

Abstract
Background
Drug resistance is one of the most important causes for failure of anti-AIDS treatment. During therapy, multiple mutations accumulate in the HIV genome, eventually rendering the drugs ineffective in blocking replication of the mutant virus. The huge number of possible mutants precludes experimental analysis to explore the molecular mechanisms of resistance and develop improved antiviral drugs.
Results
In order to solve this problem, we have developed a new algorithm to reveal the most representative mutants from the whole drug resistant mutant database based on our newly proposed unified protein sequence and 3D structure encoding method. Mean shift clustering and multiple regression analysis were applied on genotype-resistance data for mutants of HIV protease and reverse transcriptase. This approach successfully chooses less than 100 mutants with the highest resistance to each drug out of about 10K in the whole database. When considering high level resistance to multiple drugs, the numbers reduce to one or two representative mutants.
Conclusion
This approach for predicting the most representative mutants for each drug has major importance for experimental verification since the results provide a small number of representative sequences, which will be amenable for in vitro testing and characterization of the expressed mutant proteins.
Background
AIDS (Acquired Immunodeficiency Syndrome) is one of the most severe pandemic diseases, and approximately 35.5 million people were infected in the year 2012 [1]. It has been almost three decades since the first case of AIDS was found in US and the cause of AIDS was identified as HIV (Human Immunodeficiency Virus) [2]. Currently, a total of 26 licensed drugs are used in anti-AIDS therapy [3]. These drugs target different steps during the HIV life cycle, including viral entry, reverse transcription, integration and maturation. HIV protease (PR) is the enzyme responsible for processing viral precursor proteins after budding of virus from the host cell during the maturation stage of the viral life cycle [4]. PR inhibitors block the proteolytic activity, preventing formation of the infectious virus [5,6]. HIV reverse transcriptase (RT) converts the viral RNA genome into DNA during the early stages of the HIV life cycle. The nucleoside analog zidovudine (AZT), which inhibits RT, was the first FDA approved anti-AIDS drug [7,8]. The HIV RT inhibitors can be categorized into two classes: Nucleotide analog reverse transcriptase inhibitors (NRTIs) and non-nucleoside reverse transcriptase inhibitors (NNRTIs). NRTIs are structural analogs of nucleotides, and compete with the enzyme's natural substrates during the reverse transcription step. NNRTIs specifically target a separate site on HIV-1 RT to decrease its enzymatic activities [9].
During typical anti-AIDS treatment, which is often referred to as highly active antiretroviral therapy (HAART), three or more antiretroviral drugs chosen from different categories are given to patients. Such treatment extends the lifespan of the patients [10].
However, since HIV is a member of the retrovirus family [11], its genomic information is carried by RNA [11,12]. Due to the lack of proofreading by RT [13] and the high replication rate of as many as 109 viral particles daily [14], drug resistance is one of the most severe challenges for successful long-term AIDS therapy [15,16]. Drug pressure causes the selection of resistant viral strains, which can replicate in the presence of drugs [17,18]. This drug resistance can cause the failure of antiviral therapy. Two strategies have been pursued to overcome the challenge of drug resistance. First, in the clinic, genotyping of the infecting virus is used to guide the choice of effective drugs for therapy. Drug resistance can be predicted from genotype data by a variety of algorithms [19-22], including our approach of applying a structure vector from Delaunay triangulation [23,24]. Second, research to understand the molecular mechanisms of drug resistance is important and could help in the design of new drugs for improved anti-AIDS therapy.
Several possible mechanisms have been described for drug resistance [25,26]. Laboratory studies can only be performed on a small number of mutants. However, a huge number of possible mutants can occur, since HIV has a high mutation rate of about 10-4 to 10-5 mutations per nucleotide and cycle of replication [26] and a naturally polymorphic genome. Taking HIV PR as an example, mutations of more than thirty different residues have been associated with PR inhibitors [16]. Moreover, multiple mutations accumulate as the virus evolves higher levels of resistance [27,28]. For instance, we have studied a PR mutant with 20 substitutions, which shows more than 1000-fold worse binding to inhibitors darunavir (DRV) and saquinavir (SQV) compared to wild-type PR [29]. Therefore, considering the huge number of possible mutants, can a tractably small number of mutants be identified as the most representative of high level resistance? Answering this question could save both time and money, and facilitate the study of drug resistant mechanisms.
One approach to selecting a small number of meaningful mutants uses the Mean shift clustering, which was first introduced in 1975 by Fukunaga and Hostetler [30] for the purpose of seeking the mode of a density function in the given sample set. Fukunaga and Hostetler [30] also suggested that mean shift clustering is an instance of gradient ascent by using decreasing distance functions, which often referred as a kernel, from a given point to a point in the sample set. This algorithm became more widely used after 1995 when Cheng [31] developed a more generalized formulation. By clarifying the relationship between mean shift and the optimization, the algorithm could potentially be applied on clustering and global optimization problems by declaring each mode as representative of one cluster and assigning each data point to the mode it converges to. Applications of the mean shift algorithm range over image/video segmentation, image representation/retrieval, discontinuity-preserving smoothing [32,33], higher level tasks like appearance-based clustering [34,35], tracking including blob tracking [36] and face tracking [37], shape detection and recognition [38]. Subsequently, applications of this algorithm were extended to other fields like biology. These applications include analysis of structural variation in genomes [39], DNA microarray analysis [40], and time-warped gene expression analysis [41].
In this paper, we have proposed a new algorithm based on the non-parametric iterative mean shift and our recently reported protein encoding method to extract the most representative drug resistant mutants from the Stanford HIV database [42].
Results
Mean shift clustering, multiple regression and quantile analysis were performed on the data for both HIV-1 PR and RT mutants whose sequences and structures were encoded by Delaunay triangulation.
Mean shift clustering on HIV protease inhibitor resistance
After each of the mutated sequences was represented by a 210-dimensional vector, we performed the mean shift clustering on the drug resistance data to select the most representative mutants. Data were analyzed for the PR inhibitors atazanavir (ATV), nelfinavir (NFV), ritonavir (RTV), indinavir (IDV), lopinavir (LPV), tipranvir (TPV) and saquinavir (SQV). The results show that the larger the bandwidth, the smaller number of mutants was selected. The plot for the PR inhibitor ATV is given as a representative example in Figure 1.
