

Abstract
A fascinating yet perhaps overlooked trait of the p75 neurotrophin receptor (p75NTR) is its ability to bind ligands with no obvious neurotrophic function. Using cultured basal forebrain (BF) neurons, this study demonstrates selective internalization of amyloid β (Aβ) 1–42 in conjunction with p75NTR (labelled with IgG192-Cy3) by cholinergic cells. Active under resting conditions, this process was enhanced by high K+ stimulation and was insensitive to inhibitors of regulated synaptic activity—tetrodotoxin or botulinum neurotoxins (BoNT type/A and/B). Blockade of sarco-endoplasmic reticulum (SERCA) Ca2+ ATPase with thapsigargin and CPA or chelation of Ca2+ with EGTA-AM strongly suppressed the endocytosis of p75NTR, implicating the role of ER released Ca2+. The uptake of IgG192-Cy3 was also reduced by T-type Ca2+ channel blocker mibefradil but not Cd2+, an indiscriminate blocker of high voltage-activated Ca2+ currents. A strong co-localization of IgG192-Cy3 with late endosome (Rab7) or lysosome (Lamp1) qualifier proteins suggest these compartments as the primary destination for internalized IgG192 and Aβ. Selective uptake and labeling of BF cholinergic cells with IgG192-Cy3 injected into the prefrontal cortex was verified also in vivo. The significance of these findings in relation to Aβ clearance in the cerebral cortex and pathophysiology of Alzheimer’s disease is discussed.
Keywords: p75 NTR, Basal forebrain cholinergic neurons, Lysosomal degradation, Calcium, Amyloid β clearance, Alzheimer’s disease
Introduction
The amyloid cascade hypothesis infers excessive deposition of β amyloid (Aβ) peptide in the brain as the proximal cause of Alzheimer’s disease (AD). It purports the overproduction or failure of Aβ clearance as a key factor to the pathological process, which is characterized by progressive synaptic loss and cognitive decline (Hardy and Higgins 1992; Mesulam 2004). Being one of the end products of amyloid protein precursor (APP) cleavage, Aβ when in excess, aggregates in the form of insoluble oligomers and fibrillary plaques, causing severe functional and structural disruptions, which affect primarily the limbic, paralimbic and associative areas of the cerebral cortex (Mesulam 2004; Buckner et al. 2008). The confinement of the early pathology to certain brain regions along with co-related deficits in cognitive and mnemonic (but not motor and visceral) functions are highly perplexing, given that APP is ubiquitously expressed throughout the central nervous system (Lah and Levey 2000).
Progressive depletion of cortical acetylcholine with loss of cholinergic innervations is another consistent trait of brains affected by AD (Mesulam 2004; McKinney and Jacksonville 2005). Analysis of human AD autopsies has revealed a positive correlation between the extent of cholinergic axonal loss and density of Aβ plaques in several cortical regions (Mesulam 1990; Geula et al. 1998). While an increase in Aβ loading associated with degeneration of cholinergic axons is expected, given that acetylcholine via M1 receptors promotes non-amyloidogenic cleavage of APP (Nitsch et al. 1992; Yan and Feng 2004), the mechanisms underlying greater vulnerability of cholinergic axons in AD remains unclear, despite decades of intense research (Mesulam 2004). The prevailing, neurotrophin hypothesis, implicates deregulation of trophic signalling in BF cholinergic cells as one of the primary causes (Hefti and Weiner 1986; Counts and Mufson 2005). Indeed, quantitative analysis revealed a significant reduction in the expression of the high-affinity neurotrophin receptor—tropomyosin-related kinase A (trkA) with the number of neurons immuno-reactive to trkA antibody in the BF region being greatly reduced in mild cognitive impairment (46 %) and AD (56 %) brain autopsies (Mufson 2000; Allen 2011). Single cell mRNA profiling, in addition to reduced trkA, also revealed down-regulation of trkB and trkC receptors (Ginsberg et al. 2006). Quite unexpectedly, the level of low-affinity p75 neurotrophin receptor (p75NTR), which is expressed in the adult forebrain primarily in BF cholinergic neurons, remained unchanged (Ginsberg et al. 2006). Capable of binding most of known neurotrophins, p75NTR is recognized for its trophic as well as apoptotic functions (Van der Zee et al. 1996; Naumann et al. 2002; Sotthibundhu et al. 2008), depending on its partner co-receptors or activating ligands.
An intriguing, yet neglected characteristic of p75NTR is its capacity, in addition to neurotrophins, also bind a range of collateral ligands with no apparent trophic function (Dechant and Barde 2002; Butowt and von Bartheld 2009). In the present study, the mechanisms regulating the endocytosis and transport of IgG192-Cy3-labelled p75NTR in conjunction with Alexa488-Aβ 1–42 in cholinergic neurons of primary BF cultures are analysed. Live microscopy and confocal imaging reveal late endosomes and lysosomes as primary destinations of these proteins. The pertinence of these processes to Aβ clearance via lysosomal degradation in BF cholinergic neurons, with their early loss during AD is discussed.
Materials and methods
Reagents and materials
All chemicals were purchased from Sigma (Ireland) unless specified otherwise. Reagents and materials for tissue culture were obtained from BioSciences (Ireland); Ibidi GmbH supplied 35 mm glass dishes. Alexa-488 labeled and non-labeled Aβ (1–42) and rat monoclonal anti-Aβ (1–42) antibody were obtained from Ana Spec (UK). Goat anti-ChAT polyclonal antibody and rabbit polyclonal anti-early endosome antigen-1 (EEA1) IgG were purchased from Millipore (Ireland); rat monoclonal IgG192-Cy3 came from Advanced Targeting Systems (USA). Goat polyclonal anti-Rab11 (C-19) IgG and rabbit polyclonal anti-Rab7 (H-50) IgG were obtained from Santa Cruz Biotechnology (USA). Mouse anti-Lamp1 monoclonal-and FITC-labeled rabbit anti-goat polyclonal antibodies were purchased from Abcam (Ireland). Alexa fluors 488 or 594 secondary antibodies and 12 % NuPage Bis–Tris gels were obtained from Invitrogen (BioSciences, UK). Vectashield mounting medium with DAPI was purchased from Vector Labs (UK). Recombinant botulinum neurotoxins (BoNT/A and/B) were produced and purified in-house as described (Lacy and Stevens 1997; Smith 1998). Ketamine (Velatar™) was bought from Pfizer (Ireland). Aβ oligomers were prepared according to procedures (Dahlgren et al. 2002) with the presence of oligomeric forms confirmed through dilution of the sample in NuPage sample buffer and separation on a 12 % NuPage Bis–Tris gel. The size of the bands confirmed the presence of monomers, trimers, and tetramers in the sample used for the presented experiments. Gels were scanned using a standard gel imaging scanner (SynGene).
Primary basal forebrain neuronal cultures
All procedures involving animals (rats, P1 or P60–70 days old) conformed to guidelines approved by the Research Ethics Committee of the University and the Ministry of Health and Children, Republic of Ireland and the U.S. Public Health Service Policy on Human Care and Use of Laboratory Animals with the National Institute of Health, approved by the Institutional Board of the Rutgers University. BF neurons were cultured according to the protocol described (Mudd et al. 1998; Auld et al. 2000). In brief, under deep ketamine anaesthesia (120 mg/kg, i.p.), P1 rat pups were decapitated, septum/BF area extracted and placed in dissecting medium containing Hank’s Balanced Salt Solution supplemented with 25 mM glucose and 10 mM HEPES buffer. After mincing, the tissue was transferred into trypsin/EDTA solution (7–10 min; 37 °C) followed by centrifugation (5 min, 10,000 rpm). Pellet was re-suspended in medium containing 1:1 mixture of modified Eagle Medium (DMEM):F12 (1:1) and 5 % fetal bovine serum, followed by gentle triturating and centrifugation (5 min, 1,000 rpm). Cells were then plated in 35 mm poly-L-lysine-coated dishes at a density of 0.5–1.0 × 105 cells/cm2 in serum-free DMEM:Neurobasal (1:1) plus B27 supplement containing growth medium with antibiotic mixture (1 % Gibco, 10,000 units penicillin, 10 mg streptomycin and 25 mg amphotericin B per ml) and NGF (50 ng/ml) (mouse, 2.5S). 24 h after plating antimitotic cytosine arabinofuranoside (10 μM final concentration) was added to the cultures for 48 h to inhibit the proliferation of non-neuronal cells. Neurons were kept in a humidified incubator (5 % CO2) for max. 2 weeks at 37 °C, with half of the growth medium being replaced every 5 days.
