

Abstract
The mechanical properties of cortical bone, which is largely comprised of collagen, hydroxyapatite, and water, are known to hinge on hydration. Recently, the characteristics of water in bone have drawn attention as potential markers of bone quality. We report on the dynamics, diffusion, population, and exchange of water in cortical bone by NMR relaxation and diffusion methodologies. Relaxation measurements over timescales ranging from 0.001 to 4.2 s reveal two distinguishable water environments. Systematic exposure to ethylenediaminetetraacetic acid or collagenase reveals one peak in our 2D relaxation map belonging to water present in the hydroxyapatite rich environment, and a second peak with shorter relaxation times arising from a collagen rich site. Diffusion-T2 measurements allowed for direct measurement of the diffusion coefficient of water in all observable reservoirs. Further, deuterium relaxation methods were applied to study cortical bone under an applied force, following mechanical wear or fracture. The tumbling correlation times of water reduce in all three cases, indicating that water dynamics may be used as a probe of bone quality. Lastly, changes in the relative populations and correlation times of water in bone under an applied force suggest that load bearing occurs largely in the collagen rich environment and is reversible.
Keywords: Cortical bone, 2H Relaxation, NMR, Diffusion
Introduction
Bone is a composite biomaterial comprised of collagen which is surrounded by hydroxyapatite crystals 1,2. Water is the third major component of bone and contributes approximately 20% to its wet weight3. Collagen provides elasticity4, the mineral component confers strength5, and water is believed to impart shock absorbing capacity1. Because of this unique makeup, the fracture toughness of bone is 3 to 10 times higher than silicon4. Early bone mineral density (BMD) tests were considered a standard method for accessing bone quality and risk of fracture6. In the last three decades, however, studies have shown that BMD tests alone are not a good predictor of fracture risk7,8. Hence, there has been an ongoing need for additional parameters to assess bone quality.
The strength, toughness, and stiffness of bone is known to be associated to the degree of hydration9. Solid state nuclear magnetic resonance (SSNMR) spectroscopy has proved to be a popular technique to study bone structure as it can probe both hard (mineral, mineralized collagen) and soft matter (water) unlike X-ray methodologies10–16. Water and its role in bone has been extensively studied by NMR spectroscopy and relaxation methodology12,16–21. Many studies have been performed to observe or distinguish bone water (i.e., mobile or bound)16,18 or have addressed the presence of different water layers19 or reservoirs12,16,17,21 in cortical bone. SSNMR has also been applied to indirectly measure the thickness of a water layer in bone to be 9Å, by measuring the distance of the organic and inorganic components10. Apart from these approaches, NMR relaxation methods have allowed for the study of bone porosity and to determine the pore size distribution in bone21–25. Recently, it has been shown that NMR relaxation can also be employed to study bone quality by measuring the mobile and bound water components18. The diffusion characteristics of exchangeable water in cortical bone have also been measured and the results point to the presence of two different water environments, i.e. mobile and tightly bound water3.
Water should not be considered an isolated component of bone as it interacts with collagen and inorganic apatite and influences both macroscopic and microscopic characteristics26. SSNMR studies have shown that water may deeply influence the orientation of apatite crystals by forming an amorphous layer outside crystals13 and plays a key role in triple helical assembly of native collagen26. The total water concentration in bone was recently shown to exhibit a correlation with bone stiffness followed by BMD measurements, bone volume/trabecular volume and trabecular number in rat trabecular bones27. Recent NMR studies showed that bound water shows a strong correlation with bone fracture properties28,29. Although there have been studies elucidating mobile water, bound water and bound water associated with collagen, many questions remain relating to the differentiation of water related to collagen and hydroxyapatite, their associated diffusion coefficients, and how the dynamics and population of water in each reservoir are altered following fracture, mechanical wear or when bone is placed under a mechanical load.
In the present study, we have measured the diffusion, exchange, and tumbling correlation times of water in intact cortical bone. 2H two dimensional T1 – T2, T2 – T2, D–T2 experiments have been performed, based on an inverse Laplace transform (ILT)30–33. These two dimensional relaxation methods have been previously applied to characterize water in multisite systems such as elastin 34, silk35, poly(N-substituted α/β-asparagine) derivative 36, potato tissue37, cement pastes38 and saturated sedimentary rocks39. The bone matrix was systematically modified by either collagenase or EDTA (ethylenediaminetetraacetic acid) to follow the changes in water dynamics and its distribution in each resolvable reservoir observed in the two dimensional relaxation data. These experiments allowed for identifying different water reservoirs present in the bone extracellular matrix (i.e., collagen and mineral apatite). Diffusion-T2 experiments by pulsed gradient field NMR methods were also performed and the molecular diffusion coefficients of different water reservoirs were directly measured in fractured cortical bone. Our studies of bone under a mechanical load, following fracture or mechanical wear indicate that the largest changes in dynamics and population of water occur in the organic component (collagen) of cortical bone and are reversible.
Materials and Methods
Sample Preparation
Cortical bone from goat femora were collected from a local slaughter house. The bone was cut into a right-angled parallelepiped weighed approximately 40±5 mg. Bone samples were demineralized or deproteinized following the procedures detailed below.
EDTA (ethylenediaminetetraacetic acid) Treatment
A small piece of intact bone with a dimension of approximately 6.0 mm × 3.0 mm × 1.0 mm was immersed in a 0.1M EDTA solution and incubated for about 1 week at 37 °C. After the treatment the bone was washed several times in water and subsequently dried.
Collagenase Treatment
Collagenase from Clostridium histolyticum was purchased from Sigma Aldrich (St. Louis, MO). A solution was prepared using PBS (Phosphate Buffer Saline) and 20 mg/ml collagenase. A sample of the bone having approximate dimensions 5.0 mm × 3.0 mm × 2.0 mm was immersed in the collagenase solution and incubated at 37 °C for approximately 1 week. Afterward, the sample was washed and dried as in the EDTA treatment.
Prior to the NMR experiments each specimen (untreated, EDTA and collagenase treated) was immersed into 99.9 atom % D2O (Sigma Aldrich,USA) and sonicated for 20 min. During sonication the temperature did not exceed 40°C. For the untreated bone the soaking time was 50 days while for collagenase and EDTA treated samples the soaking time was 8 days. Fractured bone samples were prepared by striking the samples with a hammer under liquid nitrogen and consisted of several small fragments of bone. 2H T1 – T2 measurements were performed on three different goat femurs and were reproducible (results from only one measurement are shown). Studies that involved mechanical loading, fracture, and mechanical wear were performed on a sample from one goat femur, twice.