Figure 1.
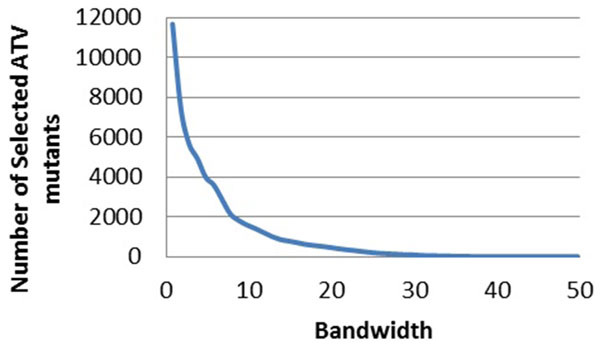
The relationship between the bandwidths and the number of selected mutants for PIs. The bandwidth is plotted against the number of selected mutants. The trend line is shown in blue. Plots show regression for drug resistance to ATV.
Mean shift clustering on HIV reverse transcriptase inhibitor resistance
Similarly, mean shift clustering was performed on the drug resistance data for HIV-1 RT inhibitors. The bandwidth and the selected numbers of mutants are compared for the RT inhibitors, including the NRTIs lamivudine (3TC) (Figure 2), abacavir (ABC), zidovudine (AZT), stavudine (D4T), didanosine (DDI) and tenofovir (TDF) (Figure 2), and the NNRTIs nevirapine (NPV) (Figure 3), delaviridine (DLV), and efavirenz (EFV).
Figure 2.
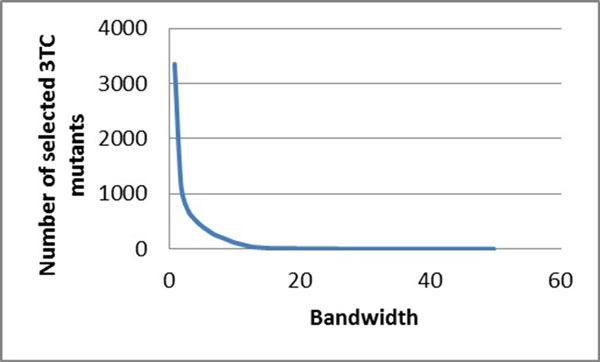
The relationship between the bandwidths and the number of selected mutants for NRTIs. The bandwidth is plotted against the number of selected mutants. The trend line is shown in blue. Plots show regression for drug resistance to 3TC.
Figure 3.
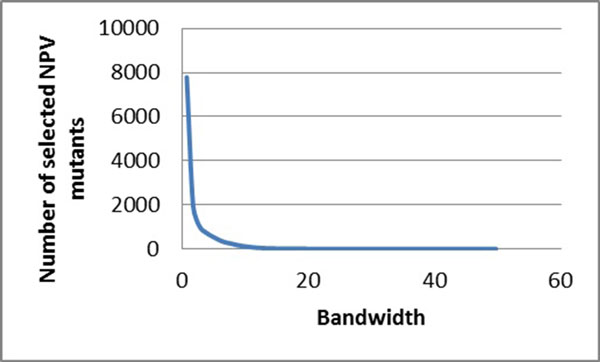
The relationship between the bandwidths and the number of selected mutants for NNRTIs. The bandwidth is plotted against the number of selected mutants. The trend line is shown in blue. Plots show regression for drug resistance to NPV.
Multiple regression on HIV protease inhibitor resistance
Afterwards, a multiple regression was applied to the selected mutants to evaluate the selected results. The R2 values for relative resistance were plotted against the number of selected mutants as shown in (Figure 4) for the PR inhibitors ATV, NFV, RTV, IDV, LPV, TPV and SQV. The x-axis is the number of selected mutants, while the y-axis is the R2 value after applying multiple linear regression on selected protein sequences with their drug resistant values.
Figure 4.
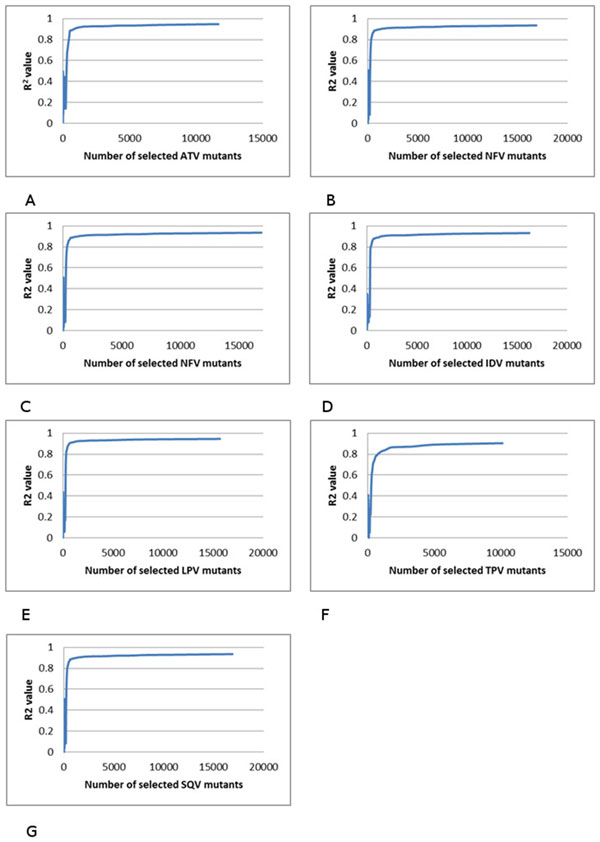
The relationship between the multiple regression results and the number of selected mutants. The R2 value is plotted against the number of selected mutants. The trend line is shown in blue. Plots show regression for resistance to drugs: (A) ATV, (B) NFV, (C) RTV, (D) IDV, (E) LPV, (F) TPV, and (G) SQV.