Immuno-cytochemistry and microscopic analysis
Procedures for pre-labelling of BF cholinergic neurons with Cy3-IgG192 in vivo followed by ChAT staining of brain sections, fluorescence microscopy, cell counting and data analysis were conducted according to protocols described elsewhere (Ovsepian et al. 2012). In three rats (~2 months old), IgG192-Cy3 was injected into the medial prefrontal cortex with its transport to BF verified. In brief, under deep anaesthesia (ketamine: 90 mg/kg; xylasine: 10 mg/kg, i.p.), IgG192-Cy3 (2 μl, 0.4 mg/ml) was slowly injected with a Hamilton syringe (22-gauge needle) into the brain, using stereotaxic coordinates as follows: 2.5 mm anterior from bregma; 0.4 mm lateral from midline and 2.3 mm below the dura mater. Three days later injected animals were overdosed with sodium pentobarbital (200 mg/kg) and killed, with the site of injection in the prefrontal cortex and labelling of neurons in BF verified with fluorescence microscopic analysis, using appropriate filters (Cy3/DsRed/Rhodamine filter set) (Axioscope, Zeiss). In different sets of experiments, IgG192-Cy3 was injected into the lateral ventricle, which was followed by staining of BF slices for ChAT, to verify the localization of IgG192-Cy3 in BF cholinergic neurons (Ovsepian et al. 2012). In tissue culture experiments, primary BF cultures of 6–9 days old were assayed for ChAT-positive cholinergic neurons, the only neuron type in the forebrain that expresses p75NTR (Hartig et al. 1998). After overnight incubation in growth medium supplemented with fluorlabelled anti-p75NTR monoclonal antibody IgG192-Cy3 (5 nM final concentration), cultures were rinsed (3 × 5 min) with phosphate buffer saline (PBS), fixed with 4 % paraformaldehyde (PFA; pH 7.4, 30 min in PBS), washed (3 × 5 min) and permeabilized (0.4 % Triton X-100, 1 h). These steps were followed by blocking of non-specific immuno-reactive sites with 5 % bovine serum albumin plus 2 % rabbit serum (1 h), followed by overnight exposure of tissue to polyclonal goat anti-ChAT antibody (1:100) in the same blocking medium also containing 0.4 % Triton X-100 (PBS, pH 7.4; room temperature). Following extensive rinses (3 × 15 min, PBS), samples were exposed to FITC-labelled secondary polyclonal antibody (2 h; 1:1,000), air-dried and coverslipped with Vectashield mounting medium (where specified also containing DAPI). Cholinergic neurons were identified based on their positive labelling for ChAT or presence of anti-p75NTR IgG192-Cy3, while DAPI (in primary cultures) was used for assessment of the total number of neurons. Negative control dishes were processed in the same way but with the omission of primary ChAT antibody. To identify the intracellular location of IgG192-Cy3, neuronal cultures were additionally labelled for early (EEA1), recycling (Rab11), and late (Rab7) endosome and (Lamp1) lysosome qualifier proteins (Nixon 2007; Saavedra et al. 2007). In brief, after overnight incubation of BF cultures in growth medium supplemented with Cy3-IgG192 (5 nM), samples were extensively rinsed, fixed, permeabilized and blocked, as specified above, and exposed overnight to anti- EEA1, Rab11, Rab7 and Lamp1 primary antibodies (1:100) in blocking medium supplemented with Triton X-100 in PBS (0.4 %, pH 7.4; room temperature). Subsequently, the tissue was exposed for 1 h to Alexa-488 labelled corresponding secondary antibody (1:1,000) at room temperature, rinsed (3 × 5 min), dried and mounted for confocal microscopic analysis.
Live microscopy and functional imaging of BF cultures
To monitor internalization of Aβ and its trafficking with IgG192-Cy3/p75NTR, 100 nM HiLyte fluor (Alexa 488) labeled Aβ (1–42) (Nazer et al. 2008) was co-applied with 5 nM IgG192-Cy3 to BF cultures for 2 h (37 °C, 5 % CO2). This was followed by extensive rinsing of cultures with growth medium and live confocal imaging (LSM710, AxioObserver, 40 × 1.3 Plan Apochromat oil-immersion objective). Argon and Helium/Neon lasers provided 488 and 543 nm lines in time series mode; emitted fluorescence signals were sampled at the resolution of 30 nm/pixels with a dwell time of 1.5 μs. Co-localization of Alexa-488 Aβ and Cy3-IgG192 is defined by the presence of two labels in the same pixel in the digitally acquired images, using a co-localization algorithm (Zen 2008, Carl Zeiss). Separation of emission spectra was ensured with appropriate cut-off filters (green 492–590 nm; red 585–734 nm). In selected cases and due to mobility of fluor-loaded bodies, co-localization of Alexa-488 Aβ and Cy3-IgG192 was verified after fixation of samples with 4 % PFA for 30 min followed by extensive washes (3 × 15 min, PBS), air drying of the samples and coverslipping with Vectashield mounting medium. Stacks of optical sections in Z-dimension (750 nm thick) were collected from cell bodies and processes with individual frames analyzed for signal intensity and co-localization. In experiments with assessment of IgG192-Cy3 internalization under high [K+] (KCl 50 mM) stimulation or blockade of Na+ channels by 0.5 μM tetrodoxin (TTX), cholinergic neurons were imaged and the signal intensity was compared to those acquired from untreated controls. The reliance of IgG192-Cy3 uptake on synaptic vesicle turnover was assessed in a similar way with neurons exposed to BoNT/A (5 nM) and BoNT/B (10 nM) neurotoxins, which cleave and inactivate SNAP-25 and VAMP, respectively, and hence, block SNARE-dependent exo-endocytosis. Similar steps were followed in experiments involving chelation of Ca2+ with EGTA (200 μM), selective blockade of high (CdCl2; 50 μM) and low (mibefradil; 10 μM) voltage-gated Ca2+ channels as well as depletion of internal Ca2+ stores with inhibitors of the sarco-endoplasmic reticular Ca2+ ATPase (SERCA)—thapsigargin (1 μM) or cyclopiazonic acid (CPA, 10 μM). In experiments for testing the effects of Aβ 1–42 on the internalization of IgG192-Cy3, a mixture of soluble mono- and oligomeric forms of this peptide were prepared and added in various concentrations to cultured BF neurons with incubation for 2 h in a humidified incubator at 37 °C in 5 % CO2 before addition of 5 nM IgG192-Cy3 for 2 h or overnight. Afterwards, cultures were washed with PBS (pH 7.4), fixed with 4 % PFA and processed for microscopy as described above. Similar procedures were followed in control experiments, but with omission of tested compounds. For two-colour time lapse series, a dichroic filter was used for simultaneous imaging of IgG-Cy3 and Alexa-488-Aβ1–42, with pinhole kept fully open. Images were acquired in continuous scanning mode.
Data analysis and statistical significance
All settings used for data acquisition were kept constant throughout experiments (Zen 2008). For cell counting, neurons with fluorescence signal intensity exceeding three times the average intensity of the background were considered suitable for including in the analysis (O’Leary et al. 2011). Numerical values of fluorescence intensity were normalized over the regions of interest (ROI) pulled from at least 25 frames, collected from 20 to 30 cells for each experimental condition. These were tabulated and analysed using Zen 2008 and Excel analytical software. Data are reported as mean ± SE with statistical significance assessed using paired or unpaired Student’s t test, with p < 0.05 defining a significant (*) difference.
Results
BF cholinergic neurons in culture retain a high level of p75NTR expression
The majority of ChAT-positive profiles in medial septum-diagonal band Broca (MS-DBB) as well as in more caudal BF nuclei were also immuno-reactive for IgG192-Cy3 (Fig. 1a) with punctuate Cy3 labelling of intracellular compartments visible in neurites and soma (Fig. 1a, a2 inset). Counting of double-labelled neurons revealed 94.3 ± 5 % of cholinergic cells (350–400 neurons, from 3 rats) being positive for IgG192-Cy3 in MS-DBB nuclei, while in more caudal BF structures the percentage of double-labelled cells was lower (85.6 ± 5.7 %, 350–400 neurons, from 3 rats) (Fig. 1a, c). With no exception, all Cy3-IgG192 positive cells were immuno-reactive for ChAT (Fig. 1 panel a2 and a3), an observation that confirms IgG192-Cy3 as a reliable marker for BF cholinergic neurons (Hartig et al. 1998; Ovsepian et al. 2012). Extensive labelling of ChAT-positive cells with Cy3-IgG192 was also evident in BF primary neuronal cultures (Fig. 1b3, d) with punctuate Cy3 fluorescence visible in somata and neurites (Fig. 1b3). Assessment of the time course of IgG192-Cy3 uptake showed that the bulk of it is internalized within the first 2 h of exposure (78.1 %), with prolonged exposure of cultures to IgG192-Cy3 (24 h) causing only a modest (~20 %) further gain in Cy3 labelling (not shown). It is worth noting that similar to brain slices, a notable fraction of ChAT-positive profiles remained non-labelled with IgG192-Cy3 (25–30 % of cells, 300–400 neurons counted from 3 culture dishes) (Fig. 1b, d). Overall, these findings demonstrate that the majority of BF cholinergic cells in primary cultures retain high expression of p75NTR, which at the resting state is constitutively turned over together with associated ligands.
Fig. 1.
