

Abstract
The first direct evidence is provided for the presence of an interstitial carbide in the Fe–V cofactor of Azotobacter vinelandii vanadium nitrogenase. As for our identification of the central carbide in the Fe–Mo cofactor, we employed Fe Kβ valence-to-core X-ray emission spectroscopy and density functional theory calculations, and herein report the highly similar spectra of both variants of the cofactor-containing protein. The identification of an analogous carbide, and thus an atomically homologous active site in vanadium nitrogenase, highlights the importance and influence of both the interstitial carbide and the identity of the heteroatom on the electronic structure and catalytic activity of the enzyme.
Keywords: cofactors, nitrogen fixation, nitrogenases, vanadium, X-ray spectroscopy
The biological fixation of atmospheric dinitrogen (N2) to ammonium ions is exclusively promoted by nitrogenases (N2ases), multicomponent metalloenzymes that occur in diazotrophic bacteria and archaea.[1, 2] These enzymes utilize highly complex Fe–S clusters to effect N2 reduction, and in the case of the more widely studied Mo-dependent N2ase, the redox centers of the protein have been characterized by X-ray crystallography.[3] The active-site-containing MoFe protein, an α2β2 heterotetramer, uses an [8Fe–7S] cluster (P-cluster) as an electron-transfer relay, and the [Mo–7Fe–9S–C] Fe–Mo cofactor (FeMoco) as the catalytic site of N2 reduction (Figure 1). Despite the identification of an interstitial light atom at the center of FeMoco in 2002,[4] it was not until 2011 that the identity of the so-called “X” atom was definitively shown to be carbon by a combination of high-resolution X-ray crystallography, pulsed electron paramagnetic resonance (EPR) spectroscopy,[3] and Fe Kβ valence-to-core (VtC) X-ray emission spectroscopy (XES).[5]
Figure 1.
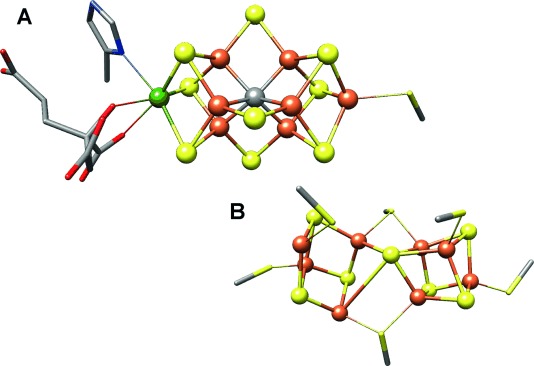
Representation of A) the FeMo cofactor and B) the P-cluster from Azotobacter vinelandii Mo nitrogenase, adapted from PDB 3U7Q. Color scheme for atoms: Fe=orange, S=yellow, C=gray, Mo=green, N=blue, O=red. Cysteinate residues are shown as sticks, and inorganic sulfides as spheres.
This fully valent carbide, ligated by six Fe atoms, is unprecedented and unique in biology. Genetic, spectroscopic, and isotopic radiolabeling experiments have determined the source of this carbon atom to be a radical S-adenosylmethionine (SAM), and have shown that the C atom is inserted into the cofactor through methyl group transfer in the cofactor assembly protein NifB.[6, 7] The effect of this carbide ion on the electronic structure of the cofactor, and its role in promoting N2 reduction, is still unknown.
Vanadium was first discovered as a promoter of nitrogen fixation in 1933,[8] and subsequently a V-dependent N2ase was identified in 1986.[9] In contrast to the well-characterized Mo N2ase, relatively little is known about the V N2ase despite nearly 30 years of research. At ambient conditions the enzyme is a much poorer nitrogen fixation catalyst than the Mo analogue, capable of N2 reduction at a rate of only 660 nmol mg−1 N2ase min−1 compared to 1040 nmol mg−1 N2ase min−1 for the Mo N2ase in the A. vinelandii system.[10] Furthermore, the V N2ase requires 4 more reducing equivalents, an additional 24 adenosine triphosphate (ATP) molecules per turnover, and has a significantly lower turnover number (112) than Mo N2ase (2230).[11]
In 2010 it was shown that in addition to N2 reduction, the V N2ase can perform reductive C–C bond coupling using CO and protons.[12] Intriguingly, it also does so at roughly 700 times the activity of the native Mo N2ase.[13] Engineered Mo N2ases have been shown to catalyze CO reduction as well, and at rates comparable to V N2ase, albeit with the loss of N2 reduction ability.[14, 15] The dramatic differences in the reactivity of these enzymes, and the ability of the vanadium enzyme to promote Haber–Bosch and Fischer–Tropsch chemistry at ambient conditions, has led to renewed interest in understanding the differences between the active-site cofactors in the Mo and V N2ases.