Multiple regression on HIV reverse transcriptase inhibitor resistance
Multiple regression analysis was performed similarly on genotype-phenotype data for drugs inhibiting HIV-1 RT. The R2 values for relative resistance were plotted against the number of selected mutants as shown in for the RT inhibitors including NRTIs 3TC, ABC, D4T, DDI, TDF and AZT (Figure 5), and NPV, DLV and EFV for NNRTIs (Figure 6).
Figure 5.
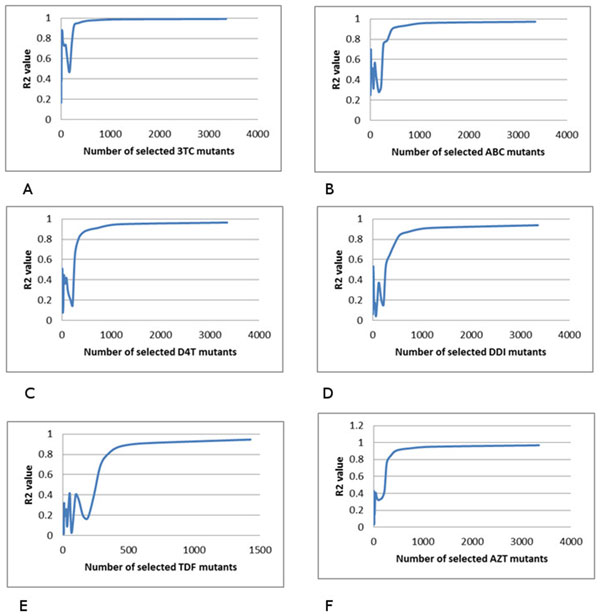
The relationship between the multiple regression results and the number of selected mutants. The R2 value is plotted against the number of selected mutants. The trend line is shown in blue. Plots show regression for resistance to drugs: (A) 3TC, (B) ABC, (C) D4T, (D) DDI, (E) TDF and (F) AZT.
Figure 6.
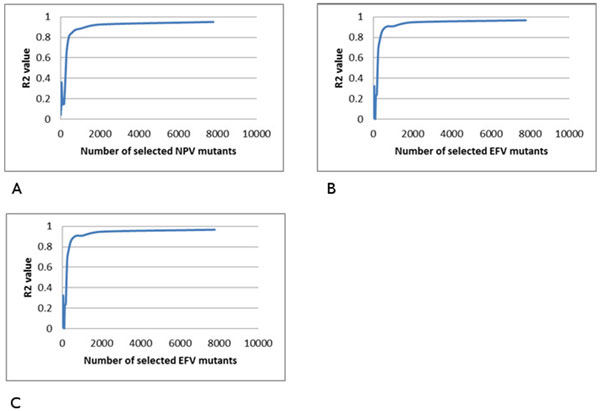
The relationship between the multiple regression results and the number of selected mutants. The R2 is plotted against the number of selected mutants. The trend line is shown in blue. Plots show regression for resistance to drugs: (A) NPV, (B) DLV and (C) EFV.
Bandwidth selection and multiple regression on HIV-1 PR and RT inhibitor resistance
The following experiments were performed to test the accuracy of the selected mutants with different R2 results. According to the results of the above experiments, it could be inferred that the larger the bandwidth is, the fewer representative mutants are selected, and therefore the R2 would be lowered. Moreover, with different R2 values, the selected mutants of lower R2 need to be a subset or have a large intersection with the selected mutants of the higher R2. Based on the above results, in this experiment, the higher R2 was set to be above 0.80 while the lower R2 was set to be 0.60. With this goal, the overlap group was then calculated.
The overlap group of mutants is a significant fraction of those selected for the lower R2, which suggests the procedure selects meaningful representative mutants correctly. The fractional overlap ranges from 0.79 to 0.94 for HIV PR inhibitors, and 0.89-0.94 for NNRIs (Tables 1, 2). For NRTIs, the results in Figure 5 show that when the number of resistant mutants increases, the R2 value does not increase smoothly. There are many ripples in the plots, making it difficult to select the R2 cutoff in this experiment. Therefore, analysis of NRTIs was not possible.
Table 1.
The number of selected mutants and R2 for HIV-1 PR Inhibitors.
| Selected for higher R2 | Higher R2 | Selected for lower R2 | Lower R2 | Overlap | Overlap Ratio (%) | |
|---|---|---|---|---|---|---|
| ATV | 412 | 0.8089 | 289 | 0.6004 | 260 | 89.97 |
| NFV | 353 | 0.8085 | 253 | 0.6246 | 235 | 92.89 |
| RTV | 281 | 0.8027 | 243 | 0.5915 | 228 | 93.83 |
| IDV | 420 | 0.8018 | 252 | 0.5749 | 217 | 86.11 |
| LPV | 307 | 0.8006 | 258 | 0.6073 | 242 | 93.80 |
| TPV | 826 | 0.8080 | 288 | 0.5837 | 227 | 78.82 |
| SQV | 453 | 0.8151 | 243 | 0.6188 | 215 | 88.48 |
Table 2.
The number of selected mutants and R2 for HIV-1 RT NNRTIs.
| Selected for higher R2 | Higher R2 | Selected for lower R2 | Lower R2 | Overlap | Overlap Ratio (%) | |
|---|---|---|---|---|---|---|
| NPV | 429 | 0.8067 | 273 | 0.6050 | 242 | 88.64 |
| DLV | 337 | 0.8162 | 257 | 0.6047 | 242 | 95.16 |
| EFV | 337 | 0.8073 | 243 | 0.6449 | 222 | 91.36 |
Quantile information analysis on HIV-1 protease inhibitor resistance
In order to further analyze the mutants selected by mean shift, quantile information analysis was performed and the result indicates that the proposed algorithm could successfully cluster the datasets, and pick the potentially most drug resistant mutants from the cluster centers (Tables 3, 4). In the tables, the numbers are given for selected/total number in each bin, and R2 used here is around 0.70.
Table 3.