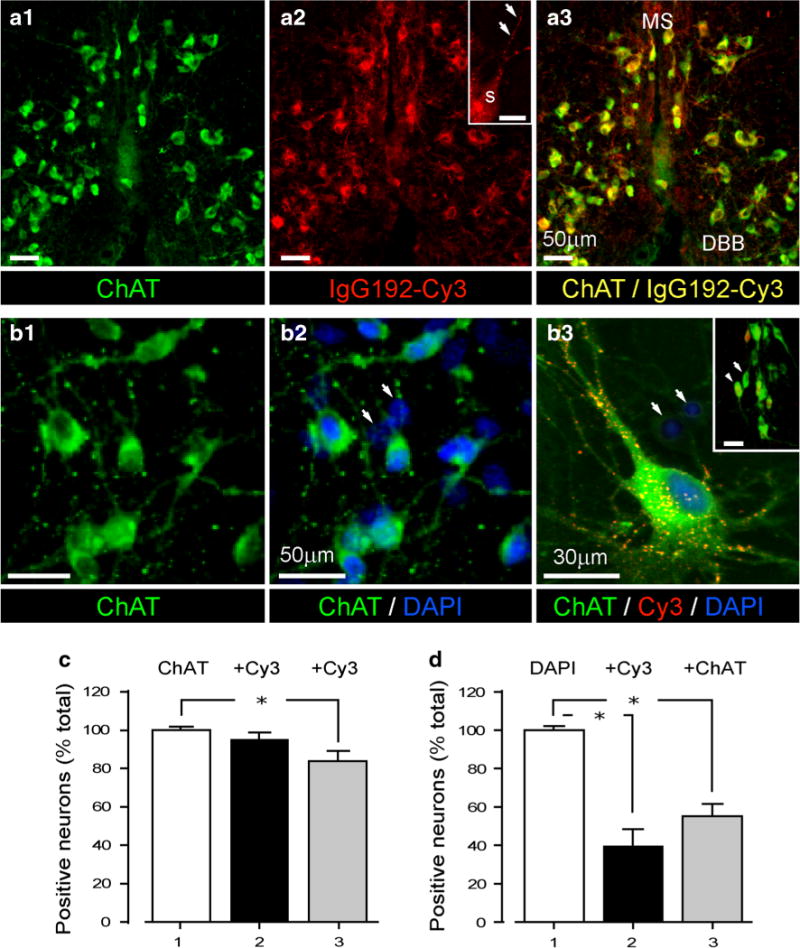
Selective labelling of BF cholinergic neurons with IgG192-Cy3. a1–3 IgG192-Cy3-labelled neurons also immuno-reactive to ChAT antibody in the vertical limb of DBB: (a1) ChAT positive (green) (a2) IgG192-Cy3 positive (red) and double (a3) ChAT + IgG192-Cy3-labelled neurons (yellow). (MS medial septum, DBB diagonal band Broca). Inset (a2) illustrates high magnification IgG192-Cy3-labelled bodies in soma (S) and dendrites (arrows) of BF cholinergic neuron. Scale corresponds to 10 μm. b1–2 ChAT-labelled BF cholinergic neurons (b1) and DAPI stained cells in the same field of view (b2). Note, only a fraction of cells are double-labelled with DAPI and ChAT. Arrows point to DAPI-labelled non-cholinergic cells. b3 High power triple labelled (ChAT/IgG192-Cy3/DAPI) BF cholinergic neuron with punctuate IgG192-Cy3 labelling along with two non-cholinergic cells also in the field (arrows) (inset b3—larger field of view of cultured BF neurons with some (arrowhead) being immuno-reactive for ChAT and IgG192-Cy3). Scale corresponds to 35 μm. c Summary plot of the fraction (%) of Cy3-positive neurons in MS/DBB (black bar) and caudal BF nuclei (grey bar) of total population of ChAT-positive cells (white bar). d Summary plot of the fraction (%) of Cy3 (black bar) and ChAT (grey bar) positive cells of the total DAPI-positive neurons in BF cultures (white bar)
IgG192-Cy3 and Aβ share entry routes and intracellular carriers
The p75NTR was suggested as a neuronal acceptor for Aβ (Yaar et al. 1997). To establish if interactions of Aβ with p75NTR are confined to the plasmalemma, or similar to neurotrophins, Aβ undergoes p75NTR mediated internalization and trafficking within ‘signalling’ endosomes, the loading of Alexa-488-Aβ (1–42) and IgG192-Cy3 were monitored in cultured cholinergic cells with live microscopy. Due to known hydrophobicity of Aβ, the background florescence of cultures exposed to Alexa-488-Aβ 1–42 remained high even after rigorous washes, making the clear-cut distinction between surface-bound and internalized Aβ in distal neurites impossible, hence, limiting our microscopic analysis to cell bodies and proximal neurites. Figure 2 shows a reconstructed 3D Z-stack of the soma and proximal neuritis of a representative live BF cholinergic neuron co-labelled with IgG192-Cy3 and Alexa-488 Aβ 1–42 (Fig. 2a). Analysis of randomly defined ROI revealed strong co-localization of Alexa-488 and Cy3 in both perikaryon and neurites, with closely matching co-localization coefficients (0.91 ± 0.07 vs. 0.96 ± 0.1; neurites vs. soma, respectively) (Fig. 2a, c). Reversed assessment of Cy3 and Alexa-488 co-localization revealed significantly lower coefficients (0.36 ± 0.05 vs. 0.48 ± 0.08, neurites and cell bodies, respectively), suggestive of the crucial role of p75NTR for the uptake of Alexa-488 labelled Aβ 1–42 by cholinergic neurons. Similar to IgG192-Cy3, Alexa-488-Aβ labelling of intracellular compartments became evident within ~30 min of exposure (Fig. 2b). Of note, trafficking of double-labelled compartments occurred in both somatopetal and somato-fugal directions at widely varying speed ranges (0.05–4.2 μm/s), with, however, closely matching average movement velocity of Cy3 and Alexa-488-labelled elements (3.18 ± 0.7 μm vs. 2.81 ± 0.8 μm, p = 0.81) (Fig. 2b, d and suppl. video 1). Comparison of the relative sizes of Cy3 and Alexa-488-labelled organelles also revealed their close correspondences (0.8 ± 0.06 vs. 0.71 ± 0.08, respectively; p = 0.34) (Fig. 2e). It is worth noting that in addition to mobile compartments there was a large amount of stationary or oscillating double-labelled elements within both neurites and perikaryon. Together with the results of earlier reports (Yaar et al. 1997, 2002), our findings strongly suggest a significant role for p75NTR in uptake and loading of Aβ onto intracellular carriers.
Fig. 2.
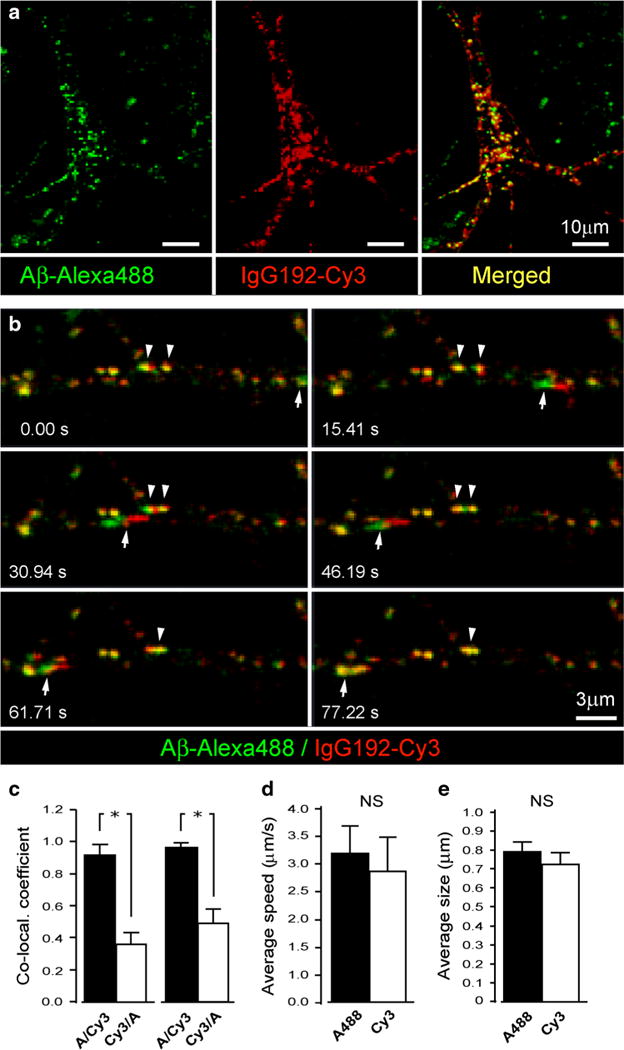
Co-labelling of stationary and mobile endosomes with IgG192-Cy3 and Alexa-488-Aβ in BF cholinergic neurons. a Representative live BF cholinergic neuron labelled with Alexa-488 Aβ1-42 (green), Cy3-IgG192 (red) and merged (yellow, right panel): a Z-stack projection. b Time lapse series of a neurite with IgG192-Cy3 and Alexa-488 Aβ containing endosomes. Insets indicate the times of acquisition of individual frames. Note rapid (arrow) and slow (arrowheads) moving vesicles in addition to the large amount of stationary double-labelled structures. c–e Summary plots of Alexa-488/IgG192-Cy3 (c, left) and IgG192-Cy3/Alexa-488 (c, right) co-localization in intracellular compartments of BF cholinergic neurons (c, neuritis and soma —left and right bar pairs, respectively), trafficking velocity of Alexa-488 and IgG192-Cy3-labelled vesicles (d) and their estimated diameters (e)
Internalization of p75NTR is independent of synaptic activity but depends on Ca2+
While much progress has been made in defining the mechanisms of the release of neurotrophins (Hartmann et al. 2001; Dean et al. 2009), little is know about events governing their uptake. To gain new insights, the effects of stimulants and blockers of synaptic activity on IgG192-Cy3 uptake by cholinergic neurons were analysed (Fig. 3). After incubating cultures for 10 min in growth medium containing 50 mM KCl or 0.5 nM TTX (CaCl2/MgCl22/1 mM), Cy3-IgG192 was supplemented (final concentration 5 nM) for 2 h, which was followed by washes and fixation of neurons for microscopic analysis. As shown (Fig. 3a), Cy3-IgG192 uptake was enhanced under high [K+] stimulation, with signal intensity in both soma and neurites exceeding those in non-stimulated controls (89.4 ± 18.2 % increase; p = 0.0071) (Fig. 3a, d). Unexpectedly, blockade of action-potential driven activity by TTX did not alter the internalization of IgG192-Cy3, with fluorescence signal of neurons in treated cultures being indistinguishable from controls (2.1 ± 17.1 % increase; p = 0.24) (Fig. 3a, e). Failure of TTX to alter the basal uptake of IgG192-Cy3 suggests its independence from regulated synaptic activity. To prove this directly, the effects of potent inhibitors of regulated neurotransmission BoNT/A (5 nM) or/B (10 nM) on IgG192-Cy3 uptake were examined. As shown, the loading of cholinergic neurons with IgG192-Cy3 was not compromised by either BoNT/A or/B proteases (4.8 ± 7.2 % and 9.2 ± 12 % reduction, respectively; p = 0.53 and p = 0.16) (Fig. 3b, e). Because Ca2+ is a ubiquitous regulator of membrane turnover at both axon terminals and dendrites (Ovsepian and Dolly 2011), we assessed if the uptake of IgG192-Cy3 relies on the increase of intracellular [Ca2+]. Indiscriminate blockade of high-voltage gated Ca2+ channels with cadmium (CdCl2, 50 μM) (Tsien et al. 1988) marginally inhibited the loading of cholinergic neurons with IgG192-Cy3 (9.6 ± 6.3 % reduction, p = 0.41) (Fig. 3c, f), an observation that contrasts with pronounced reduction of IgG192-Cy3 uptake by mibefradil—a selective blocker of low-voltage gated Ca2+ channels (75.4 ± 3.1 % reduction, p = 0.004) (Fig. 3c, f). Inhibition of Cy3-IgG192 internalization was also evident under global chelation of Ca2+ with EGTA-AM (61.2 ± 2.1 % decrease, p = 0.0081) as well as after depletion of internal Ca2+ stores by thapsigargin (59.2 ± 1.4 %, p = 0.0036) or CPA (51.2 ± 6.4 %, p = 0.021) (Fig. 3c, f). In summary, while depolarization greatly promotes the loading of cholinergic cells with IgG192-Cy3, the basal uptake appears not to rely on regulated synaptic activity but entails low-threshold calcium influx and Ca2+ release from internal stores.