Mechanical Wear and Application of an External Force
To perform mechanical wear, a small piece of intact bone with dimensions of approximately 4.5 mm × 3.0 mm × 2.0 mm was used. The sample was mechanically worn on the surface by a scalpel by carving a hole approximately 1 mm wide × 1 mm thick. Prior to the NMR experiments the sample was immersed in D2O for 17 days. To apply an external force on the bone sample the following procedure was performed. A cylinder machined from delrin having dimensions 7 mm in diameter and 2 cm in length was tapped through to accommodate an 8-32 screw. A bone sample approximately 6.5 mm × 1.5 mm × 2.0 mm was placed in the cylinder and compressed using nylon screws. The torque on the screws was measured using a digital torque wrench and the force applied was then estimated knowing the lever arm length on the torque wrench; the applied force was estimated to be approximately 10 to 25N. Before applying the force the sample was immersed in D2O for 24 days. Experiments on samples under an applied force made use of a magic angle spinning probe with a stator that can accommodate the delrin cylinder (samples were static).
1H, 2H Relaxation Measurements
All the 2H (1H) 2D T1 – T2 and 2H 2D T2 – T2 NMR experiments were carried out at a magnetic field strength of 4.699 T using a Varian Unity NMR spectrometer at a frequency of 30.716 (200.093) MHz with a Varian liquids NMR probe and DOTY MAS (magic angle spinning) probe. The experiments were performed at room temperature unless otherwise stated. The variable temperature T1 – T2 experiments were carried out in the temperature range between 2 to 40 °C and controlled by an FTS systems TC-84 Air Jet temperature controller to within ± 1 °C. The pulse sequences31,40,41 used for T1 – T2 and T2 – T2 experiments are shown in supplementary figure S1. The 90° pulse length of 24 μs was used for all experiments and calibrated at every temperature studied. The recycle delay used was 10 s (20s) which is more than five times the largest relaxation time measured at the magnetic field used in this study. For the 2D T1–T2 experiments, the delay t1 (supplementary figure S1) was logarithmically incremented from 1 ms to 10 s in 100 steps (1.5 ms to 20 s). In the pulse sequence for T1 – T2 experiments, the initial 180° pulse inverts the magnetization and then it recovers to thermal equilibrium by T1 relaxation during the variable delay time t1. After the 180° pulse, the 90° pulse following the delay t1 transforms the magnetization into the transverse plane and the NMR signal is detected by a Carr-Purcell-Meiboom-Gill (CPMG) pulse train with τ =0.350 ms42. The CPMG pulse train refocuses field inhomogeneity effects, and averages out any heteronuclear dipolar interactions (e.g. 2H-1H) in the T2 domain. The T2 – T2 experiment allows for probing the exchange time of water molecules between different water reservoirs resolved. Referring to supplementary figure S1, the 90° pulse applied after the first CPMG pulse train stores the magnetization along the azimuthal axis for variable mixing time tm. The second CPMG pulse train is applied after the time period tm and the T2 time is again measured. In the 2D ILT map a cross peak in this experiment corresponds to water molecules exchanging between different water reservoirs during time tm. In the T1 – T2 and T2 – T2 experiments n = 6000 points were collected to measure T2 in each dimension.
Diffusion-relaxation Measurements
In addition to the relaxation correlation experiments, diffusion-relaxation correlation experiments were performed. The experiments were carried out at a magnetic field of 4.214 T and 2H resonance frequency of 27.546 MHz using a homemade probe equipped with magnetic field gradients for solid state studies. The gradient coil was composed of a superposition of Maxwell coils and was capable of delivering a gradient along the azimuthal axis of the sample of up to 90G/cm· A for a sample with a diameter of 2.1 mm43. These strong magnetic field gradients allow for measuring diffusion during time intervals where the magnetization decay is dominated by strong quadrupolar relaxation as discussed later in the text. The pulse sequence used in this work is shown in supplementary figure S2 and is composed of a bipolar gradient stimulated echo pulse sequence44 in the indirect dimension for encoding diffusion and a CPMG train in the direct dimension for T2 encoding. Trapezoidal bipolar gradient pulses with duration of 300 μs were used for phase encoding and decoding as well as to mitigate Eddy currents and ring down effects. Referring to supplementary figure S2, the pre and post gradient delay τ=100μs, Δ=4ms, δc=120μs, and the amplitude of the crushing gradient was approximately 12G/cm. The π pulses had duration of 20 μs and were used to refocus any background gradients45. The maximum gradient strength in the experiment was 1120G/cm using approximately 12A. The gradient strength was calibrated using D2O, a substance with a known diffusion coefficient46. The kernel for the signal attenuation due to diffusion differs from that provided in the work by Wu44 due to the presence of trapezoidal pulses and was derived explicitly for the pulse sequence shown in supplementary figure S2 using the cumulant expansion47,48. In the T2 dimension, the delay τ′=300 μs and 1280 echoes were acquired. For all diffusion-relaxation measurements the temperature was set to 22°C and was regulated digitally to within 0.5 °C.
13C MAS NMR
The 1H-13C CP spectra at magic angle spinning were recorded on solid-state Avance Bruker NMR spectrometer operating at 600.156 MHz for 1H and 150.924 MHz for 13C frequencies with a 3.2 mm DVT probe. All the experiments were performed at 10 kHz Magic Angle Spinning (MAS). A Bruker MAS pneumatic unit controlled the spinning speed with accuracy of ± 2 Hz. The 1H 90° pulse length was 2.28 μs. For the cross-polarization measurements we used a 1.0 ms contact time with 10k transients and a 5s recycle delay.
Inverse Laplace Transformation
All 2D correlation maps were obtained by a 2D inverse Laplace transform (ILT) using an algorithm described elsewhere 31. In this study we used the BRD method49 for determining a converged regularization parameter (α). Additionally, we checked that the results obtained which include the T1, T2, and relative signal intensities did not change significantly with the value of α. In supplementary materials figure S3 we show the resulting ILT on one 2H T1 – T2 2D data set for different values of α ranging from 0.5 to 0.0005. While the resolution in the map is reduced with large value of α, the T1, T2, and relative signal intensities did not vary significantly. The converged value of α in all our 2H, 1H T1 – T2 and D – T2 2D data sets was approximately 10−4. Lastly, when documenting the measured T1 and T2 times we included the half-width of the peak of the peak observed in the ILT map and reported this in Tables 1 and 2.
Table 1.
Measured T1, T2, correlation times (τc), and relative populations of water reservoirs obtained from the untreated or treated (collagenase or EDTA) intact bone. The numbers shown after the ± symbol for any of the T1 and T2 relaxation times represent the half-width of the peak observed in the 2D ILT maps and have been propagated to the correlation times.
| Assignments | untreated intact bone | EDTA treated intact bone | collagenase treated intact bone | |
|---|---|---|---|---|
| Peak A | T1 [ms] | 475 ± 22.1 | 413 ± 19.2 | 433 ± 20.1 |
| T2 [ms] | 147 ± 15.4 | 81.1 ± 5.6 | 86.9 ± 6.0 | |
| τc [s] ×10−8 | 0.75 ± 0.11 | 1.09 ± 0.1 | 1.07 ± 0.1 | |
| Relative population | 0.62 | 0.23 | 0.54 | |
| Peak B | T1 [ms] | 413 ± 19.2 | 305 ± 21.3 | 394 ± 18.3 |
| T2 [ms] | 13.6 ± 1.4 | 13.2 ± 1.8 | 9.3 ± 1.3 | |
| τc [s] ×10−8 | 3.19 ± 0.27 | 2.75 ± 0.34 | 3.8 ± 0.41 | |
| Relative population | 0.23 | 0.19 | 0.37 | |
| Peak C | T1 [ms] | 385 ± 26.9 | 183 ± 12.8 | 376 ± 17.5 |
| T2 [ms] | 2.9 ± 0.5 | 3.1 ± 0.3 | 2.4 ± 0.5 | |
| τc [s] ×10−8 | 6.85 ± 0.95 | 4.48 ± 0.43 | 7.33 ± 1.1 | |
| Relative population | 0.15 | 0.58 | 0.09 | |
Table 2.