Number of selected mutants in each bin for PIs.
| ATV | NFV | RTV | IDV | LPV | TPV | SQV | |
|---|---|---|---|---|---|---|---|
| I | 189/9454 | 183/13711 | 151/12220 | 246/14885 | 152/11630 | 366/9921 | 223/14746 |
| II | 36/1179 | 55/2126 | 34/1589 | 35/1101 | 62/2087 | 16/87 | 19/910 |
| III | 18/844 | 22/540 | 11/918 | 10/511 | 31/1393 | 0/0 | 0/107 |
| IV | 9/200 | 7/357 | 6/300 | 14/216 | 13/200 | 0/0 | 3/28 |
| V | 10/39 | 4/21 | 7/304 | 4/14 | 8/333 | 1/1 | 2/94 |
| VI | 1/3 | 1/256 | 0/0 | 0/0 | 1/26 | 0/0 | 2/132 |
| VII | 3/34 | 0/2 | 2/22 | 1/8 | 3/153 | 0/0 | 0/0 |
| VIII | 1/129 | 0/0 | 0/0 | 1/12 | 2/3 | 0/0 | 1/1 |
| IX | 0/0 | 1/9 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 |
| X | 24/202 | 15/523 | 59/1299 | 10/99 | 12/444 | 29/219 | 28/1100 |
Table 4.
Selected ratios in each bin for PIs (%).
| ATV | NFV | RTV | IDV | LPV | TPV | SQV | |
|---|---|---|---|---|---|---|---|
| I | 2.00 | 1.33 | 1.24 | 1.65 | 1.31 | 3.69 | 1.51 |
| II | 3.05 | 2.59 | 2.14 | 3.18 | 2.97 | 18.4 | 2.09 |
| III | 2.13 | 4.07 | 1.20 | 1.96 | 2.23 | N/A | 0.00 |
| IV | 4.50 | 1.96 | 2.00 | 6.48 | 6.50 | N/A | 10.7 |
| V | 25.6 | 19.1 | 2.30 | 28.6 | 2.40 | 100 | 2.13 |
| VI | 33.3 | 0.391 | N/A | N/A | 3.85 | N/A | 1.52 |
| VII | 8.82 | 0.00 | 9.09 | 12.5 | 1.96 | N/A | N/A |
| VIII | 0.775 | N/A | N/A | 8.33 | 66.7 | N/A | 100 |
| IX | N/A | 11.1 | N/A | N/A | N/A | N/A | N/A |
| X | 11.9 | 2.87 | 4.54 | 10.1 | 2.70 | 13.2 | 2.55 |
Bin I includes the mutants with least resistance to each inhibitors, while Bin × has the mutants with the highest resistance to the inhibitors. As shown in Table 4, the selected ratio in bin × is larger than that of bin I. This result suggests that the mutants vary more in the drug resistant category than in the non-drug resistant one.
Quantile information analysis on HIV-1 reverse transcriptase inhibitor resistance (NRTIs)
In order to further analyse the mutants selected by mean shift, all the drug resistant mutants were grouped and separated into 10 bins based on their drug resistance value. Both the total number of mutants and the selected number of mutants were counted and recorded in each corresponding table (Tables 5, 6). In the tables, the numbers are selected/total number in each bin, and R2 used here is around 0.70. Similar to PIs results, as shown in Table 6, the selected ration in bin × is larger than that of bin I.
Table 5.
Number of selected mutants in each bin for NRTIs.
| 3TC | ABC | D4T | DDI | TDF | AZT | |
|---|---|---|---|---|---|---|
| I | 11/2711 | 241/4780 | 188/3791 | 314/4603 | 265/2001 | 142/4079 |
| II | 0/14 | 13/65 | 51/948 | 17/194 | 1/1 | 19/94 |
| III | 0/1 | 0/0 | 10/23 | 5/25 | 0/0 | 13/253 |
| IV | 0/1 | 0/0 | 7/14 | 1/4 | 0/0 | 4/27 |
| V | 0/73 | 0/0 | 4/37 | 2/7 | 0/0 | 3/7 |
| VI | 0/57 | 0/0 | 2/17 | 2/9 | 0/0 | 5/30 |
| VII | 1/54 | 0/0 | 1/1 | 0/2 | 0/0 | 5/19 |
| VIII | 0/45 | 0/0 | 2/4 | 0/1 | 0/0 | 3/164 |
| IX | 0/88 | 0/0 | 1/8 | 1/3 | 0/0 | 0/6 |
| X | 14/1806 | 1/1 | 0/2 | 1/1 | 1/2 | 35/168 |
Table 6.
Selected ratios in each bin for NRTIs (%).
| 3TC | ABC | D4T | DDI | TDF | AZT | |
|---|---|---|---|---|---|---|
| I | 0.406 | 5.04 | 4.50 | 6.82 | 13.24 | 3.48 |
| II | 0.00 | 20.0 | 5.38 | 8.76 | 100 | 20.2 |
| III | 0.00 | N/A | 43.5 | 20.0 | N/A | 5.14 |
| IV | 0.00 | N/A | 50.0 | 25.0 | N/A | 14.8 |
| V | 0.00 | N/A | 10.8 | 28.6 | N/A | 42.9 |
| VI | 0.00 | N/A | 11.77 | 22.2 | N/A | 16.7 |
| VII | 1.85 | N/A | 100 | 0.00 | N/A | 26.32 |
| VIII | 0.00 | N/A | 50.0 | 0.00 | N/A | 1.83 |
| IX | 0.00 | N/A | 12.5 | 33.3 | N/A | 0.00 |
| X | 0.775 | 100 | 0.00 | 100 | 50.0 | 20.8 |
Quantile information analysis on HIV-1 reverse transcriptase inhibitor resistance (NNRTIs)
In order to further analyze the mutants selected by mean shift, all the drug resistant mutants were grouped and separated into × bins based on their drug resistance value. Both the total number of mutants and the selected number of mutants are counted and recorded in each corresponding table. In the tables, the numbers are selected/total number in each bin, and R2 used here is around 0.70. The Tables 7, 8 show the total number of mutants in the bin before and after selection.
