

Abstract
To shed light on cell-adhesion-related molecular pathways, synthetic cells offer the unique advantage of a well-controlled model system with reduced molecular complexity. Herein, we show that liposomes with the reconstituted platelet integrin αIIbβ3 as the adhesion-mediating transmembrane protein are a functional minimal cell model for studying cellular adhesion mechanisms in a defined environment. The interaction of these synthetic cells with various extracellular matrix proteins was analyzed using a quartz crystal microbalance with dissipation monitoring. The data indicated that integrin was functionally incorporated into the lipid vesicles, thus enabling integrin-specific adhesion of the engineered liposomes to fibrinogen- and fibronectin-functionalized surfaces. Then, we were able to initiate the detachment of integrin liposomes from these surfaces in the presence of the peptide GRGDSP, a process that is even faster with our newly synthesized peptide mimetic SN529, which specifically inhibits the integrin αIIbβ3.
Keywords: cell adhesion, integrin, liposomes, peptide mimetics, quartz crystal microbalance
Cell adhesion is a fundamental process that is crucial for the development and functionality of multicellular organisms. Recent studies have shown that the coordinated behavior of tissue cells, including their proliferation, migration, and differentiation, is regulated in time and space by cell–cell and cell–ECM adhesion sites (ECM=extracellular matrix). Amongst various types of cell adhesion, the adhesion integrin family plays a central role in tissue physiology.[1] The functions and signaling of these transmembrane proteins have already been studied extensively in living cells.[2] Their interaction is mainly governed by molecular crowding effects that originate from the complex interplay between densely packed intracellular macromolecules.[3] Therefore, lipid vesicles with reconstituted proteins are ideal candidates to study cellular adhesion mechanisms in a spherical, cell-like unit with densely packed proteins of the cell adhesion complex.
In recent years, various proteins of the focal adhesion complex were incorporated into lipid vesicles, enabling the biochemical and biophysical elucidation of the molecular nature of cell adhesion.[5] In particular, the integrin αIIbβ3 from blood platelets was reconstituted into small liposomes using a detergent dialysis method.[6] The biological activity of the reconstituted integrin αIIbβ3 was confirmed by fibrinogen (Fg) binding assays.[7] In general, integrin can be activated in the extracellular β domain by the addition of bivalent ions[2b, 8] even if no intracellular binding partners of the adhesome (e.g., talin) are present,[9] for instance, in synthetic cell systems with reconstituted integrins.[15]
A powerful, label-free technique to follow the adhesion of lipid vesicles is the use of a quartz crystal microbalance with dissipation monitoring (QCM-D), which measures the adsorbed wet mass in real time and enables the analysis of the viscoelastic properties of the adhered layer.[16] Furthermore, its flow setup allows for easy rinsing and inhibitor presentation. Over the past years, the immobilization of bare liposomes on various crystal coatings has been extensively studied.[17] QCM-D experiments were also used to analyze the mechanisms of vesicle rupture and their transformation into supported lipid bilayers (SLBs).[18] The underlying kinetics of this process depend on many parameters, including vesicle size,[17e, 19] surface chemistry,[17e] temperature,[17e, 20] lipid charge,[21] osmotic pressure,[17e, 22] membrane fluidity,[17b] electrostatic interactions, and the presence of calcium ions.[23] In combination with QCM-D, ellipsometry, surface plasmon resonance measurements, and atomic force and fluorescence microscopy have contributed to the manifold insights into vesicle adhesion and SLB formation.[17a, 21, 23, 24]
Owing to this versatility, QCM-D has become the approach of choice for the analysis of different cellular molecular recognition processes. It has, for instance, been employed to study the physicochemical properties of hyaluronan films on SLBs as a model system for pericellular sugar coats.[25] The molecular recognition between biotinylated liposomes (simulating integrins) and avidin-coated crystals (simulating the ECM) was also studied by QCM-D.[17d] Herein, we present the first study of the synthetic adhesion of integrin-functionalized liposomes and its modulation by specific soluble inhibitors using QCM-D and SiO2 sensors coated with different ECM proteins. The newly synthesized peptide mimetic SN529 (see the Supporting Information) demonstrated superior activity against platelet integrin αIIbβ3 compared to RGD peptides.