The atomic structure of the V N2ase protein is not known. Instead, the available structural information on the protein and its redox centers has come from spectroscopic and genetic studies. The latter have shown that while the two N2ases share common precursors in their biosynthetic pathways, including the nifB gene responsible for radical SAM-dependent carbon insertion, distinct genetic factors promote final cofactor maturation.[16] Additionally, the V N2ase protein contains additional δn subunits, where n=2 or 4 depending on the species.[17, 18]
Much of the spectroscopic data collected on V N2ase has also indicated substantial differences from Mo N2ase. As early as 1987, Arber et al. and George et al. independently reported vanadium extended X-ray absorption fine structure (EXAFS) data indicating that the V atom, in contrast to expected changes based on periodic trends, was more displaced from the remainder of the cofactor than the Mo atom.[19, 20] More recent studies employing Fe X-ray absorption spectroscopy (XAS), EXAFS, and EPR spectroscopy have also shown marked differences in the electronic structures of the V N2ase metallocofactors. These differences are significant enough to prompt the authors to interpret their data as indicating the presence of a cubically symmetric Fe–V cofactor (FeVco) and two separated [4Fe–4S] clusters in place of the trigonal FeMo cofactor and fused P-cluster shown in Figure 1.[18, 21–23] Despite this, some recent reports have shown graphical models of the redox centers of the V N2ase as being structurally analogous to those of Mo N2ase known from X-ray crystallography, in contradiction to the published data.[24, 25] No direct evidence for an analogous cofactor structure has been presented in support of such models. This has led to the somewhat widespread belief that the structure of FeVco is identical to FeMoco, and that similarities in atomic composition and geometry are a foregone conclusion.
In an effort to provide more definitive evidence for the structure of FeVco, we report herein the Fe Kβ VtC XES spectrum of the VFe protein of A. vinelandii N2ase (Figure 2). We have also concomitantly re-measured the analogous spectrum of the MoFe protein, and note that the Kβ′′ feature at approximately 7100 eV, previously shown to be indicative of fluorescent emission from the 2s atomic orbital of the central carbide in FeMoco, is identically placed in the spectrum of VFe. Additionally, the difference spectrum shows only minute deviations in the intensities and energies in all spectral regions.
Figure 2.
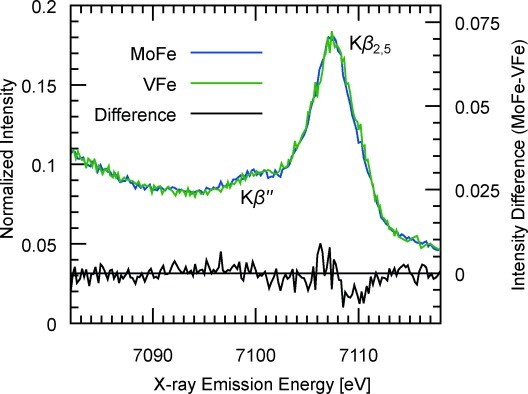
Fe Kβ VtC XES spectra of MoFe and VFe proteins from A. vinelandii. The Kβ′′ peak at 7099.8 eV has been previously shown to be attributable to the interstitial carbon in the FeMo cofactor of MoFe. The absence of an intensity difference (MoFe −VFe) at the Kβ′′ energy indicates an interstitial carbon is also present in FeVco.
We have previously shown that the energy of the distinctive Kβ′′ peak in FeMoco is inconsistent with any interstitial light atom besides carbon, and that in the absence of such a carbon atom, for example in the P-cluster, the intensity of the Kβ′′ feature is significantly diminished.[5] Given the virtually identical VtC spectra of the MoFe and VFe proteins, we believe these data strongly indicate the presence of an analogous interstitial carbide in FeVco, thereby providing, to our knowledge, the first direct evidence for a structurally homologous cofactor in the V N2ase.