Measured T1, T2, correlation times (τc), relative populations and diffusion coefficients obtained from fractured bone, intact bone following mechanical wear, under an applied force and after releasing pressure. As discussed in the text, the estimated applied force to the bone was approximately 10 to 25 N. The numbers shown after the ± symbol for any of the T1 and T2 relaxation times or diffusion coefficients represent the half-width of the peak observed in the 2D ILT maps.
| Assignments | Fractured Bone | Following mechanical wear | Under an applied force | After releasing pressure | |
|---|---|---|---|---|---|
| Peak A | T1 [ms] | 385 ± 26.9 | 404 ± 28.2 | 377 ± 35.0 | 433 ± 20.1 |
| T2 [ms] | 116 ± 24.2 | 128 ± 22.4 | 104 ± 18.0 | 73.1 ± 7.6 | |
| τc [s] ×10−8 | 0.77 ± 0.22 | 0.73 ± 0.18 | 0.83 ± 0.21 | 1.2 ± 0.13 | |
| Relative population | 0.51 | 0.43 | 0.26 | 0.50 | |
| Diffusion Coefficient [μm2/ms] | 2.10 ± 0.07 | - | - | - | |
| Peak B | T1 [ms] | 291 ± 20.3 | 351 ± 24.5 | 115 ± 18.8 | 360 ± 16.7 |
| T2 [ms] | 22.4 ± 3.9 | 22.4 ± 3.9 | 16.3 ± 1.7 | 12.3 ± 0.8 | |
| τc [s] ×10-8 | 2.0 ± 0.31 | 2.2 ± 0.34 | 1.4 ± 0.25 | 3.1 ± 0.19 | |
| Relative population | 0.27 | 0.34 | 0.23 | 0.30 | |
| Diffusion Coefficient [μm2/ms] | 1.76 ± 0.06 | - | - | - | |
| Peak C | T1 [ms] | 215 ± 10 | 285 ± 13.2 | 41.5 ± 4.8 | 298 ± 13.9 |
| T2 [ms] | 5.1 ± 0.5 | 4.6 ± 0.32 | 4.3 ± 0.6 | 3.1 ± 0.3 | |
| τc [s] × 10-8 | 3.8 ± 0.30 | 4.6 ± 0.28 | 1.7 ± 0.28 | 5.8 ± 0.45 | |
| Relative population | 0.22 | 0.23 | 0.51 | 0.20 | |
| Diffusion Coefficient [μm2/ms] | 0.62 ± 0.07 | - | - | - | |
Results and Discussion
This section is organized as follows: we first describe our two dimensional T1 – T2 relaxation experiments that were performed in deuterium hydrated cortical bone and following EDTA and collagenase treatments. Second, we highlight results from relaxation exchange experiments, by T2 – T2 methodology, which provide insight to the connectivity of the sites resolved in our T1 – T2 data. Lastly, we discuss findings from our diffusion-relaxation measurements and T1 – T2 measurements on bone under an applied force, following fracture, or mechanical wear.
T1 – T2 Experiments on 2H Hydrated Untreated, EDTA, and Collagenase Treated Bone Samples
The 2H 2D ILT T1 – T2 map of 2H2O hydrated untreated intact bone acquired at 22°C is shown in figure 1. In the 2D ILT map of untreated intact bone, apart from the bulk water (T1 and T2 value of about 464 ms and 433 ms), three water peaks (labeled A, B and C) are observed each pointing to environments where water molecules experience different dynamical characteristics distinguishable over the time scale ranging from approximately 1ms to 4.2s (range of values probed in the T2 dimension). Similar findings were observed with the same number of reservoirs in a 1H 2D T1 –T2 experiment, with cortical bone hydrated in H2O (data shown in the supplementary materials figure S4). Measurements on either 1H or 2H preclude the observation of water molecules having a relaxation time T2 less than our sampling time which was 0.35 ms for 2H and 1.5 ms for 1H. The 2H relaxation measurements, as we discuss in detail below, allow for a probe of the rotational correlation times as the relaxation pathway for 2H nuclei is quadrupolar in origin. Furthermore, as this work focuses on investigating the dynamics of water only, 2H detection allows us to readily separate the signal from 1H nuclei which relax by 1H-1H dipolar coupling (note that 1H relaxation would be mediated by 1H nuclei in the surrounding water as well as that of the neighboring protein nuclei 50).
Figure 1.
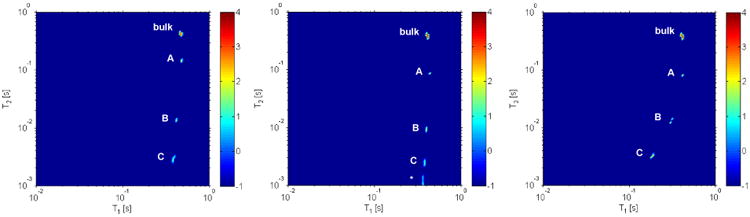
2D ILT map of the 2H T1 – T2 experiment performed on untreated (left), collagenase (middle), and EDTA (right) treated intact bone at 22 °C. In all three measurements four peaks are observed. Note that in the middle figure the peak denoted with a * is likely an artifact. In this figure the color bar to the right, denoting signal intensity in arbitrary units, is on a logarithmic scale.
Bone has a complex composite structure allowing for different environments for water, which include pores in between the mineral and collagen matrix. The peak denoted A, with T1 and T2 values of 475 ms and 147 ms respectively was assigned to the water molecules which are present at the surface of the bone sample. To support our assignment an additional experiment was performed by removing the bulk water from the sample tube (supplementary figure S5). This measurements revealed that peak A is greatly suppressed leaving peaks B and C and some residual bulk water (likely from water on the surface the NMR tube). The observation that the relaxation times are shifted with the bulk water removed is described below, and is due to exchange that occurs over times scales longer than 10 ms. Table 1 summarizes the relaxation times observed as well as the relative peak intensity of peaks B, C, and A relative to the sum of their combined intensity.