Fig. 3.
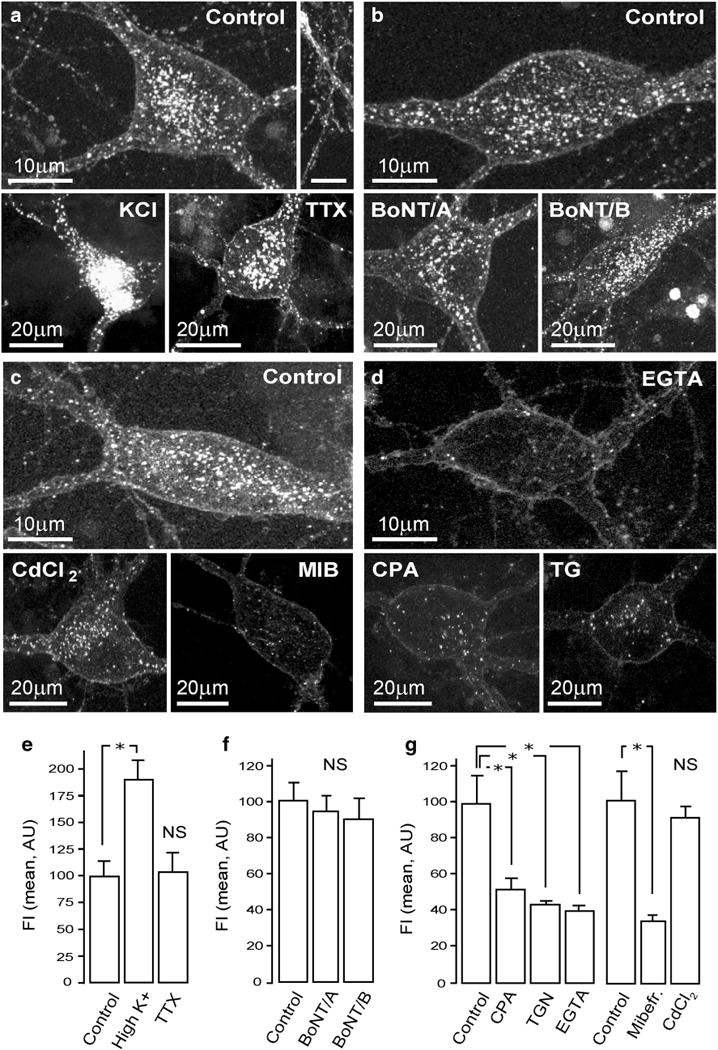
Internalization of IgG192-Cy3 is enhanced by depolarization, is independent of SNARE-driven vesicle turnover and requires intracellular Ca2+. a Representative micrographs of BF cholinergic cells showing significant basal uptake of IgG192-Cy3 (top panel), uptake under stimulation with high [K+] (lower left, KCl) and in presence of TTX (lower right). b Neither BoNT/A nor/B (lower left and right) caused notable alteration in the uptake of IgG192-Cy3 in BF cholinergic neurons (control, on the top). These concentrations of BoNT/A and/B proteases are sufficient for complete blockade of neuro-secretion. c Blockade of high-voltage activated Ca2+ channels with Cd2+ only slightly reduced the endocytosis of IgG192-Cy3, while T-type Ca2+ channel blocker mibefradil (MEB) strongly inhibited its uptake (lower right). d Chelating of Ca2+ with EGTA and blockade of SERCA ATPase by thapsigargin or CPA caused strong decrease of IgG192-Cy3 uptake by BF cholinergic cells. e–g Summary plots illustrating the average fluorescence intensities (FI) pooled from various experimental groups (mean and ± SEM)
Amyloid β inhibits the endocytosis of p75NTR in cholinergic BF neurons
The competitive binding of Aβ and NGF to p75NTR is well documented (Yaar et al. 2002; Arevalo et al. 2009) and contrasts to the non-competitive interaction of IgG192 with this receptor, attributed to binding of the latter to a different extra-cellular epitope (Taniuchi and Johnson 1985). The trait renders IgG192-Cy3 ideally suitable for studying the mechanisms of uptake and the intracellular journey of p75NTR, without interfering with its trophic functions. Taking advantage of this, we examined the effects of sub-cytotoxic concentrations of Aβ on the internalization of p75NTR in BF cholinergic neurons. After 24 h exposure of cultures to non-labelled Aβ (1–42) in growth medium (6–600 nM, mono- and oligomers; Fig. 4a), IgG192-Cy3 was supplemented (5 nM) for 2 h, which was followed by fixation of the samples and microscopic analysis. As shown, Aβ dose-dependently reduced the basal uptake of p75NTR, with over 70 % decrease in IgG192-Cy3 loading of cholinergic cells caused by the highest concentration of this peptide tested (75.2 ± 14.2 %, 600 nM; p = 0.008) (Fig. 4b, c).
Fig. 4.
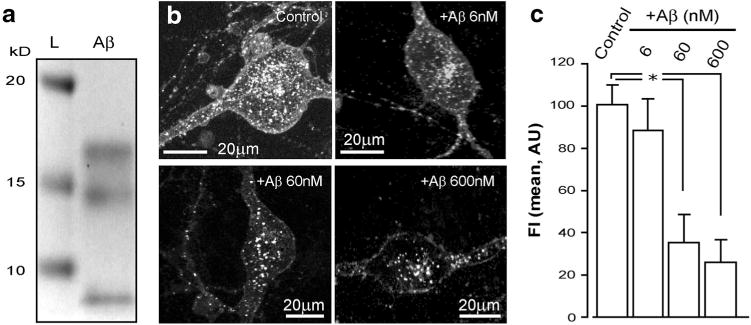
Aβ inhibits the internalization of p75NTR by BF cholinergic neurons. a Proof of the presence of mono- and oligomeric forms of the Aβ in sample applied to BF cholinergic neurons: coomassie stained (12 % Bis–Tris SDS page) gel showing 3 different fractions of Aβ in the experimental material applied to BF neuronal cultures: L molecular ladder. The gel is truncated at 25 kD. b IgG192-Cy3 uptake by representative BF cholinergic cells exposed to 3 different concentrations of Aβ (6, 60 and 600 nM top right, bottom left, and right, respectively) compared to non-exposed control (top left). c A summary plot of the effects of various concentrations of Aβ on Cy3-IgG192 internalization by BF neurons
Bulk of internalized IgG192-Cy3 accumulates in lysosomes of BF cholinergic neurons
To define the intracellular journey of p75NTR and its ligands, after 2 h exposure of BF cultures to IgG192-Cy3 (5 nM), neurons were fixed and stained for various endosomal proteins. EEA1 staining with two-colour microscopy revealed that only a small fraction of IgG192-Cy3 was present in early endosomes (7.7 ± 6.7 %) (Fig. 5a, e). Congruently, assessment of the co-localization of IgG192-Cy3 with EEA1 revealed a predominant fraction of Cy3-labelled profiles lacking EEA1 (Fig. 5e). Similar experiments revealed notably higher co-labelling of IgG192-Cy3 containing profiles with Rab11, a qualifier protein for recycling endosomes, with 25.1 ± 11 % of endosomes being positive for Rab11 and IgG192-Cy3 (Fig. 5b, e). This observation suggests that a substantial fraction of IgG192-Cy3-p75NTR carrying endosomes in BF cholinergic cells are sorted to trans-Golgi networks and recycled. Because NGF or its signalling components have been identified in lysosomes of other neurons (Butowt and von Bartheld 2009), we examined if some of the IgG192-Cy3 positive compartments advance into late endosomes and lysosomes in BF cholinergic neurons. As shown in Fig. 5c and d, both Rab7- and Lamp1-containing profiles are also strongly co-labelled with IgG192-Cy3 (Rab7/IgG192-Cy3 52.9 ± 2.8 % and IgG192-Cy3/Rab7 56.1 ± 3.2 %; Lamp1/IgG192-Cy3 60.5 ± 8.8 % and IgG192-Cy3/Lamp1 82.1 ± 5.9 %). These observations suggest late endosomes and especially lysosomes as key destinations of internalized IgG192-Cy3 in BF cholinergic neurons (Fig. 5c, d, f and g). Overall, while a fraction of internalized IgG192-Cy3-p75NTR is recruited to EEA1 and Rab11 positive compartments for recycling, the bulk of it appears to be guided for degradation in late endosomes and lysosomes.
