

Abstract
As some of the oldest organic chemical reactions known, the ionic additions of elemental halogens such as bromine and chlorine to alkenes are prototypical examples of stereospecific reactions, typically delivering vicinal dihalides resulting from anti-addition. Whilst the invention of enantioselective variants is an ongoing challenge, the ability to overturn the intrinsic anti-diastereospecificity of these transformations is also a largely unsolved problem. In this Article, we describe the first catalytic, syn-stereospecific dichlorination of alkenes, employing a group transfer catalyst based on a redox-active main group element (i.e., selenium). Thus, with diphenyl diselenide (PhSeSePh) (5 mol %) as the pre-catalyst, benzyltriethylammonium chloride (BnEt3NCl) as the chloride source, and an N-fluoropyridinium salt as the oxidant, a wide variety of functionalized cyclic and acyclic 1,2-disubstituted alkenes, including simple allylic alcohols, deliver syn-dichlorides with exquisite stereocontrol. This methodology is expected to find applications in streamlining the synthesis of polychlorinated natural products such as the chlorosulfolipids.
Since the seminal report of the 1,2-addition of molecular chlorine to carbon-carbon double bonds in 1877 (i.e., the dearomatizing addition of two equivalents of Cl2 to 1,5-dichloronaphthalene),1 the vicinal dichlorination of alkenes has continued to challenge the ingenuity of synthetic organic chemists in providing creative solutions to fundamental problems of selectivity. Owing largely to the high reactivity of Cl2 and the difficulties associated with controlling the stoichiometry of a gaseous reactant, the reactions of alkenes with elemental chlorine are frequently plagued by side reactions (ionic and/or radical),2 and the extremely toxic and corrosive nature of Cl2 gas renders it experimentally unappealing. Accordingly, somewhat milder and more practical electrophilic chlorinating agents for alkene dichlorination have been developed, including SO2Cl2,3 PhICl2,4 Et4NCl35 (Mioskowski’s reagent) and 2:1 NCS-PPh36 (Yoshimitsu’s reagent). Alternatively, Cl2 (or its formal equivalent) may be generated in-situ from the oxidation of chloride sources with strong oxidants, and reagent systems such as H2O2-HCl,7 KMnO4-Me3SiCl-BnEt3NCl8 (Markó-Maguire reagent) and Oxone®-NaCl9 have been tailored for this purpose.
However, whereas the advent of new reagents for alkene dichlorination has largely solved the practicality and reactivity issues surrounding the use of Cl2, solutions have been less forthcoming to the problems of control over the relative and absolute configurations of the dichloride products. In recent years, the state-of-the-art in stereoselective chlorination methods have been showcased in synthetic efforts toward the chlorosulfolipids (e.g., 1–5)10,11,12,13,14 – a class of stereochemically-complex, polychlorinated natural products isolated from marine sources (Figure 1, left). The daunting synthetic challenge of constructing such densely functionalized arrays of chlorinated stereogenic centers has provided impetus for the study of (external) diastereocontrol in the dichlorination of chiral alkene substrates,12 as well as more recent efforts to effect enantioselective dichlorinations of allylic alcohols.15
Figure 1. The vicinal dichloride motif in natural products.
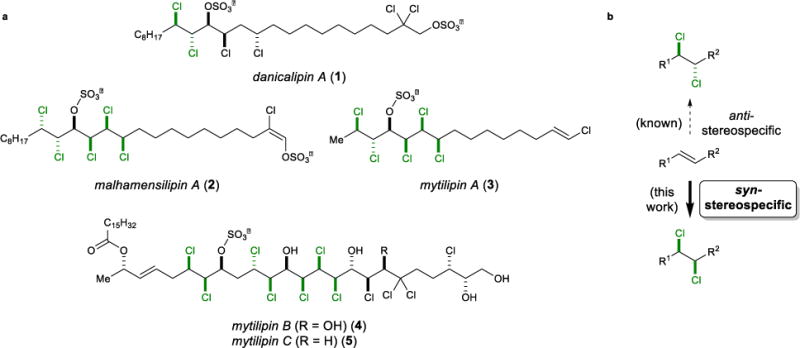
a, Selected chlorosulfolipids (with vicinal dichloride motifs highlighted). b, Most dichlorinations of alkenes (and all of those that are catalytic) exhibit anti-stereospecificity, and no generally applicable, direct syn-stereospecific alkene dichlorination has been reported. Such a method would complement current chlorination strategies employed in the synthesis of polychlorinated natural products such as the chlorosulfolipids.