Table 7.
Number of selected mutants in each bin for NNRTIs.
| Bin | NPV | DLV | EFV |
|---|---|---|---|
| I | 157/9898 | 198/9476 | 172/9907 |
| II | 17/157 | 24/241 | 14/116 |
| III | 9/114 | 12/587 | 10/166 |
| IV | 7/56 | 7/35 | 1/24 |
| V | 9/94 | 4/155 | 2/42 |
| VI | 7/169 | 3/20 | 2/132 |
| VII | 1/1 | 0/0 | 4/26 |
| VIII | 30/293 | 3/73 | 6/48 |
| IX | 0/1 | 3/9 | 1/2 |
| X | 66/584 | 43/703 | 32/891 |
Table 8.
Selected ratios in each bin for NNRTIs (%).
| NPV | DLV | EFV | |
|---|---|---|---|
| I | 1.59 | 2.09 | 1.74 |
| II | 10.8 | 10.0 | 12.0 |
| III | 7.90 | 2.04 | 6.02 |
| IV | 12.5 | 20.0 | 4.17 |
| V | 9.57 | 2.58 | 4.76 |
| VI | 4.14 | 15.0 | 1.52 |
| VII | 100 | N/A | 15.4 |
| VIII | 10.2 | 4.11 | 12.5 |
| IX | 0.00 | 33.3 | 50.0 |
| X | 11.3 | 6.12 | 3.59 |
Analysis of multi-drug resistance information for the most highly resistant mutants of HIV-1 PR to NRTIs
In order to further analyze the mutants selected by mean shift in the most drug resistant category (bin X), those mutants having resistance to multiple drugs were picked and compared. The results show that the more inhibitors a mutant is resistant to, the fewer representative mutants appear (Figure 7). The number of mutants representing high resistance to three or more PIs falls to low values of one to three, which becomes eminently verifiable by in vitro experiments.
Figure 7.
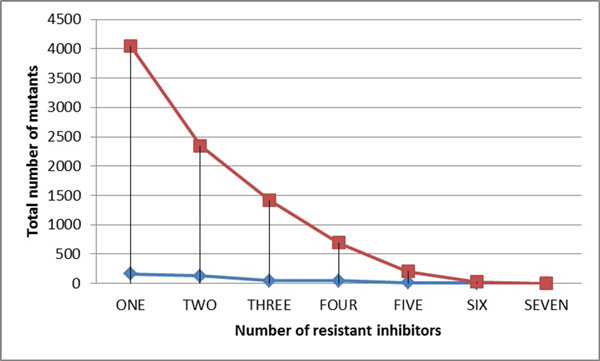
The relation between total number of mutants and number of resistant inhibitors for PIs. The red line shows the result for the original data in bin X; while the blue line shows the result for the selected mutants in bin X.
Analysis of multi-drug resistant information for the most highly resistant mutants of HIV-1 reverse transcriptase
Similar results are also obtained for NNRTIs. As shown in Figure 8, the number of mutants representing high resistance to two NNRTIs falls to values of 9-12, and when all three drugs are considered, only four mutants are representative of multidrug resistance. These low numbers of mutants can be verified by in vitro experiments.
Figure 8.
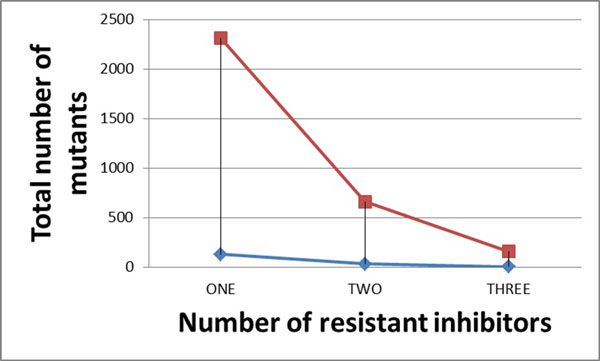
The relation between total number of mutants and number of resistant inhibitors for NNRTIs. The red line shows the result for the original data in bin X; while the blue line shows the result for selected mutants in bin X.
Discussion
The serious problem of drug resistance arising during therapy of HIV-infected individuals can cause failure of the treatment. Many scientists are working on revealing the drug resistance mechanisms using a variety of experimental techniques. However, since there are an extremely large number of mutants, it is difficult to choose representative mutants for detailed research in the laboratory.
In this experiment, we have developed new selection algorithm based on a simple graph representation of protein structure to solve this problem. The protein structure is 3-D and can be efficiently represented by Delaunay triangulation [44]. Based on this encoding method, a mean shift was applied to select the most representative mutants. Multiple linear regression was performed to evaluate the selection results.
This selection algorithm works well on selecting drug resistant mutants from both HIV PR and RT inhibitor genotype/phenotype data. Among all the mutants, around 250 most representative mutants were selected with numbers in the range of 215 to 360 [PIs+NNRTIs] mutants selected for the different drugs. Such selection was based on the kernel bandwidth, and the goal R2 value. In this experiment, the R2 value was set to be above 0.60 to be considered as a successful selection. During the experiments, after selection, the multiple linear regression was applied on these selected mutants' drug resistance values, and the R2 values fall in the range of 0.65 to 0.83, indicating excellent correlation. This high correlation suggests that the selected number of mutants can be further decreased if a lower target R2 value was applied.
Identifying a small number of representative mutants will enable laboratory studies of the molecular mechanisms of resistance, which is currently impossible due to the huge number of possible mutants.
Materials and methods
Datasets and data preparation
All the genotype-phenotype datasets were downloaded from the Stanford HIV drug resistance database [42](http://hivdb.stanford.edu/pages/genopheno.dataset.html). The proposed algorithm was tested on both HIV-1 PR and HIV-1 RT resistance data sets. For HIV-1 PR, the inhibitors atazanavir (ATV), nelfinavir (NFV), ritonavir (RTV), indinavir (IDV), lopinavir (LPV), tipranvir (TPV) and saquinavir (SQV) were tested. While for HIV RT, NNRTIs nelfinavir (NPV), delaviridine (DLV), efavirenz (EFV), and NRTIs lamivudine (3TC), abacavir (ABC), zidovudine (AZT), stavudine (D4T), didanosine (DDI) and tenofovir (TDF) were tested.