Fig. 5.
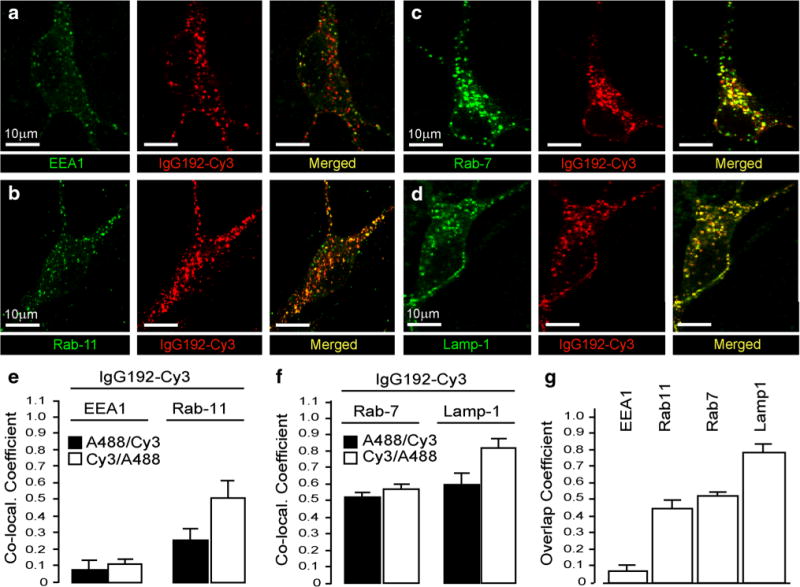
Localization of IgG192-Cy3 in intracellular compartments of BF cholinergic neurons. a, b Double staining of BF cholinergic neurons in primary cultures reveals only a small fraction of intracellular IgG192-Cy3 co-localized with early or recycling endosomal marker proteins (EEA1 and Rab11, respectively). e Summary plots demonstrating the average co-localization coefficients of IgG192-Cy3 with EEA1 (left) and with Rab11 (right). c, d Similar experiments with late endosomal markers Rab7 and lysosomal protein Lamp1 reveals much stronger co-localization of these proteins with IgG192-Cy3. f, g Higher level of double-labelling of Rab7 and Lamp1 containing compartments with IgG192-Cy3 reflected in average co-localization graphs. g A histogram of the overlap coefficients of IgG192-Cy3 positive vesicles with EEA1, Rab11, Rab7, and Lamp1 proteins in cholinergic neurons
Injection of IgG192-Cy3 into the prefrontal cortex labels BF cholinergic cells in vivo
Having shown the uptake and co-trafficking of Alexa488-Aβ and IgG192-Cy3 in cultured BF cholinergic neurons, we set out to establish the pertinence of this process to in vivo conditions. As illustrated (Fig. 6), injection of IgG192-Cy3 in the medial prefrontal cortex selectively labels neurons in the BF area, with fluorescence signal being strongest within the horizontal limb of the DBB, inferior extension of ventral pallidum and the rostral substantia innominata (Fig. 6a, b). This contrasts to widespread Cy3 labeling of cholinergic neurons throughout the entire BF area following the intra-ventricular injection of IgG192-Cy3, with highest levels of labeling in the MS nucleus and DBB. Under higher magnification, a distinctly punctuate pattern of intracellular Cy3 became evident, which is consistent with the uptake and accumulation of IgG192-Cy3 within endosomes (Hartig et al. 1998; Kacza et al. 2000). Immuno-staining of MS and DBB containing slices from animals injected with IgG192-Cy3 revealed highly specific Cy3 labeling of ChAT-positive profiles (97.4 ± 1.9 %; Fig. 6c). Along with above presented data, these in vivo observations are consistent with the constant internalization of p75NTR (and its ligands) at axon terminals of cholinergic neurons followed by its transport to the BF nuclei, where the cell bodies of these neurons reside. Quite disappointingly, similar experiments with intra-ventricular injection of Alexa488-Aβ1–42 (2 μl, 0.5 mg/ml) revealed poor and variable labelling of BF cholinergic cells. Although the basis for the discrepancy between these in vivo observations from those obtained in BF primary cultures remains unclear, the lower mobility of Aβ peptide (1) and its possible binding to other known receptors/acceptors, including NMDA, metabotropic glutamate, nicotinic, muscarinic and insulin growth factor receptors (2) (Patel and Jhamandas 2012) could contribute and attenuated labelling of BF cholinergic cells by Aβ-Alexa488 in the intact brain.
Fig. 6.
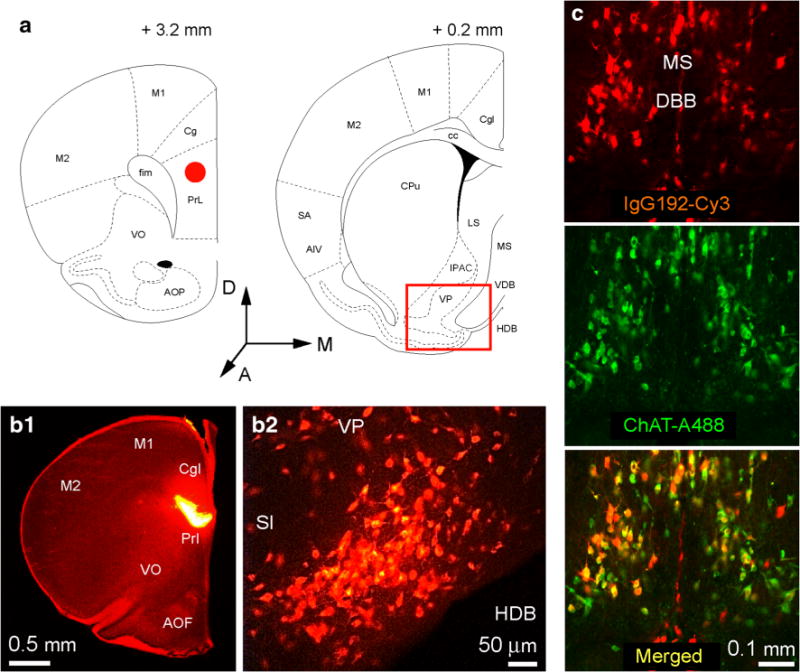
Retrograde labelling of basal forebrain cholinergic neurons with anti-p75NTR antibody IgG192-Cy3. a and b Coronal sections of the rat brain (bregma +3.2 mm and +0.2 mm) highlighting the site of injection (red circle in panel a: left schematic and b1) of IgG192-Cy3 (0.4 mg/ml, 0.5 μl cingulate/pre-limbic cortices) and retrogradely labeled cholinergic neurons in the horizontal limb of the diagonal band Broca (red box in the panel a: right schematic and B2). Anatomical references: VP ventral pallidum, VDB and HDB vertical and horizontal limb of diagonal band Broca, respectively, LS lateral septum, Cg, PrL, M1, and M2 (as defined above), respectively; cc corpus callosum, CPu caudate putamen, SA somato-sensory areas, AIV agranular insular cortex, IPAC interstitial nucleus of the posterior limb of anterior commissure. c Low power fluorescence micrographs illustrating cholinergic neurons of the medial septum (MS) and diagonal band Broca (DBB) labeled in vivo with IgG192-Cy3 injected into the lateral ventricle; 48 h after injection of Cy3-IgG192 (0.4 mg/ml; 3 μl), rat was perfused and fixed with 4 % paraformaldehyde, the brain containing MS and DBB areas was sectioned in coronal plane (30 μm) and stained for ubiquitous cholinergic neuronal marker ChAT. Note high level of neuronal labelling in both MS and DBB areas with IgG192-Cy3 (top panel), with majority of profiles being labelled also positive for ChAT (middle and lower panels)
Discussion
The survival and differentiation of neurons in central and peripheral nervous systems depend on neurotrophic support. After binding and internalization at axon terminals, neurotrophins give rise to ‘signalling’ endosomes, which are retrogradely transported to the cell body to influence an array of biological processes, including nuclear signalling, gene expression and protein phosphorylation (Chao 2003; Huotari and Helenius 2011). Over recent years, it has become evident that certain neurotoxins, peptides and viral particles are also capable of binding neurotrophin receptors, with some exploiting the sorting and long-range transport machinery of neurons to carry out their deadly mission (Salinas et al. 2010). A model example of the highjacking of neurotrophin receptors by non-neurotrophic ligands is provided by the interaction of p75NTR with the Aβ peptide (Baldwin and Shooter 1995; Yaar et al. 1997). With a reported dissociation constant of Aβ close to that of NGF, p75NTR has been shown to mediate the detrimental effects of Aβ in both neurons and non-neuronal cells (Yaar et al. 2002; Sotthibundhu et al. 2008). Using primary BF cultures, herein we demonstrate for the first time the rapid and robust internalization of Aβ (1–42) along with its trafficking in conjunction with the anti-p75NTR antibody IgG192 in BF cholinergic cells. While active at rest, this process is stimulated by high [K+] induced depolarization, requires Ca2+ influx via T-type channels and Ca2+ release from internal stores. Curiously, our data suggest that the internalization of p75NTR is independent of regulated exocytosis (and evidently coupled to it endocytosis), as it is insensitive to TTX, BoNT/A and/B proteases. Finally, we provide evidence for late endosomes and lysosomes as principal destinations of p75NTR (and presumably Aβ) in cholinergic cells and verify the pertinence of described in vitro processes to the intact brain in vivo.
