

Abstract
Hypercalcemia of malignancy affects up to one in five cancer patients during the course of their disease. It is associated with both liquid malignancies, commonly multiple myeloma, leukemia, and non-Hodgkins lymphoma and solid cancers, particularly breast and renal carcinomas as well as squamous cell carcinomas of any organ. The clinical manifestations of hypercalcemia are generally constitutional in nature and not specific to the inciting malignancy. Such physical manifestations can range from malaise to lethargy and confusion. Constipation and anorexia are common. Acute kidney injury is likely the most frequently encountered manifestation of end organ damage. Symptomatology is closely linked to both the absolute elevation of serum calcium levels and the rapidity of calcium rise. The majority of cases are humoral in etiology and related to parathyroid hormone-related protein (PTHrP). Approximately 20% of cases are the result of direct bone metastasis with extra-renal 1,25-dihydroxyvitamin D (calcitriol) and ectopic parathyroid hormone production likely accounting for less than 1% of cases. The diagnosis of hypercalcemia of malignancy is confirmed either by an elevated PTHrP or by an evidence of bone metastasis in the appropriate clinical setting. Treatment is predicated on the patient’s symptoms and absolute serum calcium level. Interventions are aimed at lowering the serum calcium concentration by inhibiting bone resorption and increasing urinary calcium excretion, the former accomplished via bisphosphonate therapy and the latter with aggressive hydration. Novel therapies for refractory disease include denosumab, a monoclonal antibody against the receptor activator of nuclear factor κB ligand, and the calcimimetic cinacalcet. Finally, anti-PTHrP antibodies have been successfully deployed in animal models of disease. Despite the efficacy of the above therapies, hypercalcemia of malignancy portends an ominous prognosis, indicating advanced and often refractory cancer with survival on the order of months.
Keywords: hypercalcemia of malignancy parathyroid hormone, parathyroid hormone-related protein, calcitonin, bisphosphonates, denosumab, cinacalcet
Video abstract
Objectives
A comprehensive review of all aspects of hypercalcemia of malignancy is presented herein to improve the physician’s understanding and management of this frequent disease state. The goals of this paper include educating the clinician on the etiology, clinical presentation, and pathogenesis of hypercalcemia among cancer patients. Thereafter, the evaluation and management of such patients is reviewed for the practicing physician. Finally, a detailed summary of previous, current, and novel therapeutic options is described.
Epidemiology
First described in 1921, hypercalcemia of malignancy now occurs in upward of 20% of cancer patients during the course of their disease.1–3 While exact estimates vary as a function of the population studied and the serum calcium cutoff used, hypercalcemia of malignancy is both the most common cause of hypercalcemia in cancer patients and the leading cause of hypercalcemia in the inpatient setting.2,4 Among all cancers, multiple myeloma appears to be the cancer with the highest prevalence of hypercalcemia.4–6 With respect to solid cancers, breast and renal carcinomas followed by squamous carcinomas of any origin are the most common culprits.1,4 Among liquid malignancies, multiple myeloma is the most prevalent hematologic cancer associated with hypercalcemia followed by leukemia and non-Hodgkins lymphoma.4–6 Tumors rarely inciting hypercalcemia include central nervous system malignancies and prostate cancer, as well as stomach and colorectal adenocarcinoma.7
Clinical manifestations
The clinical manifestations of hypercalcemia are protean, non-specific, and independent of etiology.8 Symptomatology is closely linked to both the absolute elevation of serum calcium levels and the rapidity of rise such that moderate hypercalcemia (serum calcium 12–14 mg/dL, 3–3.5 mmol/L) occurring over a period of months may be well tolerated and only vaguely symptomatic whereas similar levels of hypercalcemia occurring over a period of weeks can result in florid symptoms.8 Severe hypercalcemia (serum calcium >14 mg/dL, >3.5 mmol/L) is nearly always symptomatic both because of the absolute level of serum calcium and because such hypercalcemia is most often associated with malignancy, an elevation that occurs over a period of weeks to months. Non-specific neuropsychiatric symptoms include malaise and lassitude with progression to lethargy, confusion, and coma in those with severe elevations.7 Muscle weakness has also been reported. Constipation, anorexia, and nausea are frequent gastrointestinal expressions of disease with pancreatitis and peptic ulcer disease infrequently encountered.8,9 Cardiovascular manifestations include a shortening of the QT interval and dysrhythmias. Renal dysfunction appears to be the most clinically important sequelae of hypercalcemia. Patients often report polyuria consistent with nephrogenic diabetes insipidus, a result of the kidney’s impaired concentrating ability in the hypercalcemic milieu. Acute kidney injury, while not a symptom, is common and the product of direct renal vasoconstriction and natriuresis-induced volume contraction.10 Depressed oral intake from nausea and malaise also contribute to a state of volume depletion. Nephrolithiasis, while frequently cited, is an uncommon acute manifestation of hypercalcemia, and nearly always only found in those with longstanding disease.