All the datasets were pre-processed using the methods and the cutoff values described previously in[24]. The results of the expansion for each of the HIV-1 PR inhibitors were: a total of 16846 sequences were obtained from 1622 isolates with assays for IDV resistance; a total of 16269 sequences from 1322 isolates for LPV; a total of 10228 sequences from 744 isolates for TPV; a total of 17118 sequences from 1640 isolates for SQV; a total of 12084 sequences from 1012 isolates for ATV; a total of 17545 sequences from 1674 isolates for NFV; and a total of 16652 sequences from 1589 isolates for RTV.
For each of the HIV-1 RT inhibitors the expansion resulted in: a total of 11367 sequences were obtained from 746 isolates with assays for NPV resistance; a total of 11299 sequences from 732 isolates for DLV; a total of 11354 sequences from 734 isolates for EFV; a total of 4850 sequences from 633 isolates for 3TC; a total of 4846 sequences from 628 isolates for ABC; a total of 4847 sequences from 630 isolates for AZT; a total of 4845 sequences from 630 isolates for D4T; a total of 4849 sequences from 632 isolates for DDI; and a total of 2004 sequences from 353 isolates for inhibitor TDF.
Encoding structure and sequence with Delaunay triangulation
The sequence and structure of the protein were represented using a graph-based encoding as described in [43]. Delaunay triangulation was used to define a graph which spanned the protein structure and defined structurally adjacent pairs of amino acid residues. Adjacent pairs of amino acids were summarized into a vector of the 210 unique pairs of the 20 standard amino acids by calculating the distance for each adjacent pair in the structure and tabulating by the types of amino acids in that adjacent pair. Only the sequences of the mutated proteins are needed and only one protein structure is necessary. As a result, all mutants are represented as vectors of the same dimensionality, which is a desired property for most of the pattern recognition algorithms. The X-ray crystal structures 3OXC for HIV-1 PR, and 2WOM for HIV-1 RT (from http://www.pdb.org) were used as templates for Delaunay triangulation.
Regression analysis for drug resistance prediction
The genotype-phenotype datasets provide an experimentally measured drug resistance value, with respect to a certain type of drug, with each genotype. The mutations relative to a standard sequence are denoted as where N is the total number of mutations and R210 is the structure vector. Also the corresponding drug resistance values are denoted as the real numbers including 0 for the resistance value of the wild type virus. We then seek a linear model between the xi 's and yi's by minimizing the cost function E:
| (1) |
with respect to the 210 dimensional vector A and scalar b.
Furthermore, in order to better utilize the available data set, we performed a k-fold cross-validation (in this work, k = 5). Specifically, the training set of size N is randomly divided into k groups. Among them, k-1 groups are utilized for constructing the linear model as in Equation (1). Then, the linear model is used to predict the drug resistance for the remaining group with N/kmutations. The predicted resistances are compared with the measured ones and the R2 values are recorded. Finally, the average and standard deviation of the k R2 values are computed.
Mean shift clustering and bandwidth selection
The mathematical deviation of mean shift algorithm was first introduced by Fukunaga and Hostetler [30], then adapted by Cheng [31], and later extended by Comaniciu, Meer, and Ramesh [45]. The procedure of the mean shift is that, for each data point in the feature space, a gradient ascent procedure is performed until convergence. The stop points of the procedure are the local maxima of the kernal density function, which could also be considered as the center of the clustering.
Given N data points, which could be considered as the kernel density function with Gaussian kernel for t ≥ 0:
Where is the mixing proportion of point i (satisfying), is its covariance matrix (positive definite), is a normalization constant and is the Mahalanobis distance.
Among all the data points, the dense regions of these could be treated as the local maxima of p(x) and could be found by seeking stationary points. The mean-shift update is applied with the rule:
This rule corresponds to a fixed point iteration to find the expected value for the centre of a Gaussian kernel, and is computationally more efficient than a gradient based numerical optimization for this problem. The rule maps any point to a weighted mean of the points in the dataset denoted as f(x). The difference f(x)-x is the mean shift vector and is clearly of zero magnitude at convergence.
The mean shift algorithm is non-parametric and the resolution of the clustering is determined by the kernel bandwidth σ. The initial step is to find the range of the bandwidth. Following that, by choosing different bandwidths, different numbers of mutants were selected. A multiple regression was performed to evaluate the selected results.
Quantile information analysis
All the drug resistant mutants were grouped and separated into 10 bins based on their drug resistance value. For example, about ATV, their resistance values range from 0 to 700. Therefore, those mutants with resistance value between 0 and 70 were put into bin I, those with resistance value between above 70 and below 140 were put into bin II, and so on.
After splitting all the data into ten bins, both the total number of mutants and the selected number of mutants were counted and recorded in each corresponding table. For each bin, the number of mutants before and after the selection was calculated and compared. Moreover, the selected ratio is also calculated.
k-fold validation
In order to fully use all the data, a k-fold cross-validation was performed in all the experiments for all the drugs. Specifically, we randomly choose (k-1)/k of all the sequences (some are drug resistant, while others are non-drug resistant) for training the classifier and the remaining 1/k data are used for testing. These tests used k = 5. Independent randomly selected k-folds were chosen throughout the study to avoid bias in the results. The apparent polymorphism in the original sequence data requires extra care when generating k-fold data sets for testing or training. When a sequence was removed from a k-fold in generating a testing or training dataset, all derived instances of that sequence were removed as well. This ensures that the individual k-fold datasets are truly independent from each other and thus ensures that the estimated accuracies are meaningful. The R2 values were averaged over the k-folds.
Competing interests
Authors declare that they have no competing interests.
Authors' contributions
All authors designed the experiments. XY and RWH designed the algorithms. XY implemented the algorithms and ran the predictions. All authors interpreted the results and wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Xiaxia Yu, Email: xiaxia.yu@stonybrook.edu.