First, we compared the interaction of integrin liposomes and non-functionalized (“pure”) liposomes with SiO2 coated sensors (Figure 1 and Table 1).[21, 23, 26] Directly after injection, both the pure liposomes and the integrin liposomes showed a strong binding to the SiO2 sensors as indicated by the respective decrease in frequency and increase in dissipation (see Figure 1 a). After approximately 30 min, the resonance frequency of the pure liposome channel reached a minimum and then increased again owing to the release of trapped aqueous buffer to reach a stable value of Δffinal=−27±3 Hz (Table 1). The corresponding dissipation signal showed a similar but less pronounced response of the opposite sign. These signal changes indicate that pure liposomes ruptured and formed an SLB as depicted in Figure 1 b because of the insufficient mechanical stability of the pure liposomes. In contrast, the binding of integrin liposomes on SiO2 sensors resulted in a continuous frequency decrease and dissipation increase (Table 1). This observation indicates that the integrin liposomes stayed intact on the SiO2 sensors. In the subsequent washing step, the frequency slightly increased again and the dissipation decreased, which indicates that integrin liposomes only adhere non-specifically to SiO2 and detach again when buffer is added. In contrast, the SLB formed from pure liposomes could not be removed again.
Figure 1.

a) Frequency and dissipation recordings for liposomes on SiO2 sensors. After a 90 min washing step (step I), liposomes and integrin liposomes were loaded onto the sensors for 3.5 h (step II), followed by an additional 30 min washing step (step III). b) Schematic representation of intact integrin vesicles and formation of an SLB from pure liposomes. It may well be that there are also oppositely oriented integrins reconstituted in the liposomes. As these do not contribute to adhesion, they are not included in the schemes throughout the manuscript. c, d) Changes in viscoelasticity with the attachment of integrin liposomes (c) and pure liposomes (d). The color code in (c) and (d) represents the time dependence.
Table 1.
QCM-D studies of the binding of liposomes, integrin liposomes, and different ECM proteins to SiO2 sensors
| Protein coating[a] | Protein binding | |
|---|---|---|
| Δf [Hz] | ΔD [10−6] | |
| 1. liposomes | −27±3 | 0.49±0.09 |
| 2. integrin liposomes | −125.1±0.4 | 35.89±0.11 |
| 3. Fg | −98.8±2.2 | 3.46±0.06 |
| 4. Fn | −74.3±2.2 | 3.04±0.07 |
| 5. Col | −151±4 | 34±1 |
1. Pure liposomes yielded frequency and dissipations signals that are characteristic of SLB formation. 2. Integrin liposomes led to a frequency decrease and a large dissipation change, which shows that these vesicles stayed intact. 3.–5. Δf and ΔD after coating SiO2 sensors with different ECM proteins (Fg, Fn, Col) for 2.5 h and an additional 30 min washing step. Frequency decreases and dissipation increases indicate successful ECM protein binding to SiO2 sensors.
According to Sauerbrey’s model for the adhesion of rigid thin layers, there is a linear relationship between the frequency decrease (−ΔF) and the mass increase per unit area (Δm/A).[27] As this model was not developed for soft organic films, it only serves as an approximation in our synthetic cell model. However, ΔD/Δf plots can be used to identify conformational changes of the adhered layer.[28] Figure 1 c and d show the ΔD/Δf analysis for integrin liposomes and pure liposomes on uncoated SiO2 sensors. For the integrin liposomes, we obtained an almost linear relationship after the equilibration period, which indicates that the liposomes did not rupture on the SiO2 sensors. In contrast, for pure liposomes, a reverse ΔD/ΔF trajectory was observed, confirming SLB formation. Therefore, the reconstitution of integrin into intact liposomes enabled us to further study their adhesion on different ECM proteins.
The experimental setup of the QCM-D adhesion studies is schematically depicted in Figure 2 a. First, the SiO2 sensors of the QCM-D device were coated with Fg, fibronectin (Fn), or collagen type I (Col) by monitoring frequency and dissipation changes (Figure 2 b–d and Table 1). From these data, the Sauerbrey and Voigt models enable an estimation of the film thickness of the protein coatings (Supporting Information, Table S1). In all cases, the thickness was greater than 10 nm, indicating full coverage of the SiO2 sensor. Dynamic light scattering measurements yielded an average diameter of 100 to 200 nm for pure liposomes and integrin liposomes. Using these liposomes and pure integrin, we studied the binding to Fg-, Fn-, or Col-coated SiO2 sensors (Figure 2 b–d; Table 2).
Figure 2.