However, almost invariably, all of the aforementioned reagents or reagent combinations that react via ionic reaction pathways afford vicinal dichloride products resulting from stereospecific anti-addition to alkenes – an inescapable stereoelectronic consequence of the nucleophilic attack of chloride ion on chloriranium ion16 or alkene-Cl2 π-complex17 intermediates. A complementary, direct syn-dichlorination is notably lacking from the synthetic chemist’s repertoire (Figure 1, right). Although several examples of direct, syn-stereospecific dichlorinations involving the treatment of unfunctionalized alkenes (often in excess) with stoichiometric amounts of high-valent metal chlorides based on antimony18 and molybdenum19,20,21 are on record, the harsh Lewis acidity, high oxidation potential, and toxicity of these reagents detracts significantly from their utility. Consequently, the only current solution to the syn-dichlorination problem with functionalized alkene substrates is to orchestrate indirect, two step oxidation-deoxochlorination sequences via the intermediacy of epoxides22,23 or chlorohydrins.24
In this work, we describe the first catalytic, syn-stereospecific dichlorination of alkenes, employing a group transfer catalyst based on a redox-active main group element (i.e., selenium). On the basis of the known ability of PhSeCl3 to chloroselenylate alkenes in an anti-stereospecific fashion,25,26 and the invertive nucleophilic displacement of the high-valent selenium(IV) moiety27 by chloride ions in such adducts to afford syn-dichlorides,28,29 we surmised that a direct alkene syn-dichlorination that is catalytic in selenium may be possible. However, a key challenge in formulating such a catalytic cycle is the identification of a suitable stoichiometric oxidant to regenerate Se(IV) from Se(II). Although electrophilic chlorine sources may seem obvious candidates, both Sharpless30 and Tunge31 have reported that the selenium-catalyzed reaction of alkenes with N-chlorosuccinimide generates allylic chlorides as the major products. Alternatively, a non-chlorine-based oxidant could be employed in the presence of an exogenous chloride source. Specifically, the oxidant selected must fulfill the following criteria: (1) it must not react (or must react only very slowly) with the alkene substrate, (2) it must not oxidize chloride ions to molecular Cl2 or any other active “Cl+” equivalent over the timescale of the reaction, (3) it must not contain or release nucleophiles that might outcompete chloride in the reaction, and (4) it must not lead to the formation of selenoxide intermediates that are capable of rapid syn-elimination.32 With these restrictions in mind, cationic N–F reagents were considered as possible candidates – a choice which was inspired by the use of “F+” reagents as oxidants in transition metal-catalyzed processes33 and, more significantly, in a PhSeSePh-catalyzed allylic amination of alkenes.34
Results
Reaction Development
To probe the feasibility of a syn-dichlorination process that is catalytic in selenium, orienting experiments were carried out employing cyclohexene 6 (1.0 equiv) as the substrate, 5 mol % of PhSeSePh as the pre-catalyst, n-Bu4NCl as the chloride source (3.0 equiv), and N-fluoropyridinium tetrafluoroborate 11 (CAS# 107264-09-5) as the oxidant, with MeCN-d3 as the solvent (Table 1). 1,1,2,2-Tetrachloroethane (1.0 equiv) was added as an internal standard, and 1H NMR peaks for species 6–10 were compared against authentic samples in MeCN-d3. Under these conditions at ambient temperature for ca. 20 h, an encouraging 19% yield of the desired syn-dichloride 7 was observed, although 50% of starting material 6 remained (Table 1, entry 1). It was surmised that fluoride ions generated as a by-product from oxidant 11 may be interfering with catalytic turnover, and that the addition of a fluoride scavenger may be advantageous. Chlorotrimethylsilane (Me3SiCl) was selected for this purpose on the basis that silicon has a high affinity for fluoride35 and that the by-products of such scavenging would simply be additional chloride ion and unreactive fluorotrimethylsilane (Me3SiF). Gratifyingly, the addition of 1.0 equiv of Me3SiCl gave a substantial improvement in catalytic turnover (61% yield of 7 by 1H NMR spectroscopic analysis) (entry 2), and complete consumption of alkene 6 occurred when 2.0 equiv of Me3SiCl was added, affording 7 in 81% yield (entry 3). However, increasing the amount of Me3SiCl to 3.0 equiv led to no further enhancement (entry 4). The reaction also proved sensitive to the quantity of n-Bu4NCl – an attempt to lower the amount to 2.5 equiv led to an erosion in the yield of 7 (entry 5) and none of 7 was produced when the n-Bu4NCl was omitted altogether (entry 6). The effect of solvent was also briefly investigated, with both CD2Cl2 and THF-d8 giving inferior results (entries 7 and 8). Pleasingly, the reaction could also be carried out with Selectfluor® 12 as an alternative stoichiometric oxidant, albeit with a slightly diminished yield of 7 (71%) (entry 9). Finally, n-Bu4NCl was replaced with BnEt3NCl, which is far less expensive, and the results were essentially identical (entry 10). Notably, in no case was any of the diastereomeric anti-dichloride product detected; an observation which is particularly surprising given that Selectfluor® 12 is known to oxidize chloride ions to a “Cl+” equivalent (possibly Cl2) in MeCN.36 In fact, a control experiment with cyclohexene 6 in which PhSeSePh was omitted from the reaction gave approximately 35% yield of the anti-dichloride after 20 h, implying that a background anti-dichlorination process is possibly operative but is significantly slower than the catalyzed reaction.
Table 1.
Reaction Development with Cyclohexene
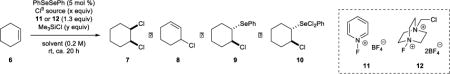
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| entry | Cl− source (equiv) | oxidant | Me3SiCl equiv | solvent | NMR yield (%) a
|
||||
| 6 | 7 | 8 | 9 | 10 | |||||
| 1 | n-Bu4NCl (3.0) | 11 | 0.0 | MeCN-d3 | 50 | 19 | 3 | 9 | 0 |
| 2 | n-Bu4NCl (3.0) | 11 | 1.0 | MeCN-d3 | 12 | 61 | 10 | 10 | 0 |
| 3 | n-Bu4NCl (3.0) | 11 | 2.0 | MeCN-d3 | 0 | 81 | 10 | 0 | 0 |
| 4 | n-Bu4NCl (3.0) | 11 | 3.0 | MeCN-d3 | 0 | 81 | 8 | 0 | 0 |
| 5 | n-Bu4NCl (2.5) | 11 | 2.0 | MeCN-d3 | 0 | 74 | 10 | 0 | 0 |
| 6b | n-Bu4NCl (0.0) | 11 | 2.0 | MeCN-d3 | 54 | 0 | 2 | 0 | 8 |
| 7 | n-Bu4NCl (3.0) | 11 | 2.0 | CD2Cl2 | 0 | 73 | 12 | 4 | 0 |
| 8 | n-Bu4NCl (3.0) | 11 | 2.0 | THF-d8 | 55 | 17 | 2 | 0 | 0 |
| 9 | n-Bu4NCl (3.0) | 12 | 2.0 | MeCN-d3 | 0 | 71 | 10 | 0 | 0 |
| 10 | BnEt3NCl (3.0) | 11 | 2.0 | MeCN-d3 | 0 | 83 | 10 | 0 | 0 |
Measured by 1H NMR spectroscopy with 1,1,2,2-tetrachloroethane (1.0 equiv) as an internal standard.
