acute lung inflammation. J Immunol. 2000;165:2950–2954. doi: 10.4049/jimmunol.165.6.2950. [DOI] [PubMed] [Google Scholar]
- 20.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 21.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 22.Harris HE, Raucci A. Alarmin(g) news about danger: workshop on innate danger signals and HMGB1. EMBO Rep. 2006;7:774–778. doi: 10.1038/sj.embor.7400759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 24.Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, Janson A, Kokkola R, Zhang M, Yang H, et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med. 2000;192:565–570. doi: 10.1084/jem.192.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levy RM, Mollen KP, Prince JM, Kaczorowski DJ, Vallabhaneni R, Liu S, Tracey KJ, Lotze MT, Hackam DJ, Fink MP, et al. Systemic inflammation and remote organ injury following trauma require HMGB1. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1538–R1544. doi: 10.1152/ajpregu.00272.2007. [DOI] [PubMed] [Google Scholar]
- 26.Sundén-Cullberg J, Norrby-Teglund A, Rouhiainen A, Rauvala H, Herman G, Tracey KJ, Lee ML, Andersson J, Tokics L, Treutiger CJ. Persistent elevation of high mobility group box-1 protein (HMGB1) in patients with severe sepsis and septic shock. Crit Care Med. 2005;33:564–573. doi: 10.1097/01.ccm.0000155991.88802.4d. [DOI] [PubMed] [Google Scholar]
- 27.Gibot S, Massin F, Cravoisy A, Barraud D, Nace L, Levy B, Bollaert PE. High-mobility group box 1 protein plasma concentrations during septic shock. Intensive Care Med. 2007;33:1347–1353. doi: 10.1007/s00134-007-0691-2. [DOI] [PubMed] [Google Scholar]
- 28.Angus DC, Yang L, Kong L, Kellum JA, Delude RL, Tracey KJ, Weissfeld L. Circulating high-mobility group box 1 (HMGB1) concentrations are elevated in both uncomplicated pneumonia and pneumonia with severe sepsis. Crit Care Med. 2007;35:1061–1067. doi: 10.1097/01.CCM.0000259534.68873.2A. [DOI] [PubMed] [Google Scholar]
- 29.Yasuda T, Ueda T, Takeyama Y, Shinzeki M, Sawa H, Nakajima T, Ajiki T, Fujino Y, Suzuki Y, Kuroda Y. Significant increase of serum high-mobility group box chromosomal protein 1 levels in patients with severe acute pancreatitis. Pancreas. 2006;33:359–363. doi: 10.1097/01.mpa.0000236741.15477.8b. [DOI] [PubMed] [Google Scholar]
- 30.Goldstein RS, Gallowitsch-Puerta M, Yang L, Rosas-Ballina M, Huston JM, Czura CJ, Lee DC, Ward MF, Bruchfeld AN, Wang H, et al. Elevated high-mobility group box 1 levels in patients with cerebral and myocardial ischemia. Shock. 2006;25:571–574. doi: 10.1097/01.shk.0000209540.99176.72. [DOI] [PubMed] [Google Scholar]