Mechanism of disease
Hypercalcemia of malignancy occurs as the result of direct bone metastasis and via humoral mechanisms such as parathyroid hormone-related protein (PTHrP) or 1,25-dihydroxyvitamin D mediated pathways. Rarely, ectopic secretion of parathyroid hormone (PTH) has been implicated. Hypercalcemia due to osteolytic bone lesions is common in multiple myeloma, leukemia, and breast cancer. Humoral hypercalcemia is predominant in squamous cell, renal cell and ovarian cancers, and lymphomas are associated with 1,25-dihydroxyvitamin D mediated hypercalcemia.11
Identified in the 1930s by Gutman et al, osteolytic metastases was the first of the aforementioned mechanisms observed among hypercalcemia individuals with extensive skeletal tumor burden.7 At present, it accounts for 20% of cases of hypercalcemia of malignancy and is frequently encountered in multiple myeloma, metastatic breast cancer, and to a lesser extent in leukemia and lymphoma.7 Local osteolytic hypercalcemia was initially attributed to the direct physical destruction of bone by malignant cells; however, current insights suggest that the presence of tumor cells in the bone marrow is insufficient to cause hypercalcemia.7,12
Rather, understanding the pathogenesis of metastasis-induced hypercalcemia requires an appreciation of bone metabolism. Physiologic bone turnover requires the complementary activity of osteoblasts – mesenchymal stem cell-derived bone-forming cells – and bone-resorbing cells of monocyte and macrophage lineage known as osteoclasts.3,13 During times of bone homeostasis, osteoclast activity is in part regulated by the interaction between its receptor activator of nuclear factor κB (RANK) surface receptor and that of the receptor activator of nuclear factor κB ligand (RANKL) protein expressed on osteoblasts and other bone marrow stromal cells. RANKL then binds to the RANK receptor on osteoclast precursors allowing for the maturation and proliferation of osteoclasts (Figure 1).12,14 In local osteolytic hypercalcemia, the RANKL/RANK interaction results in excessive osteoclast activation leading to enhanced bone resorption and thus hypercalcemia.15 In addition, osteoclast activation is also mediated by malignancy secreted cytokines, including interleukin-1, initially termed “osteoclast stimulating factor”, by Mundy et al in the 1970s.16 In the above cases, the degree of hypercalcemia correlates with the tumor burden such that the most severe hypercalcemia occurs in those patients with the most widely disseminated disease.3
Figure 1.
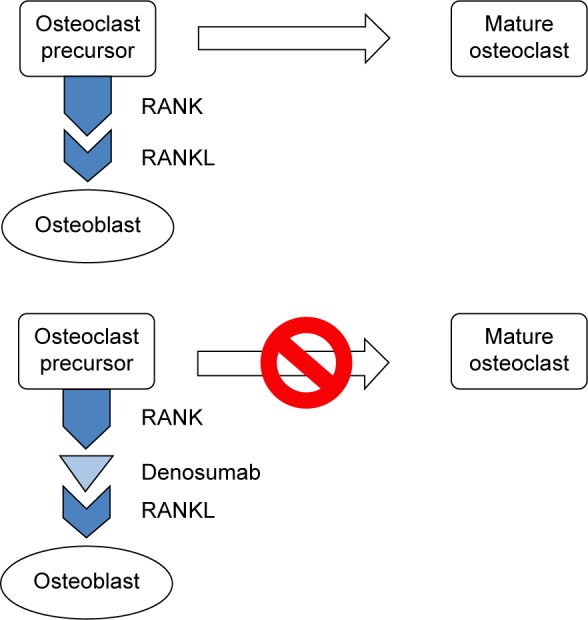
Osteoclast maturation activation and denosumab inhibition of osteoclast maturation.