11% of an unidentified species was also observed by 1H NMR spectroscopy.
Prior to further optimization efforts aimed at minimizing the formation of allylic chloride 8, it was first necessary to establish the mechanistic provenance of this particular species. Although 8 is a known by-product in the reaction of 6 with Cl2 under both ionic and radical reaction manifolds,2 the absence of any anti-dichloride under our conditions argues against the involvement of molecular Cl2. Another possibility is synperiplanar elimination of a selenoxide intermediate32 derived from the hydrolysis of 10 with adventitious water, although both rigorously dry conditions and the addition of trace water had no effect on the 7/8 ratio. Lastly, antiperiplanar E2 elimination of the Se(IV) moiety in intermediate 10 could also furnish 8. To unambiguously determine which elimination pathway is in operation, an acyclic alkene substrate capable of generating (E)- or (Z)-configured vinylic chloride by-products was considered (N.B. for cyclohexene 6, antiperiplanar elimination to give a vinylic chloride is geometrically impossible from 10). Dichlorination of (E)-tert-butyl(hex-4-en-1-yloxy)diphenylsilane 13 under the optimized conditions gave a 68:23:9 mixture of syn-dichloride 14, vinylic chlorides 15 + 16, and two (tentatively assigned) allylic chlorides, respectively, with 14 being isolated in 63% yield (>99:1 dr) and 15 + 16 in 14% combined yield (Figure 2). Crucially, the configurations within 15 and 16, as deduced via 1D NOESY experiments (reciprocal enhancements shown in blue), were found to be consistent with their formation via an antiperiplanar E2 elimination process (see lower section of Figure 2). Similar conclusions were drawn from an analogous experiment with the (Z)-isomer of alkene 13, which also gave vinylic chloride by-products consistent with antiperiplanar E2 elimination (see Supplementary Information).
Figure 2. Identification of E2 elimination by-products.
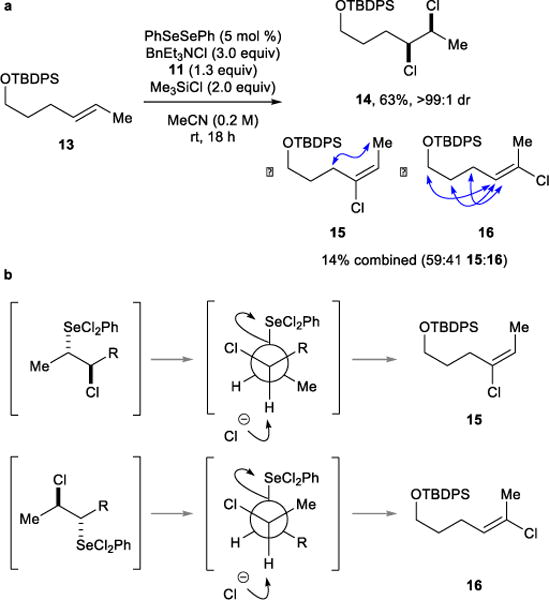
a, Formation of vinylic chloride by-products 15 and 16 from the dichlorination of alkene 13 and determination of their configurations via 1D NOESY experiments (reciprocal enhancements highlighted in blue). b, Mechanistic rationale for the formation of vinyl chlorides 15 and 16 from constitutionally isomeric anti-chloroselenylated intermediates via antiperiplanar elimination. TBDPS = tert-butyldiphenylsilyl.
With this information in hand, additional optimization experiments to minimize the competitive elimination process were conducted on (E)-1-benzyloxyl-4-hexene 17 as the substrate (Table 2). Interestingly, a three-fold rate acceleration was noted when sulfolane was used as a co-solvent (1:3 v/v) with MeCN-d3, although the extent of E2 elimination was unperturbed (compare entries 1 and 2). Based on this observation, we elected to screen a variety of Lewis basic additives (1.0 equiv) in order to search for similar acceleration effects (entries 3–8). Although the amount of elimination consistently proved insensitive to the presence of these additives, the rate enhancement effect appeared to be general, and 2,6-lutidine N-oxide 23 was identified as the optimal additive in this respect (entry 8).
Table 2.
Reaction Development with (E)-1-Benzyloxyl-4-hexene
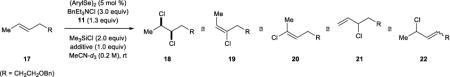
| |||||
|---|---|---|---|---|---|
| entry | (ArylSe)2 | additive | time (h) | 18:(19+20+21+22)a | 18 dra |
| 1 | PhSeSePh | – | 6 | 80:20 | 99:1 |
| 2 | PhSeSePh | sulfolaneb | 2 | 80:20 | 99:1 |
| 3 | PhSeSePh | HMPA | 3 | 82:18 | 98:2 |
| 4 | PhSeSePh | DMPU | 3.5 | 80:20 | 98:2 |
| 5 | PhSeSePh | DMI | 2.5 | 80:20 | 99:1 |
| 6 | PhSeSePh | Ph3P=O | 3.5 | 80:20 | 98:2 |
| 7 | PhSeSePh | pyridine N-oxide | 2.5 | 80:20 | 98:2 |
| 8 | PhSeSePh | 2,6-lutidine N-oxide 23 | 2 | 80:20 | 99:1 |
| 9 |
|
– | 10 | 58:42 | 88:18 |
| 10 |
|
– | 18 | 59:41 | 55:45 |
| 11 |
|
– | 3.5 | 90:10 | 99:1 |
| 12 |
|
– | 8 | 83:17 | 98:2 |
Measured by 1H NMR spectroscopy.