For 2H nucleus of 2H2O the 2H-1H and 2H-2H dipolar interactions are negligible and the NMR relaxation times are dominated by the quadrupolar interaction. The T1 and T2 are given by
| (1) |
| (2) |
In the above equation, ω0 is the Larmor frequency of the deuterium nucleus and is the effective quadrupolar coupling constant where, Γ is a motional averaging parameter. The spectral density in these equations is defined by , where τc denotes the correlation time of the nuclear spin and surrounding electric field gradient. For 2H nucleus of 2H2O, this correlation time is intramolecular in origin and allows for a probe of the rotational motion of a given water molecule. In the above formalism it is assumed that the molecular motion of the D2O exhibits fast oscillatory motion and slower diffusional process51. According to Lang et al., in the short time, reorientational motion of a given water molecule consists of fast librations about an equilibrium orientation in a random network of surrounding water molecules. In the present system the network includes the collagen protein or apatite mineral. In longer time scales, the slow diffusive motion will lead to changes in the orientation of the water molecule and thereby it will rearrange with respect to the surrounding network which includes collagen and hydroxyapatite. In this model, one assumes the cross-correlation between the two relaxation pathways is separable, as fast librational motion occurs on a much shorter time scale compared to diffusive motion51.
Using the theoretical formalism described above, the correlation time of the water molecules were determined by the ratio of the two relaxation times T1 and T2 and are summarized in Table 1. The observed relaxation times for peaks B and C are characteristic of anisotropic tumbling as T1 > T2. Additionally, the correlation time for water molecules in reservoir C (6.85 ± 0.95 × 10−8s) is approximately a factor of 2 greater than that of water molecules in reservoir B (3.19 ± 0.27 × 10−8s). We point out that the correlation time for the bulk water was not reported, as the method for determining a correlation time via the ratio of T1 and T2 is inapplicable when T1=T2. To obtain information about the activation energy for tumbling motion of different water reservoirs (i.e., peak B and C) inside the bone matrix, additional T1 – T2 experiments were carried out in the temperature range from 2 to 40 °C (data not shown). We found that the trend observed in these variable temperature experiments appeared to indicate a reduction in the correlation time of peak B with increasing temperature, whereas the data for peak C were quite scattered. Although we attempted to fit an Arrhenius expression to these data, the fit was poor and suggestive of a phase change.
Previous studies showed that water present in bone can be both mobile and tightly bound3,16,17,21,52. Horch et al. performed a1H 2D T1 – T2 experiment on human cortical bone and they observed several peaks17. Their work highlighted two short lived T2 components with T2 values of 60 and 400 μs at 4.7 T arising from collagen side chain or methylene protons and collagen bound water, respectively, whereas the long T2 component arises from pore water. They also carried out T2 – T2 relaxation exchange spectroscopy (REXSY) and observed a cross peak between the short relaxing T2 components with a mixing time of 200 ms but they remain isolated from the long lived protons. Unal et al. recently employed Raman spectroscopy to study bound and unbound bone water following drying and exchange of water by ethanol or deuterium 52. By analyzing the different Raman–OH stretch bands they were able to identify different water compartments i.e., unbound water and water bound to either the collagen or mineral sites. Their work suggested that water molecules are mainly bound to the collagen and a small portion to the mineral phase. Previous NMR studies on bone water have allowed for differentiating the details of collagen bound and mobile water16, and in addition, water in proximity to the mineral matrix53.
To provide further insight into the environments of the different water reservoirs in cortical bone we systematically exposed the samples to EDTA or collagenase. It is expected that collagenase will partially digest the collagen and EDTA treatment will demineralize the bone and thereby affect the population of water in these respective reservoirs that are observable and distinguishable on the NMR time scale. Without EDTA or collagenase treatment we find an almost equal intensity of water in sites B and C (0.23 and 0.15 respectively). The observation of almost equal concentration of water in the two sites appears to correlate with the concentration of collagen and mineral in bone which is known to be approximately equal (reported % by volume) 29.
Similar to the untreated bone sample (figure 1), four water peaks are observed including the bulk water signal in the EDTA or collagenase treated samples. Due to this treatment, a change is observed in the relative population of the different peaks resolved in the T1-T2 map (Table 1). Compared to the population present in untreated bone sample, when the collagenase treatment is performed the relative intensity of peak C is reduced by 40 %, whereas the EDTA treatment decreases the population of peak B 17.4 %. This observation indicates that the peaks denoted B and C result from water present in mineral rich and collagen rich environments, respectively. Additionally, the EDTA treatment modified the surface of the outer surface of the bone sample as well (this was observable macroscopically with the naked eye) and changes were observed for both the correlation times and populations for peak A. Furthermore, 13C MAS NMR spectra of collagenase and EDTA treated bone samples (supplementary figure S6) verified that the collagenase treatment worked well as the intensity of all the spectroscopically resolvable 13C peaks are greatly reduced after collagenase treatment. Though the cross-polarization experiment is not quantitative, the intensity of the carboxylic carbon is reduced by 20 % compared to the EDTA treated bone sample. Our measurements also indicate changes in the T1 and T2 relaxation times following EDTA or collagenase treatment. We suspect that this may be due to the additional crystallites or collagen fragments in the reservoirs denoted A, B, and C following each respective treatment and as a consequence should not be interpreted quantitatively.
T2 – T2 Experiments on Untreated Bone Samples
Figure 2 represents the 2D ILT maps of T2 – T2 exchange experiments that were performed using mixing times ranging from 100 μs to 10 ms at 22 °C. Similar to the T1 – T2 experiments, three peaks (A, B and C) and bulk are observed. This experiment allows the study of exchange between the various water sites and the appearance of cross peaks in the T2 – T2 map is a signature of exchange during the mixing time. However, no cross peaks are observed indicating no exchange over the time scales probed. Additionally, the T2 values measured here appear slightly larger than that observed in T1 – T2 experiments —this could be due to slow exchange with the bulk D2O. However, the ability to measure the exchange at longer time scales beyond 10 ms is challenging due to the short T2 relaxation times.
Figure 2.
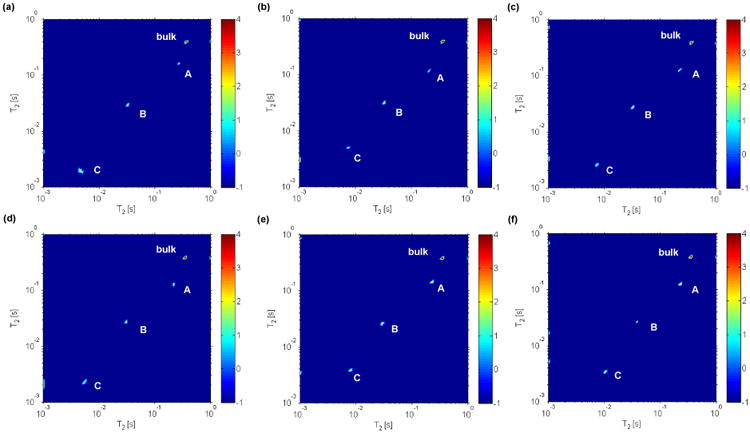
2D ILT map of 2H T2 – T2 exchange experiments performed on untreated intact bone at 22 °C. In the experiments the mixing time tm (supplementary figure S1) was experimentally varied as follows (a) 100 μs, (b) 500 μs, (c) 1 ms, (d) 2 ms, (e) 3 ms and (f) 10 ms. No cross peaks are observed over the times probed which would indicate slow or little exchange between different sites. In this figure the color bar to the right, denoting signal intensity in arbitrary units, is on a logarithmic scale.