To provide additional support for this conclusion, we have performed density functional theory (DFT) calculations on large, 225-atom models of both cofactors, based on the published crystal structure of MoFe. The FeMoco model was previously utilized in our report on the determination of the MoIII oxidation state in FeMoco,[26] and the FeVco model was calculated as being isostructural and valence isoelectronic (see the Experimental Section for details). Our optimized structure of the FeVco active-site model results in V–Fe distances that are 0.11 Å longer than the Mo–Fe distances in the optimized structure of FeMoco, in reasonably good agreement with experimental metrical parameters from EXAFS (0.08 Å longer).[20] VtC XES spectra were calculated within a one-electron approximation, and the averages of the spectral contributions from all cofactor Fe atoms are presented in Figure 3.
Figure 3.
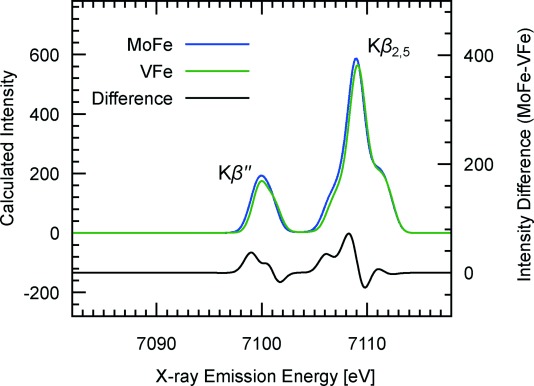
DFT-calculated Fe Kβ VtC XES spectra of 225-atom models of FeMoco and FeVco. Spectra are an average of calculated transitions from all seven Fe atoms, and a 1.5 eV broadening was applied to discrete transition moments. Features in the difference spectrum (MoFe−VFe) correspond well to the experimental data. A scalar shift of 123 eV was applied to correct the absolute transition energies.[27].
The calculations, like the experimental results, show strikingly similar spectra in both the Kβ2,5 and Kβ′′ regions, where the latter is dominated by S 3s and C 2s contributions and the former by S 3p contributions. The subtle differences in the calculated spectra are highlighted by a difference spectrum, which again agrees well with experiment. In particular, the derivative shape under the Kβ2,5 peak is reproduced by the calculations and may be attributed to the sulfur 3p orbitals being shifted to slightly higher energy in FeVco than in FeMoco. We caution, however, that because of the limited experimental resolution, a more quantitative analysis is not possible. Nonetheless, these spectra clearly establish the presence of a carbide in FeVco. A more detailed study of the electronic structural differences will likely require higher resolution spectroscopies (such as high-resolution (HERFD) XAS)[28] and will be the topic of future studies in our laboratories.
The present work takes an initial step toward directly defining the structure of FeVco. The presence of a second biological cofactor with an interstitial carbide has now been experimentally established. With this finding, we have laid the groundwork for more focused studies aimed at understanding how perturbations to electronic structure, likely engendered by the heterometal, differentially tune these remarkable enzymes, enabling reactions as diverse as N2 activation and C–C bond coupling under ambient conditions.
Experimental Section
Cell growth and protein purification: MoFe protein of N2ase was produced and isolated following established procedures.[3] To obtain the VFe protein, Azotobacter vinelandii (Lipmann 1903, ATCC 478) was cultured in molybdenum-free Burke medium[29] under nitrogen-limited conditions. The production of vanadium N2ase was monitored by activity assays.[30] All purification steps were performed under strict exclusion of dioxygen, using an anaerobic chamber or modified Schlenk techniques. Cells were disrupted using an Avestin Emusiflex at a pressure of 1000–1500 bar, and cell debris was separated by centrifugation. The supernatant was loaded onto a 5 mL HiTrapQ HP column equilibrated with 50 mm Tris/HCl buffer at pH 7.4. VFe protein eluted in a linear gradient of 0 mm to 500 mm NaCl. Pure protein was obtained after an additional size-exclusion step on a 26/60 Superdex 200 gel filtration column (GE Healthcare).
X-ray spectroscopy: Fe Kβ X-ray emission spectroscopy (XES) experiments were performed at beamline ID-26 at the European Synchrotron Radiation Facility (ESRF) in Grenoble, France. Incident photon energy was 7800 eV, selected using a Si(111) double crystal monochromator. Photon flux at the sample was approximately 1×1013 photons sec−1, with a maximum ring current of 200 mA and a ring energy of 6.03 GeV. The beam spot size on the sample was 0.1 mm×1 mm. Protein samples were in aqueous solution at approximately 100 mg mL−1, loaded in Delrin cells sealed with 38 μm Kapton tape and maintained at 10 K during measurements using a liquid He cryostat. Fe Kβ X-ray emission was analyzed with a Johann-type spectrometer, using five spherically bent Ge(620) crystals in a Rowland geometry, as described previously,[31] and detected using a dead-time-corrected Ketek Si drift diode detector. To determine the acceptable dwell time per sample spot, rapid Fe Kβ high-energy resolution fluorescence detected (HERFD) X-ray absorption spectra were recorded on the same sample spot, and the data examined for evidence of change during the course of sample dosing. For all XES measurements, the photon dose was well below the acceptable limit. Multiple scans on the same sample were normalized to incident flux and averaged using MATLAB. Data were referenced to the Kβ1,3 and Kβ2,5 features of Fe2O3[27] and normalized to a total integrated spectral intensity of 100 by numerical integration.