Shared route of uptake and trafficking of Cy3-IgG192 and Aβ in BF cholinergic cells
Cholinergic BF neurons source one of the largest modulator systems of the brain (Mesulam 1990; Zaborszky 2002). Through their widespread ascending projections, these cells supply acetylcholine to the entire cerebral mantle, regulating a wide range of cortical functions (Buzsaki and Gage 1989; Huerta and Lisman 1995; Ovsepian et al. 2004; Hasselmo and Giocomo 2006; Lawrence et al. 2006; Lawrence 2008; Ovsepian 2008). In turn, neurons of target fields provide continuous neurotrophic support to cholinergic projections, vital to their survival and stability (Swaab 1991). Aging and neurodegenerative disease-related disruptions in trophic support of the BF cholinergic system have been long recognized with a significant down-regulation of the expression of high affinity trkA, B and C receptors shown in BF along with reduction of NGF in the nucleus basalis Meynert (Counts and Mufson 2005; Ginsberg et al. 2006). Intriguingly, the level of the pan-neurotrophin receptor p75NTR (low affinity NGF receptor) known to interact with NGF and several other neurotrophins as well as Aβ remained unaltered (Ginsberg et al. 2006; Coulson et al. 2009). The latter is of special interest for neurodegenerative mechanisms during AD due to its known pro-apoptotic functions activated by Aβ (Coulson et al. 2008; Sotthibundhu et al. 2008) as well as its suspected role in regulating the production and aggregation of Aβ (Wang et al. 2011; Zeng et al. 2011). Our live and confocal microscopic data demonstrate joint internalization and trafficking of Aβ and IgG192-Cy3 in BF cholinergic cells. The high basal uptake of IgG192-Cy3 is consistent with its constitutive character, a notion supported by its persistence in the presence of potent blockers of regulated synaptic activity such as TTX or BoNT/A and/B (Ovsepian and Dolly 2011; Ovsepian and Friel 2012). Enhancement of IgG192-Cy3 loading under high K+ stimulation and its inhibition by the selective T-type Ca2+ channel blocker mibefradil (Ertel and Ertel 1997; Ertel et al. 1997) as well as by SERCA inhibitor thapsigargin or CPA (Lytton et al. 1991; Wayman et al 1996) imply the significance of low voltage-gated Ca2+ influx and Ca2+ release from internal stores. Rather unexpectedly, the indiscriminate blocker of high voltage-activated Ca2+ channels Cd2+ (Tsien et al. 1988) only marginally reduced the uptake of IgG192-Cy3, suggestive of the negligible role of high voltage-gated Ca2+ influx to the constitutive internalization of p75NTR. Taken together these data imply a stringent segregation between processes governing the uptake of neurotrophins (and presumably Aβ) from those contributing to the membrane recovery after regulated fusion of synaptic vesicles, reliant primarily on Ca2+ influx via high voltage-activated Ca2+ channels (Gad et al. 1998; Shupliakov 2009). It is worth stressing that the BF cholinergic cells express a range of voltage-gated Ca2+ channels and undergo age- and disease-related compensatory changes in their Ca2+ buffering mechanisms (Griffith et al. 1994; Murchison and Griffith 1995; Etheredge et al. 2007). In light of unaltered (Ginsberg et al. 2006) or enhanced expression of p75NTR (Chakravarthy et al. 2012) during aging and AD with the capacity of p75NTR to bind and remove Aβ from target cortical fields, the widespread basalo-cortical cholinergic projections are likely to afford the entire cerebral cortex with a powerful molecular ‘drain’ for Aβ clearance. The robust uptake of IgG192-Cy3 by cholinergic axon terminals in the prefrontal cortex with its transport to BF nuclei in vivo supports the potential homeostatic role of p75NTR in Aβ clearance.
Late endosomes and lysosomes constitute the primary destination of Cy3-IgG192 in BF cholinergic neurons
As an endogenous ligand of p75NTR (Yaar et al. 1997; Dechant and Barde 2002), Aβ is likely to undergo rapid neurotrophin-like internalization followed by retrograde trafficking to the perykarion of BF cholinergic cells. Indistinguishable kinetics of intracellular trafficking and distribution of Alexa-488-Aβ and Cy3-IgG192 along with their strong co-localization within transport organelles are consistent with such a premise. Although the precise sites and molecular machinery driving the uptake of p75NTR remain unclear, its association with caveolae-like compartments on the surface membrane has been documented in PC-12 cells and motor neurons (Huang et al. 1999; Lalli and Schiavo 2002). Indeed, specialized lipid rafts enriched with caveolin and p75NTR have been shown to bind NGF and mediate its rapid internalization (Herreros et al. 2001; Saavedra et al. 2007). Partial co-localization of Alexa488-Aβ (1–42) with caveolin containing vesicles has been also shown in cultured superior cervical ganglionic neurons, albeit the bulk of internalized Aβ in these cells was not associated with caveolin or vesicles carrying EEA1, an early endosome qualifier protein (Saavedra et al. 2007). Our microscopic data with double immuno-labelling of BF cholinergic cells shows a low level of co-localization of Cy3-IgG192 and EEA1, consistent with the negligible role of early endosomes in the internalization of p75NTR and its ligands. Similar experiments using markers for recycling (Rab11), late (Rab7) endosomes and lysosomes (Lamp1) (Cantalupo et al. 2001) revealed their much stronger co-localization with Cy3-IgG192, with estimated coefficients being highest for Lamp1. Importantly, both Rab7 and Lamp1 proteins label acidifying late endosomes and lysosomes, which constitute the primary site for degradation of metabolite and cellular debris (Ullrich et al. 1996; Jager et al. 2004). The direct relevance of acidifying late endosomes and multi-vesicular bodies (MVBs) to Aβ degradation, through their full fusion or transient ‘kiss-and-run’ interactions with lysosomes has been explicitly shown, with MVBs and lysosomes forming dynamic late endosome-lysosome hybrids, warranting effective proteolysis of Aβ (Mullins and Bonifacino 2001; Nixon 2007). It is worth noting that the level of MVBs and late endosomes and their loading with Aβ increases with age and have been demonstrated to be especially high in mouse models of AD and in AD brain autopsies (Takahashi et al. 2002b; Takahashi et al. 2002a; LaFerla et al. 2007). In APPxPS1 mice, for instance, the Aβ positive granulated structures are largely concentrated within the perinuclear regions of neurons and are immuno-reactive for key lysosomal proteins, including cathepsin-D—a major photolytic enzyme (Langui et al. 2004; Almeida et al. 2006). Taken as a whole, while internalization and removal of Aβ by p75NTR rich basalo-cortical axons are likely to keep check on the level of Aβ in the cerebral cortex, the pathological increase of Aβ production with failure of other clearance mechanisms could overwhelm the proteolytic machinery of cholinergic cells, jeopardising numerous key biological processes, thereby, perhaps, contributing towards higher vulnerability and early loss of these neurons in to course of AD.
Supplementary Material
Acknowledgments
This work was supported by the Program for Research in Third Level Institutions Cycle 4 grant from the Irish Higher Educational Authority for the Neuroscience section of ‘Targeted-driven therapeutics and theranostics’ (to Saak V. Ovsepian and J. Oliver Dolly), NIH/NINDS NS023945 grant (to Laszlo Zaborszky) and Deutsche Forschungsgemeinschaft within the framework of the Munich Cluster for Systems Neurology (EXC 1010 SyNergy) (Jochen Herms). Authors thank Liam Ryan for expressing and purifying BoNT/A and/B.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00429-013-0583-x) contains supplementary material, which is available to authorized users.
Contributor Information
Saak V. Ovsepian, Email: saak.ovsepian@gmail.com, International Centre for Neurotherapeutics, Dublin City University, Glasnevin, Dublin 9, Republic of Ireland; Department of Translational Brain Research, DZNE, Munich, Germany.
Inga Antyborzec, International Centre for Neurotherapeutics, Dublin City University, Glasnevin, Dublin 9, Republic of Ireland; Department of Translational Brain Research, DZNE, Munich, Germany.
Valerie B. O’Leary, International Centre for Neurotherapeutics, Dublin City University, Glasnevin, Dublin 9, Republic of Ireland Institute of Radiation Biology, Helmholtz Zentrum Munchen, Neuherberg, Germany.
Laszlo Zaborszky, Centre for Molecular and Behavioural Neuroscience, The State University of New Jersey Rutgers, Newark, NJ, USA.
Jochen Herms, Department of Translational Brain Research, DZNE, Munich, Germany.
J. Oliver Dolly, Email: oliver.dolly@dcu.ie, International Centre for Neurotherapeutics, Dublin City University, Glasnevin, Dublin 9, Republic of Ireland.