T1–T2 and D-T2 Measurements: Fracture, Mechanical Wear, and Load Bearing Studies
T1 – T2 experiments were performed on fractured bone and similar to the intact bone sample four water peaks were visible in the 2D ILT map (figure not shown). The observed T1 and T2 values and the correlation times (τc) are tabulated in table 2. A reduction in correlation time for peak B and C is observed compared to that of the intact bone sample. Because the correlation time appearing in equations 1 and 2 is intramolecular in origin, the reduction in τc indicates that the water tumbling motion increases when the bone is fractured. Fracturing of bone may decrease the interaction of water molecules to the collagen and mineral matrix and thereby increase water mobility. However, no change in τc is observed for peak A, which again strengthens our interpretation that peak A corresponds to surface water. We note that the population of peak A decreases slightly following fracture from 0.62 to 0.51. The changes in the relative populations in fractured bone should not be interpreted quantitatively as the size of the fractured fragments we used were smaller than that of the intact bone studied (we expect that the relative population should increase as the surface area of the fragments following fracture are greater than the initial size of the sample).
Additional T1 – T2 experiments were performed after inducing mechanical wear on the bone with a scalpel and when the bone sample was placed under an applied load. The observed T1 and T2 values, correlation times, and the relative population of the peaks deduced from these measurements are tabulated in Table 2. We note that the damage induced by mechanical wear with a scalpel is likely not a realistic model of wear that occurs physiologically, but probes the effects of acute wear or damage. As in the case of fractured bone, following mechanical wear on the surface a reduction in correlation time is observed for peak B and C compared to that of the intact bone sample. The findings resulting from measurements of bone under an applied load are highlighted graphically in Figure 3; the correlation times of peak B and C decrease (Table 2 and Figure 3). However, the reduction in τc for peak C is much higher compared to fracturing the bone or after mechanical wear. This behavior would suggest that when bone is exposed to an external force the collagen is more significantly altered or deformed. This deformation may lead to the breaking of hydrogen bonds between water molecules and collagen which result in changes in the tumbling motion of localized water. An additional key finding of these measurements is that the population of water in the collagen rich site increases from 0.15 to 0.51 when bone is placed under a mechanical load, whereas the population of the mineral rich site does not change. The same measurement revealed, as expected, that the population of surface water is altered (decreases from 0.62 to 0.26) when a force was applied. This observation resulted from the fact that two nylon screws were used to compress the bone which altered its surface area.
Figure 3.

T1, T2, τ c, and relative populations of the water reservoirs denoted A, B and C (see figure 1) in bone without an applied force (yellow), under an applied load (green) and when the applied force was removed (blue).
A T1 – T2 experiment was also performed on the bone sample 23 days after releasing pressure (Table 2 or Figure 3). We observed that changes in the correlation times and populations of water were somewhat reversible to their initial values without an applied force. The finding that the populations and dynamics of water are altered more so at the collagen rich site under mechanical load suggest that much of the load bearing occurs therein, and not at the mineral rich sites. This behavior is reversible and indicates that the flexibility of bone is largely due to collagen.
It is known that dehydration significantly affects the mechanical properties of bone9,10,18,54. Water in bone may form hydrogen bonds with the collagen or mineral matrix or hydrogen bonded water bridges. The role of water in fracture resistance of dentin, a bio-composite with structure comparable with bone, has been studied55. Following chemical dehydration with different solvents (e.g., acetone, ethanol, methanol which form weaker hydrogen bonds), Nalla et al. suggested the hydrogen bonded network or hydration states and may increase the material strength, stiffness, and fracture properties. However, the hydrogen bonding of water plasticizes collagen leading to lower strength, stiffness, and fracture resistance55. To follow the strain level in bone (tissue, fibrils and mineral particle) Gupta et al. used in situ tensile testing with combined synchroton X-ray diffraction and scattering56. Their work showed that the structural arrangement at the nanoscale leads to deformation and that the collagen matrix plays an important role in strain transfer to mineral platelets56. Our study shows that fracture and/or mechanical wear increases the mobility of water molecules inside the bone as the correlation times of peak B and C are observed to decrease. Thus, this study provides additional insight that water mobility in bone may be used as a local probe of microscopic fracture which may be challenging to observe with current methods. For example, one might envision applying these methods in a spatially resolved two dimensional 1H T1-T2 measurement to probe local microscopic fracture and mechanical wear in bone.
Two dimensional diffusion–T2 experiments were performed on the fractured bone at 22 °C. Figure 4 shows the 2D ILT map, where four water peaks are visible as observed in previous experiments. The observation of four peaks with diffusion coefficients on the order of μm2/ms eliminates the possibility that any of the peaks observed in the T1 – T2 and T2 – T2 experiments raised from 2H exchange with 1H protein nuclei. As the diffusion–T2 experiment was carried out in a different magnetic field a slightly different T2 is observed compared to T1 – T2 experiments. The measurement of diffusion for short T2 components e.g., peak C (T2 = 5 ms in fractured bone) is experimentally challenging as it requires strong gradient pulses to encode and decode the grating to track diffusion. Fernandez et al. studied the dynamics of osteoid water exchange (i.e., exchange of water by D2O) by NMR and reported the apparent diffusion coefficient of water in cortical bone of rabbit to be (3.56 ± 0.78) × 10−7 cm2/s at 25 °C3. In bone, the diffusion of water presumably takes place through the pores, i.e., Haversian canals, lacunae, canaliculi etc. However, in their study they observed a significant amount of water with slower diffusion than that of the major water fraction passing through the pores mentioned above, monitored from the slow exchange rate. The water molecules with slow apparent diffusion coefficients are associated to the collagen or mineral sites and diffuse along the micropores present in collagen-hydroxyapatite matrix. Diffusion of water molecules associated with the mineral rich environment is faster compared to that of the collagen rich environment (peak B, D=1.76 ± 0.06 μm2/ms and peak C, D=0.62 ± 0.07 μm2/ms). A possible reason for the reduction in transport may arise from diffusion being hindered due to the strong affinity between water and collagen. Given these diffusion coefficients it is worthwhile to provide a spatial dimension probed by the T1 – T2 measurements. For peak B the diffusion coefficient is approximately 1.76 μm2/ms and over a time scale of T2 (T2 = 13.6 ms) the spatial dimension we probe is . For peak C the diffusion coefficient was lower, D = 0.62 μm2/ms, and over a time scale of T2 (T2 = 2.9 ms) the spatial dimension probed is approximately Δr (≃) 3μm.
Figure 4.