Sulfolane was used as a co-solvent with MeCN-d3 in 1:3 v/v. HMPA = hexamethylphosphoramide; DMPU = N,N′-dimethylpropyleneurea; DMI = N,N′-dimethyl-2-imidazolidinone.
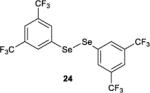
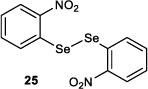
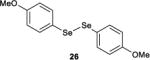
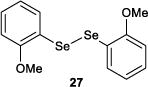
The effect of tuning the electronic nature of the aryl group on the selenium pre-catalyst was also examined (entries 9–12). Interestingly, electron-deficient pre-catalysts 24 and 25 gave longer reaction times, favored E2 elimination processes, and significantly eroded the syn:anti dichlorination ratio (entries 9 and 10). On the other hand, the electron-rich 4-methoxyphenyl pre-catalyst 26 behaved in precisely the opposite sense, giving reduced reaction times, less E2 elimination, and near complete syn-diastereoselectivity (entry 11). In contrast, the 2-methoxyphenyl pre-catalyst 27 behaved similarly to PhSeSePh, albeit with a slightly extended reaction time (entry 12).
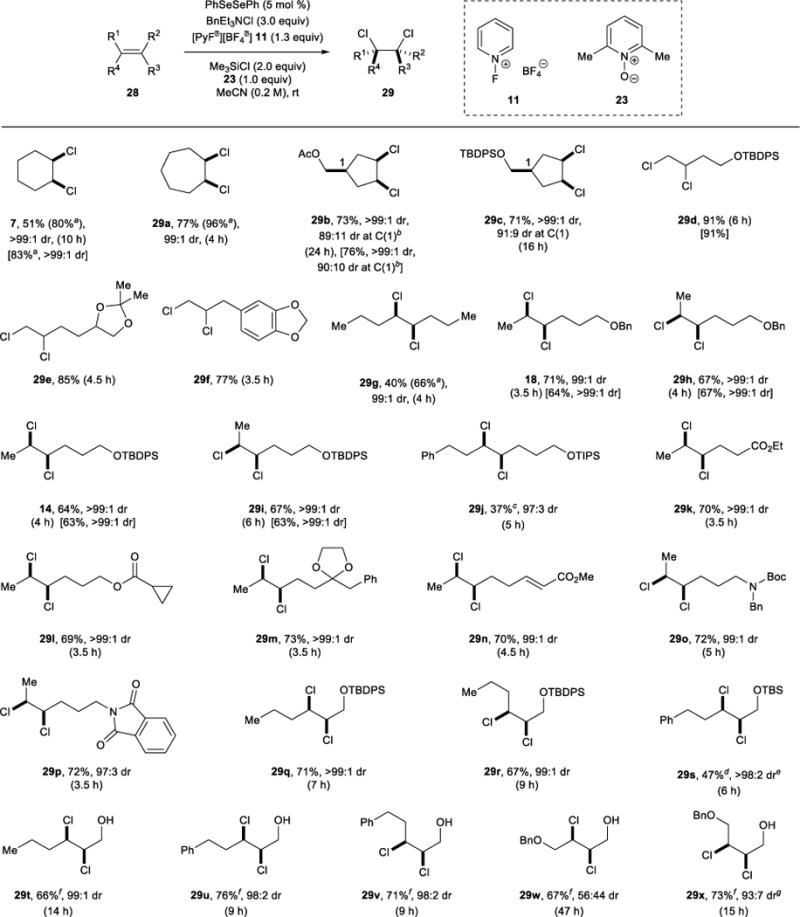
Reaction Generality
The generality of the catalytic syn-dichlorination with a range of alkene substrates was next surveyed (Table 3). Despite the fact that our optimization studies had identified 26 as the optimal pre-catalyst, its ability to reduce elimination by-products (relative to PhSeSePh) did not prove to be general with other substrates (see Supplementary Information). Consequently, PhSeSePh was employed in all preparative scale reactions. To take advantage of the rate enhancement imparted by 23, this additive was included in all preparative reactions, with the exception of allylic alcohols 28u–y, for which reduced conversions and the formation of unidentified by-products were observed in the presence of 23. However, the inclusion of 23 is not mandatory for the success of the reactions and it can generally be omitted without detriment to yields or diastereoselectivities, albeit at the expense of reaction rates (for comparison, the yields and diastereoselectivities for reactions conducted for 18 h without 23 are given in square brackets for several substrates).
Table 3.
Reaction Generality.
|
For comparison, yields and diastereoselectivities for selected reactions conducted for 18 h without additive 23 are given in square brackets. Each diastereomeric ratio (dr) was determined on the crude mixture after passing through a short plug of silica gel.
Yield was determined by 1H NMR spectroscopy with 1,1,2,2-tetrachloroethane (1.0 equiv) as an internal standard.
91:9 dr at C(1) after isolation.
24% of the free alcohol syn-dichloride was also isolated, and tentatively assigned cyclized products were also observed in the crude product mixture.
21% of the free alcohol syn-dichloride was also isolated.
The dr corresponds to the sum of the dichlorides.
Additive 23 was omitted.
97:3 dr after isolation.