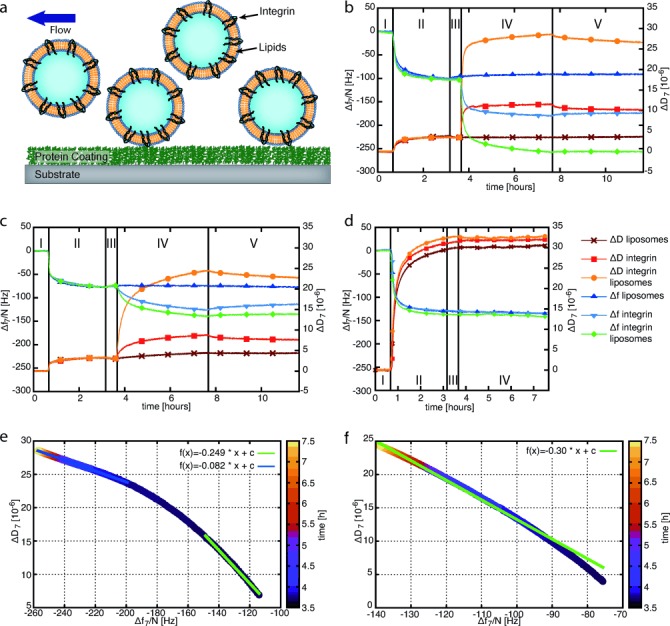
a) Schematic representation of integrin liposomes being flushed over protein-coated sensors in the QCM-D chamber. b–d) Δf and ΔD for the binding of liposomes, integrin αIIbβ3, and integrin liposomes on different ECM protein coatings. For the first 40 min, buffer A with MnCl2 and MgCl2 flowed over the sensors (step I). In the following 2.5 h, a solution containing 50 μg mL−1 of Fg (b), Fn (c), or Col (d) was loaded into the QCM chamber (step II). After a second 30 min washing step with buffer A (step III), one of three different samples was added to one QCM-D sensor: 1) pure liposomes to one sensor, 2) 50 μg mL−1 of activated integrin αIIbβ3 to another sensor, and 3) integrin liposomes to a third sensor. e, f) Changes in the viscoelasticity for the binding of integrin liposomes on Fg- (e) and Fn-coated (f) SiO2 sensors.
Table 2.
Maximum Δf and ΔD values for pure integrin, liposomes, and integrin liposomes on different ECM coatings[a]
| Protein coating | Pure integrin | Liposomes | Integrin liposomes | |||
|---|---|---|---|---|---|---|
| Δf [Hz] | ΔD [10−6] | Δf [Hz] | ΔD [10−6] | Δf [Hz] | ΔD [10−6] | |
| Fg | −73.6±0.1 | 6.78±0.04 | 4.93±0.15 | 0.14±0.04 | −153.34±0.09 | 23.20±0.04 |
| Fn | −38.84±0.14 | 4.53±0.03 | −0.02±0.08 | 0.17±0.03 | −60.79±0.15 | 19.69±0.04 |
| Col | −5.1±0.2 | 0.32±0.09 | −4.9±0.2 | 0.23±0.07 | −4.5±0.2 | −0.41±0.08 |
The frequency and dissipation shifts were determined by subtracting the average value of the last 5 min of the buffer wash before adding the samples (step III) from that of the last 5 min of the final buffer wash (step V). The errors are the sums of both standard deviations.
For the Fg coatings, pure liposomes only yielded a small increase in frequency and a very stable dissipation. Integrin binding led to a frequency reduction and an increase in dissipation. For the integrin liposomes, we measured the strongest frequency decrease and dissipation increase. Similarly, the addition of pure liposomes to Fn-coated surfaces only caused small changes in the frequency and dissipation signals. The binding of integrin and integrin liposomes to these surfaces resulted in a decrease in the resonance frequency and a dissipation increase, which were, however, less pronounced than for the Fg coatings. These signal recordings indicate that integrin liposomes and pure integrin bound very well to Fg and less efficiently to Fn coatings whereas no binding was observed for pure liposomes in both cases. For Fg coatings, it is also notable that the measured signals with pure integrin are about half of the signals with the integrin liposomes (Table 2). Considering that a dominant fraction of the latter signal is due to trapped water, it is suggested that much fewer binding sites are occupied by the liposomes than by the integrins in solution. Possibly, the integrin liposomes stay attached to the surfaces over several hours owing to their polyvalent interactions.
Unlike Fn and Fg, Col has no binding sites for integrin αIIbβ3. As shown in Figure 2 d, there were only small shifts in the resonance frequency and dissipation signal when pure integrin or integrin liposomes were loaded onto Col-coated SiO2 sensors. This observation differs only slightly from the results for pure liposomes on Fg and Fn and confirms our assumption that integrin liposomes or pure integrin do not specifically bind to Col.