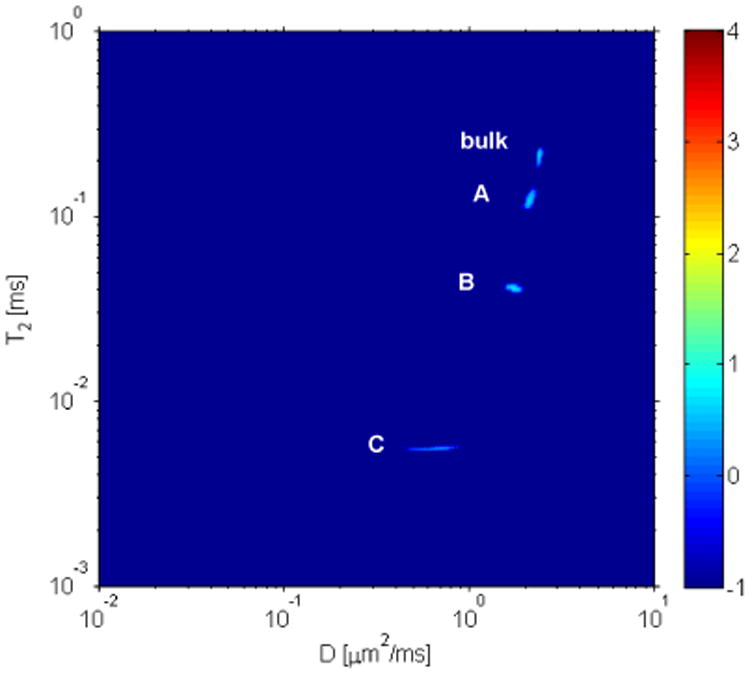
2D diffusion-T2 correlation map of fractured bone soaked in D2O for 30 days. As discussed in the text, four distinguishable components of water were observed. Note that the measured T2 values are slightly different than that in the T1 – T2 and T2 – T2 as the Larmor field was slightly different. The measured diffusion coefficients are tabulated in table 2. In this figure the color bar to the right, denoting signal intensity in arbitrary units, is on a logarithmic scale.
Water, arguably the most ubiquitous molecule found in biological tissue, is known to mediate macromolecular ordering via hydrogen bonding or electrostatic interactions, especially in collagen. Recently, Fathima et al. showed that the extent of cross linking in collagen samples modified by different chemical agents alter significantly the dynamics of bound water on the NMR time scale57. Libonati et al.58 performed molecular dynamic simulations in a collagen-hydroxyapatite nanocomposite (in both dry and wet cases). Their work showed that the hydrogen bonds between collagen and hydroxyapatite play an important role in fracture resistance. In their model, they showed that confinement leads to breaking of intramolecular hydrogen bonding, whereas inter-molecular hydrogen bonds undergo continuous formation and breakage. In their simulation, water present in bone forms a network of hydrogen bonds, and in the confined state, the progressive formation and breakage of hydrogen bonds acts as an energy dissipation mechanism. The observation that the NMR T2 relaxation times of water may be used to study the volume of bound and pore water in cortical bone and report on fracture characteristics has been reported recently elsewhere28. In our study, we have observed the changes in localized water dynamics and relative populations in bone due to fracture or mechanical wear, and when bone is under an applied force. Our observations suggest that water dynamics can be used to follow structural changes of collagen as bone undergoes alteration or deformation under stress, fracture, or mechanical wear.
Conclusions
In this work we have measured the tumbling correlation times, diffusion coefficients, population and exchange of water in goat cortical bone by 2D 2H T1 – T2, T2 – T2, and diffusion–T2 NMR methods. Collagenase or EDTA treatment allowed for assigning the different water reservoirs revealed in our relaxation measurements to a collagen or a mineral rich environment of the bone matrix. Our T2 – T2 correlation measurements indicate these sites are relatively isolated as no exchange was observed over time scales from 1 ms to 10 ms. Lastly, T1 – T2 experiments were performed on fractured bone, bone under mechanical wear, and under applied force revealing dynamical and population changes of the various sites. These measurements revealed that load bearing occurs largely in the collagen rich site, and that the changes in relative populations and dynamics of water may serve as a local probe of microscopic fracture which may be challenging to probe with other methods. While this study focused on goat cortical bone, we believe the findings would be the same across cortical bones from other mammalian subjects as the bone structures are similar59.
Supplementary Material
Acknowledgments
Research performed at Brooklyn College of the City University of New York was supported from award No. SC1GM086268-08 from the National Institute of General Medical Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health. The content is solely the responsibility of the authors and does not represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health (NIH). The authors thank S. W. Morgan for useful discussions and help with the experiments. Chandan Singh, acknowledges research fellowship from CSIR, India. Financial support from the Department of Biotechnology India (Grant No. BT/PRI2700/BRB/10/719/2009), is gratefully acknowledged. Lastly, the authors thank Yi-Qiao Song for the use of the ILT code used in this work.
Footnotes
Author Contributions: F. G., C.S., and A.P. performed measurements and analysis of experimental data and contributed to writing the manuscript. N.S. contributed to writing the manuscript. G.S.B coordinated the paper, performed measurements and analysis, and contributed to writing the manuscript.