Irene T Weber, Email: iweber@gsu.edu.
Robert W Harrison, Email: rwh@gsu.edu.
Acknowledgements
This research was supported, in part, by the National Institutes of Health grant GM062920 (ITW, RWH), and by a fellowship from the Georgia State University Molecular Basis of Disease Program (XY).
Declarations
Publication of this article was funded by the National Institutes of Health grant GM062920 (ITW, RWH).
This article has been published as part of BMC Bioinformatics Volume 16 Supplement 17, 2015: Selected articles from the Fourth IEEE International Conference on Computational Advances in Bio and medical Sciences (ICCABS 2014): Bioinformatics. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcbioinformatics/supplements/16/S17.
References
- Bakamjian L, Neggers Y, Crowe K, Geelhoed D, Lafort Y, Chissale E, Candrinho B, Degomme O, Barbiero V, Bollinger L, Global report. UNAIDS report on the global AIDS epidemic 2013. Journal of the American Board of Family Medicine. 2013;26(2):187–95. doi: 10.3122/jabfm.2013.02.120227. [DOI] [PubMed] [Google Scholar]
- Gallo RC, Salahuddin SZ, Popovic M, Shearer GM, Kaplan M, Haynes BF, Palker TJ, Redfield R, Oleske J, Safai B. Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science. 1984;224(4648):500–503. doi: 10.1126/science.6200936. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. International journal of antimicrobial agents. 2009;33(4):307–320. doi: 10.1016/j.ijantimicag.2008.10.010. [DOI] [PubMed] [Google Scholar]
- Lendeckel U, Hooper NM. Viral proteases and antiviral protease inhibitor therapy. Springer; 2009. [Google Scholar]
- Kohl NE, Emini EA, Schleif WA, Davis LJ, Heimbach JC, Dixon R, Scolnick EM, Sigal IS. Active human immunodeficiency virus protease is required for viral infectivity. Proceedings of the National Academy of Sciences. 1988;85(13):4686–4690. doi: 10.1073/pnas.85.13.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelmeier S, Schmidt H, Turk V, Von Der Helm K. Human immunodeficiency virus has an aspartic-type protease that can be inhibited by pepstatin A. Proceedings of the National Academy of Sciences. 1988;85(18):6612–6616. doi: 10.1073/pnas.85.18.6612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuya H, Weinhold KJ, Furman PA, St Clair MH, Lehrman SN, Gallo RC, Bolognesi D, Barry DW, Broder S. 3'-Azido-3'-deoxythymidine (BW A509U): an antiviral agent that inhibits the infectivity and cytopathic effect of human T-lymphotropic virus type III/lymphadenopathy-associated virus in vitro. Proceedings of the National Academy of Sciences of the United States of America. 1985;82(20):7096–7100. doi: 10.1073/pnas.82.20.7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl MA, Richman DD, Grieco MH, Gottlieb MS, Volberding PA, Laskin OL, Leedom JM, Groopman JE, Mildvan D, Schooley RT. The efficacy of azidothymidine (AZT) in the treatment of patients with AIDS and AIDS-related complex. A double-blind placebo-controlled trial. The New England journal of medicine. 1987;317(4):185–191. doi: 10.1056/NEJM198707233170401. [DOI] [PubMed] [Google Scholar]
- Sluis-Cremer N, Temiz NA, Bahar I. Conformational changes in HIV-1 reverse transcriptase induced by nonnucleoside reverse transcriptase inhibitor binding. Current HIV research. 2004;2(4):323.. doi: 10.2174/1570162043351093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahuja TS, Borucki M, Grady J. Highly active antiretroviral therapy improves survival of HIV-infected hemodialysis patients. American journal of kidney diseases. 2000;36(3):574–580. doi: 10.1053/ajkd.2000.16196. [DOI] [PubMed] [Google Scholar]
- Levy JA, Shimabukuro J. Recovery of AIDS-associated retroviruses from patients with AIDS or AIDS-related conditions and from clinically healthy individuals. Journal of Infectious Diseases. 1985;152(4):734–738. doi: 10.1093/infdis/152.4.734. [DOI] [PubMed] [Google Scholar]
- Barré-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret S, Gruest J, Dauguet C, Axler-Blin C, Vézinet-Brun F, Rouzioux C. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS) Science. 1983;220(4599):868–871. doi: 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]
- Ji J, Loeb LA. Fidelity of HIV-1 reverse transcriptase copying RNA in vitro. Biochemistry. 1992;31(4):954–958. doi: 10.1021/bi00119a002. [DOI] [PubMed] [Google Scholar]
- Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373(6510):123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- Chaix-Couturier C, Holtzer C, Phillips KA, Durand-Zaleski I, Stansell J. HIV-1 drug resistance genotyping: a review of clinical and economic issues. Pharmacoeconomics. 2000;18(5):425–433. doi: 10.2165/00019053-200018050-00002. [DOI] [PubMed] [Google Scholar]
- Johnson VA, Calvez V, Gunthard H, Paredes R, Pillay D, Shafer RW, Wensing AM, Richman DD. Update of the drug resistance mutations in HIV-1: March 2013. Top Antivir Med. 2013;21(1):6–14. [PMC free article] [PubMed] [Google Scholar]
- Drake JW. Rates of spontaneous mutation among RNA viruses. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(9):4171–4175. doi: 10.1073/pnas.90.9.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Ghosh SK, Taylor ME, Johnson VA, Emini EA, Deutsch P, Lifson JD, Bonhoeffer S, Nowak MA, Hahn BH. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 1995;373(6510):117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- Beerenwinkel N, Schmidt B, Walter H, Kaiser R, Lengauer T, Hoffmann D, Korn K, Selbig J. Diversity and complexity of HIV-1 drug resistance: a bioinformatics approach to predicting phenotype from genotype. Proceedings of the National Academy of Sciences. 2002;99(12):8271–8276. doi: 10.1073/pnas.112177799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerenwinkel N, Däumer M, Oette M, Korn K, Hoffmann D, Kaiser R, Lengauer T, Selbig J, Walter H. Geno2pheno: estimating phenotypic drug resistance from HIV-1 genotypes. Nucleic acids research. 2003;31(13):3850–3855. doi: 10.1093/nar/gkg575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Laethem K, De Luca A, Antinori A, Cingolani A, Perna CF, Vandamme A. A genotypic drug resistance interpretation algorithm that significantly predicts therapy response in HIV-1-infected patients. Antiviral therapy. 2002;7(2):123–9. [PubMed] [Google Scholar]