The specific binding of integrin liposomes to Fg- and Fn-coated sensors was further characterized by analysis of the ΔD/Δf plots (Figure 2 e, f). For both protein coatings, we obtained a linear relationship. In the case of Fg (see Figure 2 e), we split the linear fit into two parts as we observed a change in viscoelasticity from low coverage (green line) to a crowding of liposomes on the surface (blue line), which leaves less space for dissipative sideways motion on the oscillating sensor with increasing vesicle coverage.[18a] The observed linear relationship between the bound mass and dissipation after the equilibration period underlines that the liposomes did not rupture or form an SLB on Fg. For integrin liposomes adhered to Fn, we obtained a linear ΔD/Δf relationship (Figure 2 f). As the frequency and dissipation shifts reach higher values on Fg than on Fn, a denser packing of integrin liposomes on the surfaces can be assumed. This could cause rearrangement and deformation of the liposomes, which would account for the observed temporal changes in the ΔD/Δf regime on Fn.
We further analyzed how the adhesive behavior of our cell model systems could be modulated during QCM-D analysis. Initially, we studied the effect of free inhibitors in solution on the adhesion of integrin liposomes on Fg-coated SiO2 sensors. (Figure 3 a, b). The peptide mimetic SN529 with an IC=30.8 nm was synthesized for the first time (see Figure 3 a and the Supporting Information). Furthermore, we used the RGD peptide GRGDSP with an IC>1000 nm as a control inhibitor in our adhesion studies. We started by specifically adhering integrin liposomes and pure integrins to Fg-coated SiO2 sensors (Figure 3 c, d; step I). Subsequently, we added the peptide GRGDSP or the peptide mimetic SN529 to the bound integrin liposomes and integrins, respectively (step II). Both the peptide and the mimetic had been dissolved in standard buffer A containing 1 mm MgCl2 and 1 mm MnCl2. We analyzed the frequency and dissipation shifts from the end of sample binding to the end of the final washing step (see Table 3).
Figure 3.
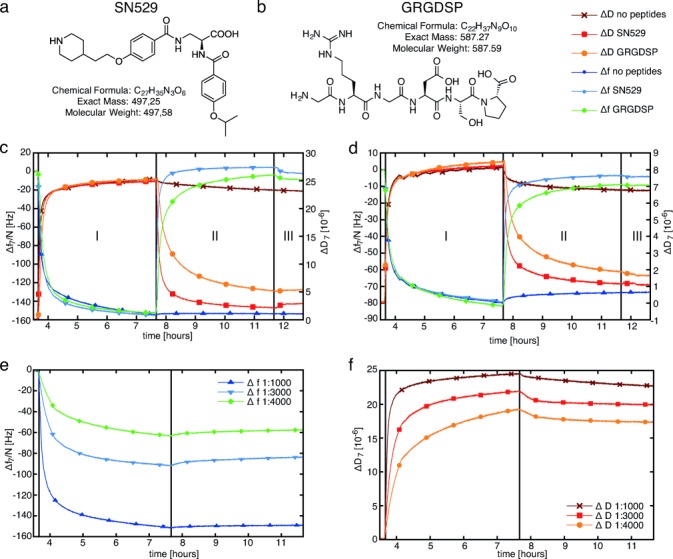
a, b) Modulation of synthetic integrin mediated adhesion by adding free inhibitors and different integrin concentrations: peptide mimetic SN529 (a) and RGD peptide GRGDSP (b). c, d) Comparison of Δf and ΔD for the competitive versus the uncompetitive unbinding of integrin liposomes (c) and integrin αIIbβ3 (d) on Fg in the presence of RGD peptides or mimetics. Integrin liposomes and 50 μg mL−1 of pure integrin αIIbβ3 were added to two Fg-coated SiO2 sensors each (step I). Then, 500 μm of the RGD peptide GRGDSP or the peptide mimetic SN529 were added (step II). A reference chamber was washed with our standard buffer A with MgCl2 and MnCl2, which does not contain any inhibitors, until all channels had been switched to this buffer in step III. e, f) Adhesion of integrin liposomes with different integrin concentrations to Fg-coated SiO2 sensors. The molar lipid to protein ratios were 1:1000, 1:3000, and 1:4000.
Table 3.