As representative cyclic alkenes, both cyclohexene 6 and cycloheptene 28a underwent efficient syn-dichlorination with >99:1 and 99:1 dr, respectively, with the disparity between the isolated yields and those calculated by 1H NMR spectroscopy being attributed to difficulties in product isolation. Cyclopentene derivatives 28b and 28c also participated, and the sense of diastereofacial selectivity in both cases was consistent with preferential attack of the selenium electrophile on the sterically more accessible alkene faces, culminating in net, contrasteric syn-dichlorinations. The relative configurations within both 29b and 29c were secured by chemical correlation to an epoxy alcohol of known configuration.
For acyclic alkenes, the reaction proved generally applicable to both terminal and 1,2-disubstituted olefins, although 1,1-disubstituted, trisubstituted, and aryl-conjugated alkenes afforded complex mixtures of products. Similarly, alkenes bearing pendant nucleophiles able to engage in cyclizations also formed complex mixtures (see Supplementary Information for a table of problematic substrates). Crucially, the stereospecific nature of the syn-dichlorination reaction was confirmed by reaction of various pairs of (E)- and (Z)-configured alkenes, in which both isomers could be dichlorinated with exceptional syn-diastereoselectivity (i.e., 17/28h, 13/28i, 28q/r, 28t/u). The relative configurations in each case were typically assigned by comparison to known (or analogous) dichlorides, or to authentic samples of the diastereomeric dichlorides prepared by an anti-selective dichlorination method. For acyclic vicinal dichlorides, the 3JHH coupling constant was diagnostic, with syn-configured dichlorides 14, 18, 29j–n and anti-configured dichlorides 29h,i,o,p giving 3JHH values of 3.0–3.3 and 6.5–6.6 Hz, respectively. A similar trend was noted for dichlorides such as 29q and 29r derived from allylic alcohol derivatives, for which the syn-configured isomer (29q) gave a 3JHH value of 2.2 Hz, whereas the anti-configured isomer (29r) gave a value of 6.3 Hz.
In terms of functional group compatibility, the reaction is tolerant of free hydroxyl groups (28t–x), benzyl ethers (17, 28h,w,x), tert-butyldiphenylsilyl (TBDPS) ethers (13, 28c,d,i,q,r), esters (28b,k,l,n), acetals (28e,m), simple or electron-rich arenes (e.g., 28f,j,s,u,v), cyclopropanes (28l), electron-poor alkenes (28n), tert-butoxy carbamates (28o), and imides (28p). However, silicon-based protecting groups other than TBDPS, including triisopropylsilyl (TIPS) (28j) and tert-butyldimethylsilyl (TBS) (28s), are partially cleaved under the reaction conditions. Whilst this methodology can accommodate silyl ether functionality at the allylic position (e.g., 28q–s), free allylic (primary) alcohols are also perfectly competent substrates (28t–x), providing a convenient functional handle for further elaboration. However, allylic secondary alcohols, as well as their silyl ether or O-acyl derivatives, afforded complex mixtures of products for reasons that are currently unclear (see Supplementary Information). Curiously, subjection of the (E)-configured 1,4-dioxygenated alkene 28w to the dichlorination conditions returned a 56:44 diastereomeric mixture of syn- and anti-dichlorides 29w, respectively, whereas the analogous (Z)-configured alkene 28x underwent syn-dichlorination with 93:7 diastereoselectivity.
Certain alkene substates unexpectedly returned dichloride products resulting from anti-addition upon exposure to the standard reaction conditions. Similarly, the presence of branching at the allylic position of acyclic (E)-alkenes led to incomplete conversion and ≥80:20 selectivity in favour of anti-addition (see Supplementary Information). The preference for anti-dichlorination in these cases is difficult to rationalize at present, and may be substrate dependent. A better understanding of this phenomenon will form part of a broader mechanistic investigation of the catalytic syn-dichlorination, to be reported in due course.
Discussion
A proposed catalytic cycle for the selenium-catalyzed syn-dichlorination process is outlined in Figure 3, albeit with omission of the 2,6-lutidine N-oxide additive 23. Initial oxidation of the PhSeSePh pre-catalyst with oxidant 11 in the presence of chloride ion could generate PhSeCl3 as the active catalyst, with Me3SiCl presumably serving to trap the 0.3 equivalents (w.r.t. alkene) of fluoride ion by-product as Me3SiF. Addition of PhSeCl3 to the alkene may then ensue, possibly via loss of chloride to generate PhSeCl2+ 30 as the active electrophile, which can then intercept the alkene to form seleniranium ion intermediate 31. Nucleophilic ring-opening of 31 with chloride ion would then furnish the (β-chloroalkyl)phenylselenium dichloride species 32,25,26 and subsequent invertive displacement of the Se(IV) nucleofuge with chloride ion28,29 would deliver the syn-dichloride product, releasing the selenium as PhSeCl. The dissociation of a chloride ligand from the selenium atom within 32 prior to nucleophilic displacement is a possibility, as this would enhance the nucleofugality of the selenium by rendering it cationic. The stereochemical outcome in the chlorodeselenylation step hinges on the fact that neighboring chloro substituents are comparatively reluctant to engage in anchimeric assistance.37
Figure 3. Proposed catalytic cycle for the selenium-catalyzed syn-dichlorination of alkenes.