Density functional theory calculations: All calculations were performed using the ORCA program package developed by Neese and co-workers.[32] 225 atom cluster models of FeMoco and FeVco active sites were based on the X-ray structure of MoFe protein,[3] and were TPSSh-optimized using our previously reported procedure.[26] Charges on the metal clusters were −1 for FeMoco ([MoFe7S9C]1−) and −2 for FeVco ([VFe7S9C]2−), to maintain a valence isoelectronic configuration and a spin of S=3/2. We note that other cofactor charges are conceivable, however our previous studies have shown that VtC spectra are relatively insensitive to changes in cofactor charge,[5] and thus we have not considered them here. Analogous broken-symmetry solutions were found. Valence-to-core XES spectra were calculated within a one-electron approximation implemented in ORCA as described in Ref. [27]. These calculations used the BP86 functional[33, 34] and the DKH relativistic approximation,[35–37] with DKH-recontracted def2-TZVP triple-zeta basis sets[38, 39] and the COSMO dielectric model (ε=4).[40]
Acknowledgments
This work was supported by the European Research Council (ERC) under the European Union′s Seventh Framework Programme (FP/2007–2013) ERC Grant Agreement number 615414 (S.D.) and the ERC N-ABLE project (O.E.). Funding was also provided by the Deutsche Forschungsgemeinschaft grants EI-520/7 and RTG 1976 (O.E.), and by the Max-Planck-Gesellschaft (S.D.). J.A.R. is supported by a graduate study scholarship from the Deutscher Akademischer Austauschdienst. The ESRF is also acknowledged for providing beamtime, and Dr. Sara Lafuerza and Dr. Pieter Glatzel at beamline ID-26 are thanked for technical assistance with measurements.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1.Burgess BK. Chem. Rev. 1990;90:1377. [Google Scholar]
- 2.Seefeldt LC, Hoffman BM, Dean DR. Annu. Rev. Biochem. 2009;78:701. doi: 10.1146/annurev.biochem.78.070907.103812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spatzal T, Aksoyoglu M, Zhang L, Andrade SLA, Schleicher E, Weber S, Rees DC, Einsle O. Science. 2011;334:940. doi: 10.1126/science.1214025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Einsle O, Tezcan FA, Andrade SLA, Rees DC. Science. 2002;297:1696. doi: 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- 5.Lancaster KM, Roemelt M, Ettenhuber P, Hu Y, Ribbe MW, Neese F, Bergmann U, DeBeer S. Science. 2011;334:974. doi: 10.1126/science.1206445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wiig JA, Hu Y, Lee CC, Ribbe MW. Science. 2012;337:1672. doi: 10.1126/science.1224603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lancaster KM, Hu Y, Bergmann U, Ribbe MW, DeBeer S. J. Am. Chem. Soc. 2013;135:610. doi: 10.1021/ja309254g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bortels H. Zentralbl. Bakteriol. Abt. II. 1933;87:476. [Google Scholar]
- 9.Robson RL, Eady RR, Richardson TH, Miller RW, Hawkins M, Postgate JR. Nature. 1986;322:388. [Google Scholar]
- 10.Hales BJ, Case EE, Morningstar JE, Dzeda MF, Mauterer LA. Biochemistry. 1986;25:7251. doi: 10.1021/bi00371a001. [DOI] [PubMed] [Google Scholar]
- 11.Stacey GS, Burris RH, Evans HJ. Biological Nitrogen Fixation. New York: Chapman & Hall; 1992. [Google Scholar]
- 12.Lee CC, Hu Y, Ribbe MW. Science. 2010;329:642. doi: 10.1126/science.1191455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu Y, Lee CC, Ribbe MW. Science. 2011;333:753. doi: 10.1126/science.1206883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang ZY, Dean DR, Seefeldt LC. J. Biol. Chem. 2011;286:19417. doi: 10.1074/jbc.M111.229344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarma R, Barney BM, Keable S, Dean DR, Seefeldt LC, Peters JW. J. Inorg. Biochem. 2010;104:385. doi: 10.1016/j.jinorgbio.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eady RR. Coord. Chem. Rev. 2003;237:23. [Google Scholar]