References
- Allen SJ, Watson JJ, Dawbarn D. The neurotrophins and their role in Alzheimer’s disease. Curr Neuropharmacol. 2011;9(4):559–573. doi: 10.2174/157015911798376190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida CG, Takahashi RH, Gouras GK. Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J Neurosci. 2006;26:4277–4288. doi: 10.1523/JNEUROSCI.5078-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arevalo MA, Roldan PM, Chacon PJ, Rodriguez-Tebar A. Amyloid beta serves as an NGF-like neurotrophic factor or acts as a NGF antagonist depending on its concentration. J Neurochem. 2009;111:1425–1433. doi: 10.1111/j.1471-4159.2009.06412.x. [DOI] [PubMed] [Google Scholar]
- Auld DS, Day JC, Mennicken F, Quirion R. Pharmacological characterization of endogenous acetylcholine release from primary septal cultures. J Pharmacol Exp Ther. 2000;292:692–697. [PubMed] [Google Scholar]
- Baldwin AN, Shooter EM. Zone mapping of the binding domain of the rat low affinity nerve growth factor receptor by the introduction of novel N-glycosylation sites. J Biol Chem. 1995;270:4594–4602. doi: 10.1074/jbc.270.9.4594. [DOI] [PubMed] [Google Scholar]
- Buckner RL, Andrews-Hanna JR, Schacter DL. The brain’s default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci. 2008;1124:1–38. doi: 10.1196/annals.1440.011. [DOI] [PubMed] [Google Scholar]
- Butowt R, von Bartheld CS. Fates of neurotrophins after retrograde axonal transport: phosphorylation of p75NTR is a sorting signal for delayed degradation. J Neurosci. 2009;29:10715–10729. doi: 10.1523/JNEUROSCI.2512-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G, Gage FH. The cholinergic nucleus basalis: a key structure in neocortical arousal. Exs. 1989;57:159–171. doi: 10.1007/978-3-0348-9138-7_16. [DOI] [PubMed] [Google Scholar]
- Cantalupo G, Alifano P, Roberti V, Bruni CB, Bucci C. Rab-interacting lysosomal protein (RILP): the Rab7 effector required for transport to lysosomes. EMBO J. 2001;20:683–693. doi: 10.1093/emboj/20.4.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy B, Menard M, Ito S, Gaudet C, Dal Pra I, Armato U, Whitfield J. Hippocampal membrane-associated p75NTR levels are increased in Alzheimer’s disease. J Alzheimers Dis. 2012;30:675–684. doi: 10.3233/JAD-2012-120115. [DOI] [PubMed] [Google Scholar]
- Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- Coulson EJ, May LM, Osborne SL, Reid K, Underwood CK, Meunier FA, Bartlett PF, Sah P. p75 neurotrophin receptor mediates neuronal cell death by activating GIRK channels through phosphatidylinositol 4,5-bisphosphate. J Neurosci. 2008;28:315–324. doi: 10.1523/JNEUROSCI.2699-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulson EJ, May LM, Sykes AM, Hamlin AS. The role of the p75 neurotrophin receptor in cholinergic dysfunction in Alzheimer’s disease. Neuroscientist. 2009;15:317–323. doi: 10.1177/1073858408331376. [DOI] [PubMed] [Google Scholar]
- Counts SE, Mufson EJ. The role of nerve growth factor receptors in cholinergic basal forebrain degeneration in prodromal Alzheimer disease. J Neuropathol Exp Neurol. 2005;64:263–272. doi: 10.1093/jnen/64.4.263. [DOI] [PubMed] [Google Scholar]
- Dahlgren KN, Manelli AM, Stine WB, Jr, Baker LK, Krafft GA, LaDu MJ. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem. 2002;277:32046–32053. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- Dean C, Liu H, Dunning FM, Chang PY, Jackson MB, Chapman ER. Synaptotagmin-IV modulates synaptic function and long-term potentiation by regulating BDNF release. Nat Neurosci. 2009;12:767–776. doi: 10.1038/nn.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dechant G, Barde YA. The neurotrophin receptor p75(NTR): novel functions and implications for diseases of the nervous system. Nat Neurosci. 2002;5:1131–1136. doi: 10.1038/nn1102-1131. [DOI] [PubMed] [Google Scholar]
- Ertel SI, Ertel EA. Low-voltage-activated T-type Ca2+ channels. Trends Pharmacol Sci. 1997;18:37–42. doi: 10.1016/s0165-6147(96)01021-8. [DOI] [PubMed] [Google Scholar]
- Ertel SI, Ertel EA, Clozel JP. T-type Ca2+ channels and pharmacological blockade: potential pathophysiological relevance. Cardiovasc Drugs Ther. 1997;11:723–739. doi: 10.1023/a:1007706022381. [DOI] [PubMed] [Google Scholar]
- Etheredge JA, Murchison D, Abbott LC, Griffith WH. Functional compensation by other voltage-gated Ca2+ channels in mouse basal forebrain neurons with Ca(V)2.1 mutations. Brain Res. 2007;1140:105–119. doi: 10.1016/j.brainres.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Gad H, Low P, Zotova E, Brodin L, Shupliakov O. Dissociation between Ca2+-triggered synaptic vesicle exocytosis and clathrin-mediated endocytosis at a central synapse. Neuron. 1998;21:607–616. doi: 10.1016/s0896-6273(00)80570-x. [DOI] [PubMed] [Google Scholar]
- Geula C, Mesulam MM, Saroff DM, Wu CK. Relationship between plaques, tangles, and loss of cortical cholinergic fibers in Alzheimer disease. J Neuropathol Exp Neurol. 1998;57:63–75. doi: 10.1097/00005072-199801000-00008. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Che S, Wuu J, Counts SE, Mufson EJ. Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer’s disease. J Neurochem. 2006;97:475–487. doi: 10.1111/j.1471-4159.2006.03764.x. [DOI] [PubMed] [Google Scholar]
- Griffith WH, Taylor L, Davis MJ. Whole-cell and single-channel calcium currents in guinea pig basal forebrain neurons. J Neurophysiol. 1994;71:2359–2376. doi: 10.1152/jn.1994.71.6.2359. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Hartig W, Seeger J, Naumann T, Brauer K, Bruckner G. Selective in vivo fluorescence labelling of cholinergic neurons containing p75(NTR) in the rat basal forebrain. Brain Res. 1998;808:155–165. doi: 10.1016/s0006-8993(98)00792-6. [DOI] [PubMed] [Google Scholar]
- Hartmann M, Heumann R, Lessmann V. Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J. 2001;20:5887–5897. doi: 10.1093/emboj/20.21.5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmo ME, Giocomo LM. Cholinergic modulation of cortical function. J Mol Neurosci. 2006;30:133–135. doi: 10.1385/JMN:30:1:133. [DOI] [PubMed] [Google Scholar]
- Hefti F, Weiner WJ. Nerve growth factor and Alzheimer’s disease. Ann Neurol. 1986;20:275–281. doi: 10.1002/ana.410200302. [DOI] [PubMed] [Google Scholar]
- Herreros J, Ng T, Schiavo G. Lipid rafts act as specialized domains for tetanus toxin binding and internalization into neurons. Mol Biol Cell. 2001;12:2947–2960. doi: 10.1091/mbc.12.10.2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CS, Zhou J, Feng AK, Lynch CC, Klumperman J, DeArmond SJ, Mobley WC. Nerve growth factor signaling in caveolae-like domains at the plasma membrane. J Biol Chem. 1999;274:36707–36714. doi: 10.1074/jbc.274.51.36707. [DOI] [PubMed] [Google Scholar]
- Huerta PT, Lisman JE. Bidirectional synaptic plasticity induced by a single burst during cholinergic theta oscillation in CA1 in vitro. Neuron. 1995;15:1053–1063. doi: 10.1016/0896-6273(95)90094-2. [DOI] [PubMed] [Google Scholar]
- Huotari J, Helenius A. Endosome maturation. EMBO J. 2011;30:3481–3500. doi: 10.1038/emboj.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen EL. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci. 2004;117:4837–4848. doi: 10.1242/jcs.01370. [DOI] [PubMed] [Google Scholar]
- Kacza J, Grosche J, Seeger J, Brauer K, Bruckner G, Hartig W. Laser scanning and electron microscopic evidence for rapid and specific in vivo labelling of cholinergic neurons in the rat basal forebrain with fluorochromated antibodies. Brain Res. 2000;867:232–238. doi: 10.1016/s0006-8993(00)02239-3. [DOI] [PubMed] [Google Scholar]
- Lacy DB, Stevens RC. Recombinant expression and purification of the botulinum neurotoxin type A translocation domain. Protein Expr Purif. 1997;11:195–200. doi: 10.1006/prep.1997.0772. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer’s disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- Lah JJ, Levey AI. Endogenous presenilin-1 targets to endocytic rather than biosynthetic compartments. Mol Cell Neurosci. 2000;16:111–126. doi: 10.1006/mcne.2000.0861. [DOI] [PubMed] [Google Scholar]
- Lalli G, Schiavo G. Analysis of retrograde transport in motor neurons reveals common endocytic carriers for tetanus toxin and neurotrophin receptor p75NTR. J Cell Biol. 2002;156:233–239. doi: 10.1083/jcb.200106142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langui D, Girardot N, El Hachimi KH, Allinquant B, Blanchard V, Pradier L, Duyckaerts C. Subcellular topography of neuronal Abeta peptide in APPxPS1 transgenic mice. Am J Pathol. 2004;165:1465–1477. doi: 10.1016/s0002-9440(10)63405-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JJ. Cholinergic control of GABA release: emerging parallels between neocortex and hippocampus. Trends Neurosci. 2008;31:317–327. doi: 10.1016/j.tins.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Lawrence JJ, Grinspan ZM, Statland JM, McBain CJ. Muscarinic receptor activation tunes mouse stratum oriens interneurones to amplify spike reliability. J Physiol. 2006;571:555–562. doi: 10.1113/jphysiol.2005.103218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytton J, Westlin M, Hanley MR. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J Biol Chem. 1991;266:17067–17071. [PubMed] [Google Scholar]