Supporting Information: Complete author list of reference 11 and 13, diffusive attenuation kernel, pulse sequences used for 2D T1 – T2 and 2D diffusion–T2 experiments, 2D ILT map of 2H T1-T2 at different values of the regularization parameter α, 2D ILT map of 1H T1-T2 experiments performed on untreated intact bone at 22°C, 2D ILT map of 2H T1-T2 experiments performed on untreated intact bone at 22°C after immersing the bone in D2O and after removal of D2O from the NMR tube, 13C MAS NMR spectra of collagenase and EDTA treated bone sample. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Davies E, Müller KH, Wong WC, Pickard CJ, Reid DG, Skepper JN, Duer MJ. Citrate Bridges Between Mineral Platelets in Bone. Proc Natl Acad Sci USA. 2014;111:E1354–E1363. doi: 10.1073/pnas.1315080111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shoulders MD, Raines RT. Collagen Structure and Stability. Annu Rev Biochem. 2009;78:929–958. doi: 10.1146/annurev.biochem.77.032207.120833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fernández-Seara MA, Wehrli SL, Wehrli FW. Diffusion of Exchangeable Water in Cortical Bone Studied by Nuclear Magnetic Resonance. Biophys J. 2002;82:522–529. doi: 10.1016/S0006-3495(02)75417-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ritchie RO, Buehler MJ, Hansma P. Plasticity and Toughness in Bone. Phys Today. 2009;62:41–47. [Google Scholar]
- 5.Ammann P, Rizzoli R. Bone Strength and its Determinants. Osteoporos Intl. 2003;14:13–18. doi: 10.1007/s00198-002-1345-4. [DOI] [PubMed] [Google Scholar]
- 6.Lips P. Epidemiology and Predictors of Fractures Associated with Osteoporosis. Am J Med. 1997;103:S3–S11. doi: 10.1016/s0002-9343(97)90021-8. [DOI] [PubMed] [Google Scholar]
- 7.Kanis JA, Borgstrom F, De Laet C, Johansson H, Johnell O, Jonsson B, Oden A, Zethraeus N, Pfleger B, Khaltaev N. Assessment of Fracture Risk. Osteoporos Intl. 2005;16:581–589. doi: 10.1007/s00198-004-1780-5. [DOI] [PubMed] [Google Scholar]
- 8.Hui SL, Slemenda CW, Johnston CC., Jr Age and Bone Mass as Predictors of Fracture in a Prospective Study. J Clin Invest. 1988;81:1804–1809. doi: 10.1172/JCI113523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nyman JS, Roy A, Shen X, Acuna RL, Tyler JH, Wang X. The Influence of Water Removal on the Strength and Toughness of Cortical Bone. J Biomech. 2006;39:931–938. doi: 10.1016/j.jbiomech.2005.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rai RK, Sinha N. Dehydration-Induced Structural Changes in the Collagen–Hydroxyapatite Interface in Bone by High-Resolution Solid-State NMR Spectroscopy. J Phys Chem C. 2011;115:14219–14227. [Google Scholar]
- 11.McElderry JDP, Zhu P, Mroue KH, Xu J, Pavan B, Fang M, Zhao G, McNerny E, Kohn DH, Franceschi RT, et al. Crystallinity and Compositional Changes in Carbonated Apatites: Evidence from 31P Solid-State NMR, Raman, and AFM Analysis. J Solid State Chem. 2013;206:192–198. doi: 10.1016/j.jssc.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson EE, Awonusi A, Morris MD, Kohn DH, Tecklenburg MM, Beck LW. Three Structural Roles for Water in Bone Observed by Solid-State NMR. Biophys J. 2006;90:3722–3731. doi: 10.1529/biophysj.105.070243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Von Euw S, Fernandes FM, Cassaignon S, Selmane M, Laurent G, Pehau-Arnaudet G, Coelho C, Bonhomme-Coury L, Giraud-Guille MM, et al. Water-Mediated Structuring of Bone Apatite. Nat Mater. 2013;12:1144–1153. doi: 10.1038/nmat3787. [DOI] [PubMed] [Google Scholar]
- 14.Zhu P, Xu J, Sahar N, Morris MD, Kohn DH, Ramamoorthy A. Time-Resolved Dehydration-Induced Structural Changes in an Intact Bovine Cortical Bone Revealed by Solid-State NMR Spectroscopy. J Am Chem Soc. 2009;131:17064–17065. doi: 10.1021/ja9081028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santos R, Wind R, Bronnimann C. 1H CRAMPS and 1H-31P HetCor Experiments on Bone, Bone Mineral, and Model Calcium Phosphate Phases. J Magn Reson Series B. 1994;105:183–187. doi: 10.1006/jmrb.1994.1120. [DOI] [PubMed] [Google Scholar]
- 16.Ong HH, Wright AC, Wehrli FW. Deuterium Nuclear Magnetic Resonance Unambiguously Quantifies Pore and Collagen-Bound Water in Cortical Bone. J Bone Miner Res. 2012;27:2573–2581. doi: 10.1002/jbmr.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horch RA, Nyman JS, Gochberg DF, Dortch RD, Does MD. Characterization of 1H NMR Signal in Human Cortical Bone for Magnetic Resonance Imaging. Magn Reson Med. 2010;64:680–687. doi: 10.1002/mrm.22459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nyman JS, Ni Q, Nicolella DP, Wang X. Measurements of Mobile and Bound Water by Nuclear Magnetic Resonance Correlate with Mechanical Properties of Bone. Bone. 2008;42:193–199. doi: 10.1016/j.bone.2007.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson EE, Awonusi A, Morris MD, Kohn DH, Tecklenburg MM, Beck LW. Highly Ordered Interstitial Water Observed in Bone by Nuclear Magnetic Resonance. J Bone Miner Res. 2005;20:625–634. doi: 10.1359/JBMR.041217. [DOI] [PubMed] [Google Scholar]
- 20.Wehrli FW, Fernández-Seara MA. Nuclear Magnetic Resonance Studies of Bone Water. Ann Biomed Eng. 2005;33:79–86. doi: 10.1007/s10439-005-8965-8. [DOI] [PubMed] [Google Scholar]
- 21.Ni Q, Nyman JS, Wang X, De Los Santos A, Nicolella DP. Assessment of Water Distribution Changes in Human Cortical Bone by Nuclear Magnetic Resonance. Meas Sci Technol. 2007;18:715–723. [Google Scholar]
- 22.Wang X, Ni Q. Determination of Cortical Bone Porosity and Pore Size Distribution Using a Low Field Pulsed NMR Approach. J Orthopaedic Res. 2003;21:312–319. doi: 10.1016/S0736-0266(02)00157-2. [DOI] [PubMed] [Google Scholar]
- 23.Fantazzini P, Bortolotti V, Brown RJ, Camaiti M, Garavaglia C, Viola R, Giavaresi G. Two 1H-Nuclear Magnetic Resonance Methods to Measure Internal Porosity of Bone Trabeculae: By Solid–Liquid Signal Separation and by Longitudinal Relaxation. J Appl Phys. 2004;95:339–343. [Google Scholar]
- 24.Ni Q, King JD, Wang X. The Characterization of Human Compact Bone Structure Changes by Low-Field Nuclear Magnetic Resonance. Meas Sci Technol. 2004;15:58–66. [Google Scholar]
- 25.Ni Q, Nicolella DP. The Characterization of Human Cortical Bone Microdamage by Nuclear Magnetic Resonance. Meas Sci Technol. 2005;16:659–668. [Google Scholar]
- 26.Rai RK, Singh C, Sinha N. Predominant Role of Water in Native Collagen Assembly Inside Bone Matrix. J Phys Chem B. 2015;119:201–211. doi: 10.1021/jp511288g. [DOI] [PubMed] [Google Scholar]