Maximal Δf and ΔD values during integrin-mediated adhesion on Fg upon addition of RGD peptides (GRGDSP) or mimetics (SN529)
| Regular buffer (control) | SN529 | GRGDSP | ||||
|---|---|---|---|---|---|---|
| Integrin liposomes | Integrin | Integrin liposomes | Integrin | Integrin liposomes | Integrin | |
| Δf [Hz] | 0.68±0.15 | 6.1±0.1 | 135±13 | 77±11 | 129±18 | 74±13 |
| ΔD [10−6] | −2.10±0.05 | −1.39±0.04 | −26.9±1.1 | −7.2±0.6 | −23.2±1.9 | −6.9±0.7 |
For specifically adhered integrin liposomes, the addition of SN529 yielded a frequency increase of  135±13 Hz and a dissipation decrease of
135±13 Hz and a dissipation decrease of 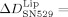 −26.9±1.1×10−6. In standard buffer only, no significant frequency and dissipation changes were recorded. These observations indicate a strong unbinding of the integrin liposomes from the Fg-coated SiO2 sensors. With the peptide GRGDSP, the corresponding signal changes were less pronounced so that a weaker unbinding of the integrin liposomes from Fg was observed with this peptide. The addition of SN529 to pure integrin bound to Fg-coated SiO2 sensors yielded a frequency increase of
−26.9±1.1×10−6. In standard buffer only, no significant frequency and dissipation changes were recorded. These observations indicate a strong unbinding of the integrin liposomes from the Fg-coated SiO2 sensors. With the peptide GRGDSP, the corresponding signal changes were less pronounced so that a weaker unbinding of the integrin liposomes from Fg was observed with this peptide. The addition of SN529 to pure integrin bound to Fg-coated SiO2 sensors yielded a frequency increase of 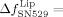 77±11 Hz and a dissipation decrease of
77±11 Hz and a dissipation decrease of 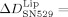 −7.2±0.6×10−6. Upon the addition of GRGDSP to bound platelet integrin, the frequency and dissipation changes were less pronounced. These recordings indicate that pure integrin was also unbound by RGD peptides and the peptide mimetic. Nevertheless, SN529 resulted in a faster unbinding effect than the peptide GRGDSP as almost all integrin and integrin liposomes were completely removed from Fg. This effect is also reflected by the respective frequency changes: 30 min after the addition of GRGDSP, the frequency returned to 77.4 % of the value before the specific binding of integrin liposomes. In comparison, SN529 led to an even higher frequency recovery of 98.6 % in the same time frame, which corresponds to an almost complete detachment of the integrin liposomes by to the peptide mimetic. In summary, the mimetic SN529 showed a drastically higher activity against the reconstituted platelet integrin αIIbβ3 than peptide GRGDSP.
−7.2±0.6×10−6. Upon the addition of GRGDSP to bound platelet integrin, the frequency and dissipation changes were less pronounced. These recordings indicate that pure integrin was also unbound by RGD peptides and the peptide mimetic. Nevertheless, SN529 resulted in a faster unbinding effect than the peptide GRGDSP as almost all integrin and integrin liposomes were completely removed from Fg. This effect is also reflected by the respective frequency changes: 30 min after the addition of GRGDSP, the frequency returned to 77.4 % of the value before the specific binding of integrin liposomes. In comparison, SN529 led to an even higher frequency recovery of 98.6 % in the same time frame, which corresponds to an almost complete detachment of the integrin liposomes by to the peptide mimetic. In summary, the mimetic SN529 showed a drastically higher activity against the reconstituted platelet integrin αIIbβ3 than peptide GRGDSP.
Second, to modulate and control the binding strength of our model cells on Fg even further, we used different molar integrin/lipid ratios of 1:1000, 1:3000, and 1:4000 during integrin reconstitution (Figure 3 and Table 4). According to DLS measurements, the average size of all of these samples was 111±2 nm. These results clearly indicate that the frequency and dissipation changes depend on the integrin/lipid ratio that is used at the start of the self-assembly-driven reconstitution process. Therefore, the reconstitution of different integrin concentrations in our synthetic cells resulted in a reduced adhesion strength at reduced integrin concentrations.
Table 4.