Abbreviations: RANK, receptor activator of nuclear factor κB; RANKL, receptor activator of nuclear factor κB ligand.
Macrophage inflammation protein 1-alpha (MIP 1-alpha) may also play a role in hypercalcemia of malignancy at least in multiple myeloma patients. MIP 1-alpha is elevated in bone marrow of patients with active multiple myeloma, and MIP 1-alpha has been shown to stimulate osteoclastic formation in murine models as well as in human bone marrow cells.17
From the identification of hypercalcemia of malignancy in the 1920s through to the first large cases series of metastasis-induced hypercalcemia in the 1930s, all hypercalcemia of malignancy was attributed to direct bone invasion.7 However, in the 1940s Fuller Albright described a patient with renal cell carcinoma and hypercalcemia but only a solitary skeletal metastasis.18 He posited that a single metastasis was insufficient to cause hypercalcemia and that other circulating factors were at play. Further credence to Albright’s “humoral hypothesis” occurred in the 1950s and 1960s when case series of patients with hypercalcemia in the absence of bony metastasis were published.19,20 In 1987, PTHrP was isolated confirming Albright’s conjecture from a half a century earlier.1
Humoral hypercalcemia of malignancy now accounts for approximately 80% of hypercalcemia in cancer patients and is often attributed to squamous cell carcinomas as well as kidney, bladder, and ovarian cancers.2 Under physiologic conditions, PTHrP is expressed in nearly all human tissue and functions within the microcirculation to regulate smooth muscle tone and transepithelial calcium transport.21 However, in humoral hypercalcemia of malignancy, PTHrP expression is enhanced and enters the systemic circulation where it exerts its pathologic effect.7
An approximately 130 amino-acid sequence, PTHrP shares 10% structural homology (at the amino-terminal end) with PTH.22,23 Owing to its comparable but non-identical structural profile, PTHrP activates similar secondary messengers such as cyclic adenosine monophosphate (cAMP) and inositol phosphate as well as protein kinases A and C and phospholipase C.24 These intracellular messengers ultimately trigger RANKL expression in osteoblasts, which upon binding with the RANK receptor in osteoclasts triggers further osteoclast generation and activation.
From this similar but non-identical structural profile, the distinct effects of PTHrP, a biochemical syndrome that shares many features with primary hyperparathyroidism, can be anticipated. For example, PTHrP agonizes PTH receptors in bone and kidney resulting in enhanced bone resorption and renal calcium reclamation.25 Specifically, by privileging osteoclast activation and osteoblast suppression, PTHrP uncouples bone resorption from formation leading to marked egress of calcium from bone into circulation. This process is, in part, mediated by cytokines such as interleukin (IL)-1, prostaglandin, lymphotoxin, and tumor necrosis factor (TNF). Both IL-1 and E series prostaglandins directly stimulate bone resorption. Moreover, lymphotoxin and TNF contribute to the recruitment, proliferation, and stimulation of osteoclasts.26 In toto, when this mobilization of calcium, estimated at up to 1,000 mg/day, is paired with depressed renal calcium excretion, profound hypercalcemia results.1 Akin to primary hyperparathyroidism, humoral hypercalcemia of malignancy also enhances renal proximal tubule phosphorus excretion leading to phosphaturia and low-normal serum phosphorus levels. (In contradistinction, metastases-induced hypercalcemia results in hyperphosphatemia). Finally, while PTHrP results in enhanced bone resorption and renal calcium reclamation – owing to the differences in structure with PTH – PTHrP does not augment 1,25-dihydroxyvitamin D production, and as a result does not increase intestinal calcium absorption.27
The third classical pathway whereby malignancy results in hypercalcemia is through extra-renal 1,25-dihydroxyvitamin D (calcitriol) production. Accounting for 1% of cases, increased production of 1,25-dihydroxyvitamin D occurs nearly exclusively in Hodgkin and non-Hodgkin lymphoma with case reports of the same in ovarian dysgerminoma.28,29 In healthy individuals, the conversion of 25-hydroxyvitamin D (calcidiol) to the most active vitamin D metabolite, 1,25-dihydroxyvitamin D (calcitriol) occurs via the enzyme 1-α-hydroxylase in the kidney, a process regulated by PTH.30 However, in 1,25-dihydroxyvitamin D induced hypercalcemia, malignant cells likely recruit and induce adjacent macrophages to express 1-α-hydroxylase, converting endogenous calcidiol into calcitriol.31 Calcitriol then binds to receptors in the intestine leading to heightened enteric calcium reabsorption with resultant hypercalcemia. While 1,25-dihydroxyvita-min D may also induce some bone resorption, this mechanism of disease is best conceptualized as an absorptive form of hypercalcemia.11 Finally, though the pathway is humoral in nature, the term “humoral hypercalcemia of malignancy” is reserved for PTHrP-mediated processes.