- 17.Lee CC, Hu Y, Ribbe MW. Proc. Natl. Acad. Sci. USA. 2009;106:9209. doi: 10.1073/pnas.0904408106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen J, Christiansen J, Tittsworth RC, Hales BJ, George SJ, Concouvanis D, Cramer SP. J. Am. Chem. Soc. 1993;115:5509. [Google Scholar]
- 19.Arber JM, Dobson BR, Eady RR, Stevens P, Hasnain SS, Garner CD, Smith BE. Nature. 1987;325:372. [Google Scholar]
- 20.George GN, Coyle CL, Hales BJ, Cramer SP. J. Am. Chem. Soc. 1988;110:4057. [Google Scholar]
- 21.Fay AW, Blank MA, Lee CC, Hu Y, Hodgson KO, Hedman B, Ribbe MW. J. Am. Chem. Soc. 2010;132:12612. doi: 10.1021/ja1019657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blank MA, Lee CC, Hu Y, Hodgson KO, Hedman B, Ribbe MW. Inorg. Chem. 2011;50:7123. doi: 10.1021/ic200636k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu Y, Corbett MC, Fay AW, Webber JA, Hedman B, Hodgson KO, Ribbe MW. Proc. Natl. Acad. Sci. USA. 2005;102:13825. doi: 10.1073/pnas.0506967102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu Y, Lee CC, Ribbe MW. Dalton Trans. 2012;41:1118. doi: 10.1039/c1dt11535a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rebelein JG, Hu Y, Ribbe MW. Angew. Chem. Int. Ed. 2014;53:11543–11546. doi: 10.1002/anie.201406863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angew. Chem. 2014;126:11727. [Google Scholar]
- 26.Bjornsson R, Lima FA, Spatzal T, Weyhermüller T, Glatzel P, Bill E, Einsle O, Neese F, DeBeer S. Chem. Sci. 2014;5:3096. [Google Scholar]
- 27.Lee N, Petrenko T, Bergmann U, Neese F, DeBeer S. J. Am. Chem. Soc. 2010;132:9715. doi: 10.1021/ja101281e. [DOI] [PubMed] [Google Scholar]
- 28.Kowalska J, DeBeer S. Biochim. Biophys. Acta Mol. Cell Res. 2015;1853:1406. doi: 10.1016/j.bbamcr.2014.11.027. [DOI] [PubMed] [Google Scholar]
- 29.Newton JW, Wilson PW, Burris RH. J. Biol. Chem. 1953;204:445. [PubMed] [Google Scholar]
- 30.Dilworth MJ. Biochim. Biophys. Acta. 1966;127:285. doi: 10.1016/0304-4165(66)90383-7. [DOI] [PubMed] [Google Scholar]
- 31.Smolentsev G, Soldatov AV, Messinger J, Merz K, Weyhermüller T, Bergmann U, Pushkar Y, Yano J, Yachandra VK, Glatzel P. J. Am. Chem. Soc. 2009;131:13161. doi: 10.1021/ja808526m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neese F. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012;2:73. doi: 10.1002/wcms.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Becke A. Phys. Rev. A. 1988;38:3098. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 34.Perdew JP. Phys. Rev. B. 1986;33:8822. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- 35.Hess BA. Phys. Rev. A. 1985;32:756. doi: 10.1103/physreva.32.756. [DOI] [PubMed] [Google Scholar]
- 36.Hess BA. Phys. Rev. A. 1986;33:3742. doi: 10.1103/physreva.33.3742. [DOI] [PubMed] [Google Scholar]
- 37.Jansen G, Hess BA. Phys. Rev. A. 1989;39:6016. doi: 10.1103/physreva.39.6016. [DOI] [PubMed] [Google Scholar]
- 38.Weigend F, Ahlrichs R. Phys. Chem. Chem. Phys. 2005;7:3297. doi: 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- 39.Pantazis DA, Chen XY, Landis CR, Neese F. J. Chem. Theory Comput. 2008;4:908. doi: 10.1021/ct800047t. [DOI] [PubMed] [Google Scholar]
- 40.Klamt A, Schüürmann G. J. Chem. Soc. Perkin Trans. 2. 1993:799. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information