Adhesion of integrin liposomes with various integrin concentrations to Fg surfaces
| Integrin concentration | |||
|---|---|---|---|
| 1:1000 | 1:3000 | 1:4000 | |
| Δf [Hz] | −153.34±0.13 | −82.8±0.3 | −54.71±0.15 |
| ΔD [10−6] | 23.20±0.03 | 19.84±0.04 | 16.9±0.03 |
Previously, biotin-functionalized liposomes have been used on avidin-coated surfaces to mimic the molecular recognition processes in cell adhesion.[17d] Nevertheless, this model system is less biorelevant as avidin–biotin binding does not occur in native cells where integrins are involved in cell adhesion.[29] Our study has overcome these limitations by reconstituting functionally active integrins into liposomes to mimic cell adhesion to ECM proteins. In such encapsulated model cells, the molecular binding rates are increased owing to reduced diffusion rates—similarly to native cells. Therefore, synthetic cells offer a powerful platform for studying cell adhesion under the influence of molecular crowding as in native cells, yet in a well-controlled environment with reduced molecular complexity.[4]
For pure liposomes on SiO2 sensors, we observed liposome rupture and SLB formation as previously reported,[21, 30] whereas integrin liposomes did not form SLBs. The protruding extracellular integrin domains, which keep the lipid head groups away from the SiO2 surface, thereby weakening the lipid–surface interaction and leaving the integrin liposomes intact, might explain this effect. On the other hand, reconstituted integrins might mechanically stabilize the liposomes.
On the RGD-containing ECM proteins Fg and Fn, we observed specific adhesion of integrin liposomes in the presence of bivalent ions. Pure integrin also specifically adhered to Fg and Fn with frequency changes comparable to the binding of integrin liposomes on both RGD-containing ECM proteins. Nevertheless, the ΔD of pure integrin on Fg and Fn was less pronounced than for integrin liposomes. This observation indicates that pure integrin forms a tight monolayer on protein-coated SiO2 sensors. In comparison, integrin liposomes enclose an aqueous solution when they bind to protein-coated sensors, which significantly contributes to the observed major dampening effect.
Furthermore, we modulated the synthetic adhesion of our model cells on Fg using different inhibitors. First, we showed that the addition of two structurally different integrin inhibitors to the buffer led to notable unbinding of integrin liposomes and pure integrin. Here, the binding sites of Fg-coated sensors competed with the much denser binding sites of the RGD peptides and mimetics in solution. Integrin liposomes were found to detach from the Fg surfaces even more strongly than pure integrins. The more pronounced unbinding of the integrin liposomes might be due to the fact that pure integrins form a tight molecular layer on the Fg surfaces and are less accessible for the free RGD peptides than the spherical integrin liposomes. In comparison, the mimetic SN529 resulted in complete and much faster unbinding of the integrin liposomes and integrins whereas GRGDSP peptides did not detach the adhered integrin liposomes and integrins as completely and quickly. This different “competitive” unbinding behavior is related to the very different activities for the platelet integrin αIIbβ3, which is mainly determined by the binding activity to the integrin: SN529 exhibits a higher binding affinity than the peptide GRGDSP, thus leading to a more pronounced unbinding of integrin liposomes and integrins.
In conclusion, we have established a new biomimetic system for studying synthetic adhesion. With these synthetic cell systems, QCM-D is an ideal method to study the involved molecular recognition processes. The next step towards functional synthetic cells that mimic and control adhesion will be the addition of further adhesion-associated proteins, such as talin, FAK, or vinculin, to encapsulated liposomes. It will be particularly exciting to visually characterize these encapsulated functional adhesion complexes also by cryo-TEM analysis and to extend the scope of these minimal synthetic cells towards more complex systems.