Ectopic production of PTH by malignant cells has been described in a handful of cases involving cancer of the ovary and lung, as well as neuroendocrine tumors and sarcoma.28,32–35 In such scenarios, immunohistochemical assays reveal elevated levels of PTH isolated from cancer tissue with subsequent decline in PTH values upon administration of chemotherapy or tumor resection.
Evaluation and diagnosis
With primary hyperparathyroidism and malignancy comprising nearly 90% of cases of hypercalcemia, a diagnostic approach that distinguishes between these two entities is most expedient. The epidemiology and natural history of both processes can assist in ascertaining the diagnosis prior to the acquisition of laboratory data. Among patients presenting to the hospital, hypercalcemia of malignancy is 2–3 times more common than primary hyperparathyroidism with the source of cancer often evident by history and physical examination.36 Moreover, owing to its rapid duration of onset and higher calcium concentrations, cancer-related hypercalcemia is more likely to provoke symptomatic disease. In contrast, findings that suggest primary hyperparathyroidism include mild elevations in serum calcium levels, an asymptomatic patient, a non-focal exam, and a patient without risk factors for cancer.36
Diagnostic algorithms exist for serologic testing and stress a step-wise approach to the identification of the etiology of hypercalcemia.37 However, given the time pressures of day-to-day clinical practice, simultaneous but focused testing is likely most efficient. As such, an initial panel consisting of PTH, PTHrP, phosphorus, 25-hydroxyvitamin D, and 1,25-dihydroxyvitamin D should be obtained. Elevated PTHrP with a depressed or low-normal PTH suggests humoral hypercalcemia of malignancy. A low to low-normal phosphorus and low 1,25-dihydroxyvitamin D confirms the diagnosis. Hypercalcemia related to direct bone invasion is characterized by a low PTH, undetectable PTHrP, and depressed 1,25-dihydroxyvitamin D. Serum phosphorus is in the high-normal range if not frankly elevated. If hypercalcemia related to bone metastasis is suspected but not apparent, then evaluation for a monoclonal gammopathy should be initiated including serum and urine protein electrophoresis, serum-free light chains, and serum and urine immune fixation. A skeletal survey may also be employed. Lymphoma, a hypercalcemia due to 1,25-dihydroxyvitamin D mediated pathways, is implied by elevations in 1,25-dihydroxyvitamin D without concomitant elevations in 25-hydroxyvitamin D. In such cases, PTH is low and PTHrP undetectable.
An important epidemiological caveat is the coexistence of hyperparathyroidism with hypercalcemia of malignancy. Affecting perhaps 10% of patients with concomitant cancer-related hypercalcemia, primary hyperparathyroidism is diagnosed via an inappropriately high-normal serum PTH level in the presence of hypercalcemia.38–40 Clinically, such patients often have sub-acute to chronic low-level hypercalcemia, attributable to primary hyperparathyroidism, with the rapid development of severe and symptomatic hypercalcemia linked to malignancy.