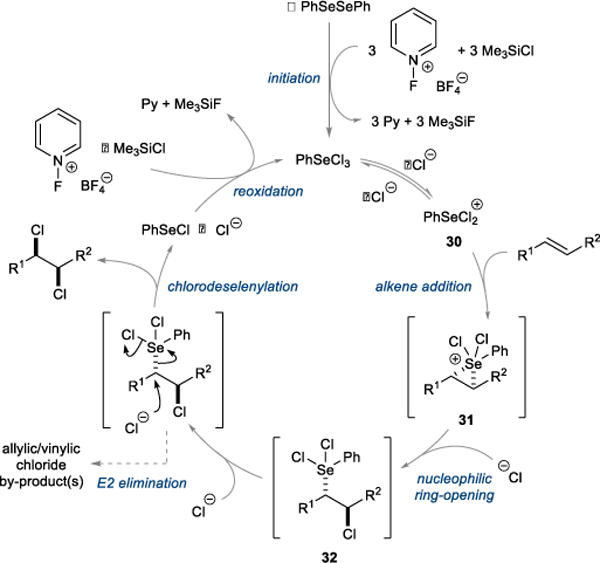
Following oxidation of the PhSeSePh pre-catalyst, the catalytic cycle commences with anti-chloroselenylation of the alkene to give 32. Invertive displacement of the selenium moiety with chloride ion furnishes the syn-dichloride product. Finally, reoxidation of Se(II) to Se(IV) closes the cycle. Py = pyridine.
To complete the cycle, oxidation of PhSeCl by 11 in the presence of chloride ions could regenerate the PhSeCl3 catalyst, although it is possible that addition of PhSeCl to the alkene could precede the oxidation of Se(II) to Se(IV). In any case, the mechanism of the reoxidation process requires further scrutiny, and the possibility that trace amounts of Cl2 (generated from the oxidation of Cl− by 11) may serve as the active oxidant cannot be ruled out at this stage.
The crucial role of Me3SiCl in the reaction also requires further investigation, but an initial hypothesis is that it may serve to trap fluoride ions (from the reduction of oxidant 11) that would otherwise interfere with catalytic turnover (N.B. Me3SiF was observed in reaction mixtures by 1H and 19F NMR spectroscopy). Specifically, it may be that halide dissociation (Cl− or F−) from PhSeX3 to generate the active PhSeX2+ electrophile (where X = Cl or F) is slower when the selenium center carries one or more fluoride ligands. In support of this assertion, it is known that the stoichiometric reaction of cyclohexene with PhSeF3 is slow, requiring 2–3 h to reach completion,38 whereas the analogous addition of PhSeCl3 requires only a few minutes.26
The observation of allylic or vinylic chloride by-products can be accounted for by a competitive E2 elimination between 32 and chloride ion, which is a weak but competent base in acetonitrile (pKa of HCl = 10.3 in MeCN39). The possibility that pyridine (generated from the reduction of oxidant 11) may serve as the base in this process cannot be excluded (pKa of pyridinium ion = 12.3 in MeCN40) although its concentration (particularly early in the reaction) will be much lower than chloride ion.
The role of the 2,6-lutidine N-oxide additive 23 is not clear, but a control experiment with alkene 17 in which the N-fluoropyridinium salt 11 was omitted (leaving 23 as the only potential oxidant for selenium) gave no reaction, indicating that 23 is not functioning as an oxidant. Similarly, omission of the PhSeSePh pre-catalyst gave no syn-dichloride, and only a slow background anti-dichlorination was observed (40% conversion after 20 h), verifying that the combination of BnEt3NCl, 11, and 23 alone is not capable of effecting syn-dichlorination via some other mechanistic pathway. As the turnover-limiting step of the catalytic cycle is yet to elucidated, it would be premature to speculate on the origin of the rate enhancement imparted by 23.
In summary, we have developed the first catalytic, syn-stereospecific dichlorination of alkenes, employing a group transfer catalyst based on a redox-active main group element (i.e., selenium). The method is applicable to a wide variety of functionalized cyclic and acyclic 1,2-disubstituted alkenes, including simple (primary) allylic alcohols, and is operationally simple to perform. As well as providing a convenient solution to the problem of direct alkene syn-dichlorination, this process could also form the basis of a conceptually new strategy for catalytic, enantioselective alkene dichlorination. Importantly, the absence of chloriranium ion intermediates can circumvent the site selectivity issue inherent in the nucleophilic trapping of such species with chloride ion: a recognised, and as yet unsolved, problem in controlling the enantioselectivity of dichlorinations of electronically-unbiased, (E)- or (Z)-configured alkenes.15
Methods
Full experimental details and characterization of compounds can be found in the Supplementary Information.