Acknowledgments
We thank Peer Fischer for his assistance with DLS measurements and Electra Gizeli for fruitful discussions. Financial support was provided by the European Research Council under the European Union’s Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement 294852 and the BMBF/MPG network MaxSynBio. J.P.S. is the Weston Visiting Professor at the Weizmann Institute of Science and is a member of the Heidelberg cluster of excellence CellNetworks. B.G. is the E. Neter Professor of Cell and Tumor Biology.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1a.Barczyk M, Carracedo S, Gullberg D. Cell Tissue Res. 2010;339:269–280. doi: 10.1007/s00441-009-0834-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b.Huttenlocher A, Horwitz AR. Cold Spring Harbor Perspect. Biol. 2011;3 doi: 10.1101/cshperspect.a005074. a005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2a.Harburger DS, Calderwood DA. J. Cell Sci. 2009;122:159–163. doi: 10.1242/jcs.018093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b.Hynes RO. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 3.Minton AP. J. Biol. Chem. 2001;276:10577–10580. doi: 10.1074/jbc.R100005200. [DOI] [PubMed] [Google Scholar]
- 4a.Soh S, Banaszak M, Kandere-Grzybowska K, Grzybowski BA. J. Phys. Chem. Lett. 2013;4:861–865. doi: 10.1021/jz3019379. [DOI] [PubMed] [Google Scholar]
- 4b.Tan C, Saurabh S, Bruchez MP, Schwartz R, Leduc P. Nat. Nanotechnol. 2013;8:602–608. doi: 10.1038/nnano.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brüggemann D, Frohnmayer JP, Spatz JP. Beilstein J. Nanotechnol. 2014;5:1193–1202. doi: 10.3762/bjnano.5.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6a.Parise LV, Phillips DR. J. Biol. Chem. 1985;260:10698–10707. [PubMed] [Google Scholar]
- 6b.Parise LV, Phillips DR. J. Biol. Chem. 1985;260:1750–1756. [PubMed] [Google Scholar]
- 7a.Müller B, Zerwes HG, Tangemann K, Peter J, Engel J. J. Biol. Chem. 1993;268:6800–6808. [PubMed] [Google Scholar]
- 7b.Erb E-M, Tangemann K, Bohrmann B, Müller B, Engel J. Biochemistry. 1997;36:7395–7402. doi: 10.1021/bi9702187. [DOI] [PubMed] [Google Scholar]
- 7c.Erb E-M, Engel J. In: Extracellular Matrix Protocols, Vol. 139. Streuli C, Grant M, editors. Totowa: Humana Press; 2000. pp. 71–82. [Google Scholar]
- 8a.Gailit J, Ruoslahti E. J. Biol. Chem. 1988;263:12927–12932. [PubMed] [Google Scholar]
- 8b.Xiong J-P, Stehle T, Goodman SL, Arnaout MA. Blood. 2003;102:1155–1159. doi: 10.1182/blood-2003-01-0334. [DOI] [PubMed] [Google Scholar]
- 8c.Xiong JP, Goodman SL, Arnaout MA. Methods Enzymol. 2007;426:307–336. doi: 10.1016/S0076-6879(07)26014-8. [DOI] [PubMed] [Google Scholar]
- 9.Das M, Subbayya Ithychanda S, Qin J, Plow EF. Biochim. Biophys. Acta Biomembr. 2014;1838:579–588. doi: 10.1016/j.bbamem.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goennenwein S, Tanaka M, Hu B, Moroder L, Sackmann E. Biophys. J. 2003;85:646–655. doi: 10.1016/S0006-3495(03)74508-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sinner E-K, Reuning U, Kök FN, Saccà B, Moroder L, Knoll W, Oesterhelt D. Anal. Biochem. 2004;333:216–224. doi: 10.1016/j.ab.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 12a.Fenz SF, Sengupta K. Integr. Biol. 2012;4:982–995. doi: 10.1039/c2ib00188h. [DOI] [PubMed] [Google Scholar]
- 12b.Stano P. Biotechnol. J. 2011;6:850–859. doi: 10.1002/biot.201100079. [DOI] [PubMed] [Google Scholar]
- 12c.Walde P, Cosentino K, Engel H, Stano P. ChemBioChem. 2010;11:848–865. doi: 10.1002/cbic.201000010. [DOI] [PubMed] [Google Scholar]