Therapy
Treatment of hypercalcemia of malignancy is aimed at lowering the serum calcium concentration by targeting the underlying disease, specifically by inhibiting bone resorption, increasing urinary calcium excretion, and to a lesser extent by decreasing intestinal calcium absorption. Because definitive serologic diagnosis takes several days, treatment is begun empirically at the time of presentation with adjustments occurring as laboratory information becomes available. The urgency and aggressiveness of treatment is predicated on both the serum calcium and importantly, the symptoms manifest. Symptomatology, in turn, is a function of both the absolute serum calcium concentration and the rate of rise of serum calcium.2 While evidence-based guidelines are lacking, in individuals with only mildly symptomatic disease, generally those with serum calcium levels <12 mg/dL (<3 mmol/L), immediate treatment can be deferred, apart supportive measures, until the exact diagnosis has been made. In patients with marked symptoms, often those with serum calcium levels above 14 mg/dL (3.5 mmol/L), prompt treatment with a full complement of therapies should be initiated.41 In those with serum calcium levels between 12 and 14 mg/dL (3–3.5 mmol/L), therapy should be based on symptoms and the clinical judgment of the physician. Given the efficacy, tolerability, and cost effectiveness of the treatments involved, it may be reasonable to treat such individuals similar to those with more severe degrees of hypercalcemia. Table 1 summarizes the various pharmacologic treatments available.42
Table 1.
Treatment of hypercalcemia of malignancy
| Agent | Regimen | Onset | Duration |
|---|---|---|---|
| 0.9% Sodium chloride | 2–4 L IV/day | Immediate | 2–3 days |
| Calcitonin | 4–8 units/kg SQ q 6–12 hours | 4–6 hours | Up to 3 days |
| Bisphosphonates | |||
| Pamidronate | 60–90 mg IV over 2–6 hours | 48 hours | 3–4 weeks |
| Zoledronic acid | 3–4 mg IV over 15–30 minutes | 48 hours | 3–4 weeks |
| Corticosteroids | 200–400 mg hydrocortisone IV/day for 3–5 days | 7 days | Unclear, perhaps 1 week |
| Gallium nitrate | 200 mg/m2 daily for 5 days | 4 days | 2 weeks |
| Denosumab | 120 mg SQ weekly for 4 weeks, then monthly thereafter | 7–10 days | 3–4 months |
Abbreviations: IV, intravenous; q, every; SQ, subcutaneous.
Patients with asymptomatic or mildly symptomatic disease (serum calcium <12 mg/dL, 3 mmol/L) should be managed supportively until a definitive diagnosis has been obtained and chemotherapy initiated. Such therapies include hydration with isotonic fluid (if admitted), avoidance of thiazide diuretics, and a low-calcium diet.
Individuals suffering from moderate (serum calcium 12–14 mg/dL, 3–3.5 mmol/L) to severe (serum calcium >14 mg/dL, >3.5 mmol/L) hypercalcemia require prompt and comprehensive therapy to categorically lower serum calcium levels. Conceptually, treatment consists of temporizing measures such as volume expansion and calcitonin with concurrent bisphosphonate therapy for targeted and definitive treatment. Volume expansion with isotonic fluid is the initial treatment of choice for hypercalcemia of malignancy, leading to a decline in serum calcium of approximately 2 mg/dL (0.5 mmol/L). Parenteral fluid administration is effective at lowering serum calcium in those with acute kidney injury as well as in patients with preserved glomerular filtration rates.43 Fluid resuscitation acts through multiple mechanisms. It corrects the decline in glomerular filtration rate mediated by the direct renal vasoconstriction of hypercalcemia as well as the natriuresis-induced volume contraction of hypercalcemia.10 In addition, it remedies impaired renal clearance of calcium associated with hypovolemia.43 Finally, it reverses volume depletion associated with vomiting and decreased oral intake characteristic of symptomatic hypercalcemia. It also addresses the polyuric state consistent with diabetes insipidus that hypercalcemia provokes. The rate and volume of infusate should take into consideration the severity of hypercalcemia, as well as the patient’s volume status, and comorbidities, particularly cardiac and renal. If oliguric renal failure or congestive heart failure is present, the addition of a diuretic should be considered. A common initial treatment regimen consists of 1–2 L normal saline bolus followed by maintenance fluids at 100–150 mL/hour titrated to a urine output of 100 mL/hour. In those with severe elevations in serum calcium and minimal comorbidities, 4–6 L can be safely administered over the first 24 hours.41 Maintenance fluids should be continued until the patient is euvolemic and adjuvant anti-hypercalcemic agents have taken effect.