- 27.Rai RK, Barbhuyan T, Singh C, Mittal M, Khan MP, Sinha N, Chattopadhyay N. Total Water, Phosphorus Relaxation and Inter-Atomic Organic to Inorganic Interface are New Determinants of Trabecular Bone Integrity. PloS One. 2013;8:e83478–e83487. doi: 10.1371/journal.pone.0083478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Granke M, Makowski AJ, Uppuganti S, Does MD, Nyman JS. Identifying Novel Clinical Surrogates to Assess Human Bone Fracture Toughness. J Bone Miner Res. 2015 doi: 10.1002/jbmr.2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Granke M, Does MD, Nyman JS. The Role of Water Compartments in the Material Properties of Cortical Bone. Calcif Tissue Intl. 2015:1–16. doi: 10.1007/s00223-015-9977-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Venkataramanan L, Song YQ, Hurlimann MD. Solving Fredholm Integrals of the First Kind with Tensor Product Structure in 2 and 2.5 Dimensions. Signal Proc IEEE Trans. 2002;50:1017–1026. [Google Scholar]
- 31.Song YQ, Venkataramanan L, Hürlimann M, Flaum M, Frulla P, Straley C. T1 – T2 Correlation Spectra Obtained Using a Fast Two-Dimensional Laplace Inversion. J Magn Reson. 2002;154:261–268. doi: 10.1006/jmre.2001.2474. [DOI] [PubMed] [Google Scholar]
- 32.Song YQ. Magnetic Resonance of Porous Media (MRPM): A Perspective. J Magn Reson. 2013;229:12–24. doi: 10.1016/j.jmr.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 33.Song YQ. Focus on the Physics of Magnetic Resonance on Porous Media. New J of Phys. 2012;14:055017. [Google Scholar]
- 34.Sun C, Mitchell O, Huang J, Boutis GS. NMR Studies of Localized Water and Protein Backbone Dynamics in Mechanically Strained Elastin. J Phys Chem B. 2011;115:13935–13942. doi: 10.1021/jp207607r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ukpebor OT, Shah A, Bazov E, Boutis GS. Inverse Temperature Transition of Elastin Like Motifs in Major Ampullate Dragline Silk: MD Simulations of Short Peptides and NMR Studies of Water Dynamics. Soft matter. 2014;10:773–785. doi: 10.1039/C3SM52001C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watanabe E, Boutis GS, Sato H, Sekine S, Asakura T. NMR Studies of Thermo-Responsive Behavior of an Amphiphilic Poly (Asparagine) Derivative in Water. Polymer. 2014;55:278–286. doi: 10.1016/j.polymer.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hills B, Costa A, Marigheto N, Wright K. T1–T2 NMR Correlation Studies of High-Pressure-Processed Starch and Potato Tissue. Appl Magn Reson. 2005;28:13–27. [Google Scholar]
- 38.McDonald P, Korb JP, Mitchell J, Monteilhet L. Surface Relaxation and Chemical Exchange in Hydrating Cement Pastes: a Two-Dimensional NMR Relaxation Study. Phy Rev E. 2005;72:011409–9. doi: 10.1103/PhysRevE.72.011409. [DOI] [PubMed] [Google Scholar]
- 39.Kleinberg R, Farooqui S, Horsfield M. T1/T2 Ratio and Frequency Dependence of NMR Relaxation in Porous Sedimentary Rocks. J of Colloid Interface Sci. 1993;158:195–198. [Google Scholar]
- 40.Lee JH, Labadie C, Springer CS, Jr, Harbison GS. Two-Dimensional Inverse Laplace Transform NMR: Altered Relaxation Times Allow Detection of Exchange Correlation. J Am Chem Soc. 1993;115:7761–7764. [Google Scholar]
- 41.Washburn KE, Callaghan PT. Tracking Pore to Pore Exchange Using Relaxation Exchange Spectroscopy. Phys Rev Lett. 2006;97:175502–175505. doi: 10.1103/PhysRevLett.97.175502. [DOI] [PubMed] [Google Scholar]
- 42.Meiboom S, Gill D. Modified SpinEcho Method for Measuring Nuclear Relaxation Times. Rev Sci Instrum. 1958;29:688–691. [Google Scholar]
- 43.Zhang W, Cory D. Pulsed Gradient NMR Probes for Solid State Studies. J Magn Reson. 1998;132:144–149. doi: 10.1006/jmre.1998.1408. [DOI] [PubMed] [Google Scholar]
- 44.Wu D, Chen A, Johnson CS. An Improved Diffusion-Ordered Spectroscopy Experiment Incorporating Bipolar-Gradient Pulses. J Magn Reson Series A. 1995;115:260–264. [Google Scholar]
- 45.Hahn E. Spin Echoes. Phys Rev. 1950;80:580–594. [Google Scholar]
- 46.Mills R. Self-Diffusion in Normal and Heavy Water in the Range 1-45°. J Phys Chem. 1973;77:685–688. [Google Scholar]
- 47.Kiselev VG. The Cumulant Expansion: an Overarching Mathematical Framework for Understanding Diffusion NMR. Diffusion MRI: Theory, Methods, and Applications. 2011:152–168. [Google Scholar]
- 48.Sodickson A, Cory DG. A Generalized k-Space Formalism for Treating the Spatial Aspects of a Variety of NMR Experiments. Prog Nucl Magn Reson Spectrosc. 1998;33:77–108. [Google Scholar]
- 49.Butler J, Reeds J, Dawson S. Estimating Solutions of First Kind Integral Equations with Nonnegative Constraints and Optimal Smoothing. SIAM J Numer Anal. 1981;18:381–397. [Google Scholar]
- 50.Gore J, Brown M, Zhong J, Armitage I. Prediction of Proton Relaxation Rates from Measurements of Deuterium Relaxation in Aqueous Systems. J Magn Reson (1969) 1989;83:246–251. [Google Scholar]
- 51.Lang E, Lüdemann HD, Piculell L. Nuclear Magnetic Relaxation Rate Dispersion in Supercooled Heavy Water Under High Pressure. J Chem Phys. 1984;81:3820–3827. [Google Scholar]
- 52.Unal M, Yang S, Akkus O. Molecular Spectroscopic Identification of the Water Compartments in Bone. Bone. 2014;67:228–236. doi: 10.1016/j.bone.2014.07.021. [DOI] [PubMed] [Google Scholar]
- 53.Seifert AC, Li C, Rajapakse CS, Bashoor-Zadeh M, Bhagat YA, Wright AC, Zemel BS, Zavaliangos A, Wehrli FW. Bone Mineral 31P and Matrix-Bound Water Densities Measured by Solid-State 31P and 1H MRI. NMR Biomed. 2014;27:739–748. doi: 10.1002/nbm.3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fernndez-Seara MA, Wehrli SL, Takahashi M, Wehrli FW. Water Content Measured by Proton-Deuteron Exchange NMR Predicts Bone Mineral Density and Mechanical Properties. J Bone Miner Res. 2004;19:289–296. doi: 10.1359/JBMR.0301227. [DOI] [PubMed] [Google Scholar]
- 55.Nalla R, Balooch M, Ager J, Iii, Kruzic J, Kinney J, Ritchie R. Effects of Polar Solvents on the Fracture Resistance of Dentin: Role of Water Hydration. Acta Biomaterialia. 2005;1:31–43. doi: 10.1016/j.actbio.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 56.Gupta HS, Seto J, Wagermaier W, Zaslansky P, Boesecke P, Fratzl P. Cooperative Deformation of Mineral and Collagen in Bone at the Nanoscale. Proc Natl Acad Sci USA. 2006;103:17741–17746. doi: 10.1073/pnas.0604237103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fathima NN, Baias M, Blumich B, Ramasami T. Structure and Dynamics of Water in Native and Tanned Collagen Fibers: Effect of Crosslinking. Intl J Biol Macromol. 2010;47:590–596. doi: 10.1016/j.ijbiomac.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 58.Libonati F, Nair AK, Vergani L, Buehler MJ. Mechanics of Collagen–Hydroxyapatite Model Nanocomposites. Mech Res Commun. 2014;58:17–23. [Google Scholar]
- 59.Locke M. Structure of Long Bones in Mammals. Journal of Morphology. 2004;262:546–565. doi: 10.1002/jmor.10282. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.