- 13a.Aimon S, Manzi J, Schmidt D, Poveda Larrosa JA, Bassereau P, Toombes GE. PLoS One. 2011;6 doi: 10.1371/journal.pone.0025529. e25529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b.Betaneli V, Petrov EP, Schwille P. Biophys. J. 2012;102:523–531. doi: 10.1016/j.bpj.2011.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14a.Girard P, Pécréaux J, Lenoir G, Falson P, Rigaud J-L, Bassereau P. Biophys. J. 2004;87:419–429. doi: 10.1529/biophysj.104.040360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14b.Dezi M, Di Cicco A, Bassereau P, Levy D. Proc. Natl. Acad. Sci. USA. 2013;110:7276–7281. doi: 10.1073/pnas.1303857110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Streicher P, Nassoy P, Barmann M, Dif A, Marchi-Artzner V, Brochard-Wyart F, Spatz J, Bassereau P. Biochim. Biophys. Acta Biomembr. 2009;1788:2291–2300. doi: 10.1016/j.bbamem.2009.07.025. [DOI] [PubMed] [Google Scholar]
- 16.Höök F, Larsson C, Fant C. In: Encyclopedia of Surface and Colloid Science. Hubbard A, editor. New York: Marcel Dekker; 2002. pp. 774–791. [Google Scholar]
- 17a.Serro AP, Carapeto A, Paiva G, Farinha JPS, Colaço R, Saramago B. Surf. Interface Anal. 2012;44:426–433. [Google Scholar]
- 17b.Vu TH, Shimanouchi T, Ishii H, Umakoshi H, Kuboi R. J. Colloid Interface Sci. 2009;336:902–907. doi: 10.1016/j.jcis.2009.04.048. [DOI] [PubMed] [Google Scholar]
- 17c.Dimitrievski K, Kasemo B. Langmuir. 2009;25:8865–8869. doi: 10.1021/la9025409. [DOI] [PubMed] [Google Scholar]
- 17d.Lüthgens E, Herrig A, Kastl K, Steinem C, Reiss B, Wegener J, Pignataro B, Janshoff A. Meas. Sci. Technol. 2003;14:1865. [Google Scholar]
- 17e.Reimhult E, Höök F, Kasemo B. Langmuir. 2003;19:1681–1691. [Google Scholar]
- 17f.Melzak K, Tsortos A, Gizeli E. Methods Enzymol. 2009;465:21–41. doi: 10.1016/S0076-6879(09)65002-3. [DOI] [PubMed] [Google Scholar]
- 18a.Keller CA, Kasemo B. Biophys. J. 1998;75:1397–1402. doi: 10.1016/S0006-3495(98)74057-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b.Keller CA, Glasmastar K, Zhdanov VP, Kasemo B. Phys. Rev. Lett. 2000;84:5443–5446. doi: 10.1103/PhysRevLett.84.5443. [DOI] [PubMed] [Google Scholar]
- 19.Reimhult E, Höök F, Kasemo B. J. Chem. Phys. 2002;117:7401–7404. [Google Scholar]
- 20.Reimhult E, Hook F, Kasemo B. Phys. Rev. E. 2002;66:051905. doi: 10.1103/PhysRevE.66.051905. [DOI] [PubMed] [Google Scholar]
- 21.Richter R, Mukhopadhyay A, Brisson A. Biophys. J. 2003;85:3035–3047. doi: 10.1016/S0006-3495(03)74722-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hain N, Gallego M, Reviakine I. Langmuir. 2013;29:2282–2288. doi: 10.1021/la304197m. [DOI] [PubMed] [Google Scholar]
- 23.Richter RP, Brisson AR. Biophys. J. 2005;88:3422–3433. doi: 10.1529/biophysj.104.053728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24a.Reimhult E, Zach M, Hook F, Kasemo B. Langmuir. 2006;22:3313–3319. doi: 10.1021/la0519554. [DOI] [PubMed] [Google Scholar]
- 24b.Richter RP, Berat R, Brisson AR. Langmuir. 2006;22:3497–3505. doi: 10.1021/la052687c. [DOI] [PubMed] [Google Scholar]
- 25.Richter RP, Hock KK, Burkhartsmeyer J, Boehm H, Bingen P, Wang G, Steinmetz NF, Evans DJ, Spatz JP. J. Am. Chem. Soc. 2007;129:5306–5307. doi: 10.1021/ja068768s. [DOI] [PubMed] [Google Scholar]
- 26a.Jackman JA, Zhao ZL, Zhdanov VP, Frank CW, Cho NJ. Langmuir. 2014;30:2152–2160. doi: 10.1021/la404582n. [DOI] [PubMed] [Google Scholar]
- 26b.Bluemmel J, Perschmann N, Aydin D, Drinjakovic J, Surrey T, Lopez-Garcia M, Kessler H, Spatz JP. Biomaterials. 2007;28:4739–4747. doi: 10.1016/j.biomaterials.2007.07.038. [DOI] [PubMed] [Google Scholar]
- 26c.Cooper MA, Singleton VT. J. Mol. Recognit. 2007;20:154–184. doi: 10.1002/jmr.826. [DOI] [PubMed] [Google Scholar]
- 27.Sauerbrey G. J. Phys. 1959;155:206–212. [Google Scholar]
- 28.Tellechea E, Johannsmann D, Steinmetz NF, Richter RP, Reviakine I. Langmuir. 2009;25:5177–5184. doi: 10.1021/la803912p. [DOI] [PubMed] [Google Scholar]
- 29.Zaidel-Bar R, Itzkovitz S, Ma’ayan A, Iyengar R, Geiger B. Nat. Cell Biol. 2007;9:858–867. doi: 10.1038/ncb0807-858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morita S, Nukui M, Kuboi R. J. Colloid Interface Sci. 2006;298:672–678. doi: 10.1016/j.jcis.2005.12.043. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information