- Wang K, Jenwitheesuk E, Samudrala R, Mittler JE. Simple linear model provides highly accurate genotypic predictions of HIV-1 drug resistance. Antiviral therapy. 2004;9(3):343–352. [PubMed] [Google Scholar]
- Yu X, Harrison RW, Weber IT. HIV drug resistance prediction using multiple regression. Computational Advances in Bio and Medical Sciences (ICCABS), 2013 IEEE 3rd International Conference on 2013; IEEE; 2013. pp. 1–2. [Google Scholar]
- Yu X, Weber I, Harrison R. Sparse Representation for HIV-1 Protease Drug Resistance Prediction. SIAM International Conference on DataMining (SDM13), 2013; SIAM; 2013. pp. 342–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman A, Cherepanov P. The structural biology of HIV-1: mechanistic and therapeutic insights. Nature Reviews Microbiology. 2012;10(4):279–290. doi: 10.1038/nrmicro2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menéndez Arias L. Molecular basis of human immunodeficiency virus type 1 drug resistance: overview and recent developments. Antiviral research. 2013;98(1):93–120. doi: 10.1016/j.antiviral.2013.01.007. [DOI] [PubMed] [Google Scholar]
- Ohtaka H, Schön A, Freire E. Multidrug resistance to HIV-1 protease inhibition requires cooperative coupling between distal mutations. Biochemistry. 2003;42(46):13659–13666. doi: 10.1021/bi0350405. [DOI] [PubMed] [Google Scholar]
- Henderson G, Lee SK, Irlbeck D, Harris J, Kline M, Pollom E, Parkin N, Swanstrom R. Interplay between single resistance-associated mutations in the HIV-1 protease and viral infectivity protease activity and inhibitor sensitivity. Antimicrobial agents and chemotherapy. 2012;56(2):623–633. doi: 10.1128/AAC.05549-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis J, Aniana A, Weber I, Sayer J. Inhibition of autoprocessing of natural variants and multidrug resistant mutant precursors of HIV-1 protease by clinical inhibitors. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(22):9072–9077. doi: 10.1073/pnas.1102278108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hostetler L. The estimation of the gradient of a density function, with applications in pattern recognition. IEEE Transactions on information theory. 1975;21(1):32–40. [Google Scholar]
- Cheng Y. Mean shift, mode seeking, and clustering. Pattern Analysis and Machine Intelligence, IEEE Transactions on. 1995;17(8):790–799. [Google Scholar]
- Comaniciu D, Meer P. Mean shift analysis and applications. Computer Vision, 1999. The Proceedings of the Seventh IEEE International Conference on, 1999; IEEE; 1999. pp. 1197–1203. [Google Scholar]
- Comaniciu D, Meer P. Mean shift: A robust approach toward feature space analysis. Pattern Analysis and Machine Intelligence, IEEE Transactions on. 2002;24(5):603–619. [Google Scholar]
- Ramanan D, Forsyth DA. Finding and tracking people from the bottom up, Computer Vision and Pattern Recognition, 2003. Proceedings 2003 IEEE Computer Society Conference on, 2003; IEEE. 2003;2:II-467–II-474. [Google Scholar]
- Ramanan D, Forsyth DA. Using temporal coherence to build models of animals. Computer Vision, 2003. Proceedings. Ninth IEEE International Conference on, 2003; IEEE; 2003. pp. 338–345. [Google Scholar]
- Collins RT. Mean-shift blob tracking through scale space. Vol. 2. Computer Vision and Pattern Recognition, 2003. Proceedings. 2003 IEEE Computer Society Conference on, 2003; IEEE; 2003. pp. II-234–40. [Google Scholar]
- Bradski GR. Computer vision face tracking for use in a perceptual user interface. 1998.
- Sclaroff S, Liu L. Deformable shape detection and description via model-based region grouping. Pattern Analysis and Machine Intelligence, IEEE Transactions on. 2001;23(5):475–489. [Google Scholar]
- Wang Ly, Abyzov A, Korbel JO, Snyder M, Gerstein M. MSB: a mean-shift-based approach for the analysis of structural variation in the genome. Genome research. 2009;19(1):106–117. doi: 10.1101/gr.080069.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barash D, Comaniciu D. Meanshift clustering for DNA microarray analysis. Computational Systems Bioinformatics Conference, 2004. CSB 2004. Proceedings. 2004 IEEE, 2004; IEEE; 2004. pp. 578–579. [Google Scholar]
- Liu X, Müller HG. Modes and clustering for time-warped gene expression profile data. Bioinformatics. 2003;19(15):1937–1944. doi: 10.1093/bioinformatics/btg257. [DOI] [PubMed] [Google Scholar]
- Rhee SY, Gonzales MJ, Kantor R, Betts BJ, Ravela J, Shafer RW. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic acids research. 2003;31(1):298–303. doi: 10.1093/nar/gkg100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Weber I, Harrison R. 2013 SIAM International Conference on Data mining. Austin, TX, USA; 2013. Sparse Representation for HIV-1 Protease Drug Resistance Prediction; pp. 342–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose P, Xiaxia Y, Harrison RW. Encoding protein structure with functions on graphs. Bioinformatics and Biomedicine Workshops (BIBMW), 2011 IEEE International Conference on: 12-15 Nov. 2011. 2011. pp. 338–344.
- Comaniciu D, Ramesh V, Meer P. Kernel-based object tracking. IEEE Trans Pattern Anal Mach Intell. 2003;25:564–577. [Google Scholar]