General Procedure for Catalytic syn-Dichlorination of Alkenes
The general procedure for the catalytic, syn-dichlorination of alkenes with 2,6-lutidine N-oxide as additive is as follows. In a typical experiment, an oven-dried, 10-mL Schlenk flask equipped with a magnetic stirrer bar was taken into the glovebox and charged sequentially with diphenyl diselenide (15.8 mg, 0.05 mmol, 5 mol %), benzyltriethylammonium chloride (690 mg, 3.03 mmol, 3.0 equiv), and N-fluoropyridinium tetrafluoroborate (240 mg, 1.30 mmol, 1.3 equiv), and was then sealed with a rubber septum, removed from the box, and placed under argon. MeCN (5.0 mL), 2,6-lutidine N-oxide (126 mg, 115 μL, 1.02 mmol, 1.0 equiv), and chlorotrimethylsilane (218 mg, 255 μL, 2.01 mmol, 2.0 equiv) were then added sequentially and stirring was commenced. After ca. 10 min, an off-white suspension was observed. At this point, the requisite alkene (1.00 mmol, 1.0 equiv) was transferred via syringe to the reaction mixture [for alkenes of unknown density, only 3.0 mL of MeCN was added initially and the remaining 2.0 mL (in two 1.0 mL portions) was used to transfer the alkene across from an oven-dried, 4-mL dram vial under argon, via syringe]. The resultant suspension was stirred at rt, and was monitored by TLC until no alkene substrate was detected. Once the reaction had reached completion, sat. aq. NaHCO3 (1.0 mL) was added to quench any unreacted chlorotrimethylsilane and stirred for ca. 10 min. The mixture was then transferred to a separatory funnel and diluted with H2O (15 mL). The aqueous layer was extracted with Et2O (3 × 15 mL), and the combined organic extracts were washed with brine (15 mL), then dried (MgSO4), filtered, and concentrated in vacuo (20–23 °C, ca. 20 mmHg for non-volatile products or 5–8 °C, ca. 20 mmHg for volatile products). The resultant residue was re-dissolved in Et2O (5.0 mL) and eluted through a short plug of silica gel (ca. 0.55 g SiO2 packed into a Pasteur pipet to a height of ca. 40 mm) in order to partially remove 2,6-lutidine N-oxide and any ammonium salts, and the plug was then rinsed through with further portions of Et2O (3 × 5 mL). The solvent was removed in vacuo (20–23 °C, ca. 20 mmHg for non-volatile products or 5–8 °C, ca. 20 mmHg for volatile products), and an aliquot of the crude mixture was dissolved in CDCl3 to measure the syn/anti diastereoisomeric ratio (dr) by 1H NMR spectroscopy. The syn-dichloride product was then isolated by flash column chromatography on silica gel and/or Kugelrohr distillation at reduced pressure. The procedure for the catalytic, syn-dichlorination of allylic alcohols and the preparations of the alkene substrates are presented in the Supplementary Section.
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of Health (GM R01-085235) for generous financial support. S. T.-C. E. thanks the Agency for Science, Technology, and Research of Singapore (A*STAR) for a postdoctoral fellowship.
Footnotes
Author Contributions
A. J. C. planned and carried out the experimental work and initial optimization. S. T.-C. E. completed the experimental work and final characterizations. S. E. D. directed and coordinated the project. A. J. C. wrote the manuscript with the assistance of the other authors.
The authors declare no competing financial interests.
Additional Information
Supplementary information accompanies this paper at www.nature.com/naturechemistry.
References
- 1.Atterberg A, Widman O. Neue Chlornaphtaline. Ber Dtsch Chem Ges. 1877;10:1841–1844. [Google Scholar]
- 2.Poutsma ML. Chlorination of unsaturated compounds in nonpolar media. Science. 1967;157:997–1005. doi: 10.1126/science.157.3792.997. [DOI] [PubMed] [Google Scholar]
- 3.Kharasch MS, Brown HC. Chlorinations with sulfuryl chloride. II The peroxide-catalyzed reaction of sulfuryl chloride with ethylenic compounds. J Am Chem Soc. 1939;61:3432–3434. [Google Scholar]
- 4.Tanner DT, Gidley GC. Mechanism of the addition of chlorine to olefins with iodobenzene dichloride. J Org Chem. 1968;33:38–43. [Google Scholar]
- 5.Schlama T, Gabriel K, Gouverneur V, Mioskowski C. Tetraethylammonium trichloride: a versatile reagent for chlorinations and oxidations. Angew Chem Int Ed Engl. 1997;36:2342–2344. [Google Scholar]
- 6.Kamada Y, Kitamura Y, Tanaka T, Yoshimitsu T. Dichlorination of olefins with NCS/Ph3P. Org Biomol Chem. 2013;11:1598–1601. doi: 10.1039/c3ob27345h. [DOI] [PubMed] [Google Scholar]
- 7.Ho TL, Gupta BGB, Olah GA. Synthetic methods and reactions; 39. Phase transfer catalyst promoted halogenation of alkenes with hydrohalic acid/hydrogen peroxide. Synthesis. 1977:676–677. [Google Scholar]
- 8.Markó IE, Richardson PR, Bailey M, Maguire AR, Coughlan N. Selective manganese-mediated transformations using the combination: KMnO4/Me3SiCl. Tetrahedron Lett. 1997;38:2339–2342. [Google Scholar]
- 9.Ren J, Tong R. Convenient in situ generation of various dichlorinating agents from oxone and chloride: diastereoselective dichlorination of allylic and homoallylic alcohol derivatives. Org Biomol Chem. 2013;11:4312–4315. doi: 10.1039/c3ob40670a. [DOI] [PubMed] [Google Scholar]
- 10.Nilewski C, Geisser RW, Carriera EM. Total synthesis of a chlorosulpholipid cytotoxin associated with seafood poisoning. Nature. 2009;547:573–576. doi: 10.1038/nature07734. [DOI] [PubMed] [Google Scholar]
- 11.Nilewski C, Carriera EM. Recent advances in the total synthesis of chlorosulfolipids. Eur J Org Chem. 2012:1685–1698. [Google Scholar]