- McKinney M, Jacksonville MC. Brain cholinergic vulnerability: relevance to behavior and disease. Biochem Pharmacol. 2005;70:1115–1124. doi: 10.1016/j.bcp.2005.05.019. [DOI] [PubMed] [Google Scholar]
- Mesulam MM. Human brain cholinergic pathways. Prog Brain Res. 1990;84:231–241. doi: 10.1016/s0079-6123(08)60908-5. [DOI] [PubMed] [Google Scholar]
- Mesulam M. The cholinergic lesion of Alzheimer’s disease: pivotal factor or side show? Learn Mem. 2004;11:43–49. doi: 10.1101/lm.69204. [DOI] [PubMed] [Google Scholar]
- Mudd LM, Torres J, Lopez TF, Montague J. Effects of growth factors and estrogen on the development of septal cholinergic neurons from the rat. Brain Res Bull. 1998;45:137–142. doi: 10.1016/s0361-9230(97)10328-8. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Ginsberg SD, Ikonomovic MD, DeKosky ST. Human cholinergic basal forebrain: chemoanatomy and neuro-logic dysfunction. J Chem Neuroanat. 2003;26(4):233–242. doi: 10.1016/s0891-0618(03)00068-1. [DOI] [PubMed] [Google Scholar]
- Mullins C, Bonifacino JS. The molecular machinery for lysosome biogenesis. BioEssays. 2001;23:333–343. doi: 10.1002/bies.1048. [DOI] [PubMed] [Google Scholar]
- Murchison D, Griffith WH. Low-voltage activated calcium currents increase in basal forebrain neurons from aged rats. J Neurophysiol. 1995;74:876–887. doi: 10.1152/jn.1995.74.2.876. [DOI] [PubMed] [Google Scholar]
- Naumann T, Casademunt E, Hollerbach E, Hofmann J, Dechant G, Frotscher M, Barde YA. Complete deletion of the neurotrophin receptor p75NTR leads to long-lasting increases in the number of basal forebrain cholinergic neurons. J Neurosci. 2002;22:2409–2418. doi: 10.1523/JNEUROSCI.22-07-02409.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazer B, Hong S, Selkoe DJ. LRP promotes endocytosis and degradation, but not transcytosis, of the amyloid-beta peptide in a blood-brain barrier in vitro model. Neurobiol Dis. 2008;30:94–102. doi: 10.1016/j.nbd.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitsch RM, Slack BE, Wurtman RJ, Growdon JH. Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science. 1992;258:304–307. doi: 10.1126/science.1411529. [DOI] [PubMed] [Google Scholar]
- Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–4091. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- O’Leary VB, Ovsepian SV, Raghunath A, Huo Q, Lawrence GW, Smith L, Dolly JO. Innocuous full-length botulinum neurotoxin targets and promotes the expression of lentiviral vectors in central and autonomic neurons. Gene Ther. 2011;18:656–665. doi: 10.1038/gt.2011.8. [DOI] [PubMed] [Google Scholar]
- Ovsepian SV. Differential cholinergic modulation of synaptic encoding and gain control mechanisms in rat hippocampus. Neurosci Res. 2008;61:92–98. doi: 10.1016/j.neures.2008.01.015. [DOI] [PubMed] [Google Scholar]
- Ovsepian SV, Dolly JO. Dendritic SNAREs add a new twist to the old neuron theory. Proc Natl Acad Sci USA. 2011;108:19113–19120. doi: 10.1073/pnas.1017235108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovsepian SV, Friel DD. Enhanced synaptic inhibition disrupts the efferent code of cerebellar Purkinje neurons in leaner Cav2.1 Ca2+ channel mutant mice. Cerebellum. 2012;11:666–680. doi: 10.1007/s12311-010-0210-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovsepian SV, Anwyl R, Rowan MJ. Endogenous acetylcholine lowers the threshold for long-term potentiation induction in the CA1 area through muscarinic receptor activation: in vivo study. Eur J Neurosci. 2004;20:1267–1275. doi: 10.1111/j.1460-9568.2004.03582.x. [DOI] [PubMed] [Google Scholar]
- Ovsepian SV, Dolly JO, Zaborszky L. Intrinsic voltage dynamics govern the diversity of spontaneous firing profiles in basal forebrain noncholinergic neurons. J Neurophysiol. 2012;108:406–418. doi: 10.1152/jn.00642.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AN, Jhamandas JH. Neuronal receptors as targets for the action of amyloid-beta protein (Abeta) in the brain. Expert Rev Mol Med. 2012;14:e2. doi: 10.1017/S1462399411002134. [DOI] [PubMed] [Google Scholar]
- Saavedra L, Mohamed A, Ma V, Kar S, de Chaves EP. Internalization of beta-amyloid peptide by primary neurons in the absence of apolipoprotein E. J Biol Chem. 2007;282:35722–35732. doi: 10.1074/jbc.M701823200. [DOI] [PubMed] [Google Scholar]
- Salinas S, Schiavo G, Kremer EJ. A hitchhiker’s guide to the nervous system: the complex journey of viruses and toxins. Nat Rev Microbiol. 2010;8:645–655. doi: 10.1038/nrmicro2395. [DOI] [PubMed] [Google Scholar]
- Shupliakov O. The synaptic vesicle cluster: a source of endocytic proteins during neurotransmitter release. Neuroscience. 2009;158:204–210. doi: 10.1016/j.neuroscience.2008.03.035. [DOI] [PubMed] [Google Scholar]
- Smith LA. Development of recombinant vaccines for botulinum neurotoxin. Toxicon. 1998;36:1539–1548. doi: 10.1016/s0041-0101(98)00146-9. [DOI] [PubMed] [Google Scholar]
- Sotthibundhu A, Sykes AM, Fox B, Underwood CK, Thangnipon W, Coulson EJ. Beta-amyloid(1–42) induces neuronal death through the p75 neurotrophin receptor. J Neurosci. 2008;28:3941–3946. doi: 10.1523/JNEUROSCI.0350-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaab DF. Brain aging and Alzheimer’s disease, “wear and tear” versus “use it or lose it”. Neurobiol Aging. 1991;12:317–324. doi: 10.1016/0197-4580(91)90008-8. [DOI] [PubMed] [Google Scholar]
- Takahashi RH, Nam EE, Edgar M, Gouras GK. Alzheimer beta-amyloid peptides: normal and abnormal localization. Histol Histopathol. 2002a;17:239–246. doi: 10.14670/HH-17.239. [DOI] [PubMed] [Google Scholar]
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002b;161:1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniuchi M, Johnson EM., Jr Characterization of the binding properties and retrograde axonal transport of a monoclonal antibody directed against the rat nerve growth factor receptor. J Cell Biol. 1985;101:1100–1106. doi: 10.1083/jcb.101.3.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RW, Lipscombe D, Madison DV, Bley KR, Fox AP. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci. 1988;11:431–438. doi: 10.1016/0166-2236(88)90194-4. [DOI] [PubMed] [Google Scholar]
- Ullrich O, Reinsch S, Urbe S, Zerial M, Parton RG. Rab11 regulates recycling through the pericentriolar recycling endosome. J Cell Biol. 1996;135:913–924. doi: 10.1083/jcb.135.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Zee CE, Ross GM, Riopelle RJ, Hagg T. Survival of cholinergic forebrain neurons in developing p75NGFR-deficient mice. Science. 1996;274:1729–1732. doi: 10.1126/science.274.5293.1729. [DOI] [PubMed] [Google Scholar]
- Wang YJ, Wang X, Lu JJ, Li QX, Gao CY, Liu XH, Sun Y, Yang M, Lim Y, Evin G, Zhong JH, Masters C, Zhou XF. p75NTR regulates Abeta deposition by increasing Abeta production but inhibiting Abeta aggregation with its extracellular domain. J Neurosci. 2011;31:2292–2304. doi: 10.1523/JNEUROSCI.2733-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman CP, McFadzean I, Gibson A, Tucker JF. Two distinct membrane currents activated by cyclopiazonic acid-induced calcium store depletion in single smooth muscle cells of the mouse anococcygeus. Br J Pharmacol. 1996;117:566–572. doi: 10.1111/j.1476-5381.1996.tb15228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaar M, Zhai S, Pilch PF, Doyle SM, Eisenhauer PB, Fine RE, Gilchrest BA. Binding of beta-amyloid to the p75 neurotrophin receptor induces apoptosis. A possible mechanism for Alzheimer’s disease. J Clin Invest. 1997;100:2333–2340. doi: 10.1172/JCI119772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaar M, Zhai S, Fine RE, Eisenhauer PB, Arble BL, Stewart KB, Gilchrest BA. Amyloid beta binds trimers as well as monomers of the 75-kDa neurotrophin receptor and activates receptor signaling. J Biol Chem. 2002;277:7720–7725. doi: 10.1074/jbc.M110929200. [DOI] [PubMed] [Google Scholar]
- Yan Z, Feng J. Alzheimer’s disease: interactions between cholinergic functions and beta-amyloid. Curr Alzheimer Res. 2004;1:241–248. doi: 10.2174/1567205043331992. [DOI] [PubMed] [Google Scholar]
- Zaborszky L. The modular organization of brain systems. Basal forebrain: the last frontier. Prog Brain Res. 2002;136:359–372. doi: 10.1016/s0079-6123(02)36030-8. [DOI] [PubMed] [Google Scholar]
- Zeng F, Lu JJ, Zhou XF, Wang YJ. Roles of p75NTR in the pathogenesis of Alzheimer’s disease: a novel therapeutic target. Biochem Pharmacol. 2011;82:1500–1509. doi: 10.1016/j.bcp.2011.06.040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.