- 12.Chung WJ, Vanderwal CD. Approaches to the chemical synthesis of the chlorosulfolipids. Acc Chem Res. 2014;47:718–728. doi: 10.1021/ar400246w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chung WJ, Carlson JS, Vanderwal CD. General approach to the synthesis of the chlorosulfolipids danicalipin A, mytilipin A, and malhamensilipin A in enantioenriched form. J Org Chem. 2014;79:2226–2241. doi: 10.1021/jo5000829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Umezawa T, Matsuda F. Recent progress toward synthesis of chlorosulfolipids: total synthesis and methodology. Tetrahedron Lett. 2014;55:3003–3012. [Google Scholar]
- 15.Nicolaou KC, Simmons NL, Ying Y, Heretsch PM, Chen JS. Enantioselective dichlorination of allylic alcohols. J Am Chem Soc. 2011;133:8134–8137. doi: 10.1021/ja202555m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roberts I, Kimball GE. The halogenation of ethylenes. J Am Chem Soc. 1937;59:947–948. [Google Scholar]
- 17.Poutsma ML. Chlorination studies of unsaturated materials in nonpolar media. IV. The ionic pathway for alkylated ethylenes. Products and relative reactivities. J Am Chem Soc. 1965;87:4285–4292. [Google Scholar]
- 18.Uemura S, Onoe A, Okano M. The chlorination of olefins with antimony(V) chloride. Bull Chem Soc Jpn. 1974;47:692–697. [Google Scholar]
- 19.Uemura S, Onoe A, Okano M. Molybdenum(V) chloride as a reagent for cis chlorination of olefins. Bull Chem Soc Jpn. 1974;47:3121–3124. [Google Scholar]
- 20.San Filippo JS, Jr, Sowinski AF, Romano LJ. Chlorination of alkenes and alkynes with molybdenum(V) chloride. J Am Chem Soc. 1975;97:1599–1600. [Google Scholar]
- 21.Nugent WA. In situ-generated molybdenum(VI) reagent for cis-chlorination of alkenes. Tetrahedron Lett. 1978;19:3427–3430. [Google Scholar]
- 22.Yoshimitsu T, Fukumoto N, Tanaka T. Enantiocontrolled synthesis of polychlorinated hydrocarbon motifs: a nucleophilic multiple chlorination process revisited. J Org Chem. 2009;74:696–702. doi: 10.1021/jo802093d. [DOI] [PubMed] [Google Scholar]
- 23.Denton R, Tang X, Przeslak A. Catalysis of phosphorus(V)-mediated transformations: dichlorination reactions of epoxides under Appel conditions. Org Lett. 2010;12:4678–4681. doi: 10.1021/ol102010h. [DOI] [PubMed] [Google Scholar]
- 24.Nilewski C, Geisser RW, Ebert MO, Carriera EM. Conformational and configurational analysis in the study and synthesis of chlorinated natural products. J Am Chem Soc. 2009;131:15866–15876. doi: 10.1021/ja906461h. [DOI] [PubMed] [Google Scholar]
- 25.Garratt DG, Schmid GH. The addition of arylselenium trichlorides vs. areneselenenyl chlorides to cis- and trans-1-phenylpropene. Can J Chem. 1974;52:3599–3606. [Google Scholar]
- 26.Engman L. Phenylselenium trichloride in organic synthesis. Reaction with unsaturated compounds Preparation of vinylic chlorides via selenoxide elimination. J Org Chem. 1987;52:4086–4094. [Google Scholar]
- 27.Paulmier C. Inter and intramolecular nucleophilic substitution of activated phenylselanyl groups. Phosphorus Sulfur Silicon Relat Elem. 2001;172:25–54. [Google Scholar]
- 28.Morella AM, Ward DA. The cis chlorination of alkenes using selenium reagents. Tetrahedron Lett. 1984;25:1197–1200. [Google Scholar]
- 29.Morella AM, Ward DA. Cis 1,2-functionalization of cyclohexane using selenium intermediates. Tetrahedron Lett. 1985;26:2899–2900. [Google Scholar]
- 30.Hori T, Sharpless KB. Selenium-catalyzed nonradical chlorination of olefins with N-chlorosuccinimide. J Org Chem. 1979;44:4204–4208. [Google Scholar]
- 31.Tunge JA, Mellegaard SR. Selective selenocatalytic allylic chlorination. Org Lett. 2004;6:1205–1207. doi: 10.1021/ol036525o. [DOI] [PubMed] [Google Scholar]
- 32.Sharpless KB, Young MW, Lauer RF. Reactions of selenoxides: thermal syn-elimination and H218O exchange. Tetrahedron Lett. 1973;14:1979–1982. [Google Scholar]
- 33.Engle KM, Mei TS, Wang X, Yu JQ. Bystanding F+ oxidants enable selective reductive elimination from high-valent metal centers in catalysis. Angew Chem Int Ed. 2011;50:1478–1491. doi: 10.1002/anie.201005142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trenner J, Depken C, Weber T, Breder A. Direct oxidative allylic and vinylic amination of alkenes through selenium catalysis. Angew Chem Int Ed. 2013;52:8952–8956. doi: 10.1002/anie.201303662. [DOI] [PubMed] [Google Scholar]
- 35.Luo YR. Comprehensive handbook of chemical bond energies. CRC Press; Boca Raton, FL: 2007. [Google Scholar]
- 36.Syvret RG, Butt KM, Nguyen TP, Bulleck VL, Rieth RD. Novel process for generating useful electrophiles from common anions using selectfluor® fluorination agent. J Org Chem. 2002;67:4487–4493. doi: 10.1021/jo020053u. [DOI] [PubMed] [Google Scholar]
- 37.Grunwald E. Acetolysis rates of the cis- and trans-2-chloro- and 2-bromocyclohexyl p-bromobenzenesulfonates. J Am Chem Soc. 1951;73:5458–5459. [Google Scholar]
- 38.Lermontov SA, Xavorin SI, Bakhtin IV, Pushin AN, Zefirov NS, Stang PS. Fluorination of olefins with PhSeF3, PhSeF5 and PhTeF5. J Fluorine Chem. 1998;87:75–83. [Google Scholar]
- 39.Raamat E, et al. Acidities of strong neutral brønsted acids in different media. J Phys Org Chem. 2013;26:162–170. [Google Scholar]
- 40.Kaljurand I, Rodima T, Leito I, Koppel IA, Schwesinger R. Self-consistent spectrophotometric basicity scale in acetonitrile covering the range between pyridine and DBU. J Org Chem. 2000;65:6202–6208. doi: 10.1021/jo005521j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.