Initially employed to augment renal calcium losses, the routine use of loop diuretics has fallen out of favor with the advent of bisphosphonate therapy. Given the attendant electrolyte complications associated with the high doses employed, the need for even more frequent monitoring and the potential for worsening hypovolemia, loop diuretics should be reserved for patients with congestive heart failure and evidence of symptomatic volume overload or in cases of oliguric renal failure with the same.44
Calcitonin, a 32 amino-acid hormone produced by the parafollicular C cells of the thyroid gland, is often an effective anti-hypercalcemic agent lowering serum calcium levels by approximately 2 mg/dL (0.5 mmol/L) for up to 72 hours.45 Calcitonin lowers blood calcium levels by inhibiting bone-resorbing osteoclasts and, to a lesser extent, by enhancing calcium excretion into the urine.46 Available as a synthetic salmon preparation within the US and as a natural porcine product in Europe, an initial dose of 4 units/kg is delivered subcutaneously or intramuscularly every 12 hours until the effects of bisphosphonate therapy are realized. If no response is obtained after initial administration, then doses of up to 8 units/kg every 6 hours can be attempted.46 Calcitonin begins to exert its effect within 4–6 hours with tachyphylaxis occurring after approximately 3 days, likely related to calcitonin receptors downregulation on osteoclasts.47 There are no dosing adjustments necessary for renal failure.42 Side effects are limited to nausea and hypersensitivity reaction in those with salmon allergies.
Since the approval of etidronate for hypercalcemia of malignancy in the late 1980s, bisphosphonates have become the standard of care in the treatment of cancer-associated hypercalcemia providing safe, effective, and sustained reductions in serum calcium levels.2 Pyrophosphate analogs, bisphosphonates, inhibit osteoclast activity in a dose-dependent fashion and stabilize the bone matrix by binding to extracellular calcium and phosphorus.3,48 Clinically, they are effective in treating hypercalcemia resulting from excessive bone resorption of any cause.49 At present, the preferred bisphosphonates for hypercalcemia of malignancy include the high potency parenteral agents pamidronate and zoledronic acid. In a randomized double blind trial of 50 patients with hypercalcemia of malignancy (mean serum calcium 13.5 mg/dL, 3.4 mmol/L) evaluating pamidronate versus placebo, a dose–response relationship vis-à-vis serum calcium was seen after a single pamidronate infusion. By day 4, serum calcium normalized in 40% of patients receiving 30 mg, 60% of patients receiving 60 mg, and 100% of patients receiving 90 mg. Reductions in calcium began 2 days after administration with normocalcemia maintained for 10–13 days after infusion.50 Zoledronate is nearly one thousand times more potent than pamidronate owing to its superior inhibition of both farnesyl diphosphate and geranylgeranyldiphosphate synthase, enzymes essential for osteoclast activity.51,52 In a double blind fashion, 275 patients with hypercalcemia of malignancy (mean serum calcium 14.0 mg/dL, 3.5 mmol/L) were randomized to either 4 or 8 mg zoledronic acid or 90 mg of pamidronate. In all treatment groups, serum calcium began to decline within 48 hours with normalization beginning to occur by day 4. By day 10, 88% of patients receiving 4 mg zoledronic acid, 87% of patients receiving 8 mg zoledronic acid, and 70% receiving pamidronate achieved normal calcium levels. The duration of sustained normocalcemia was 32 and 43 days in the zoledronic acid 4 and 8 mg groups, respectively, and 18 days in the pamidronate 90 mg arm.53 Based on the results of the above trials as well as the shorter administration infusion time, zoledronic acid, 4 mg, is the preferred bisphosphonate in patients with normal to moderately impaired renal function. With respect to pamidronate, it is administered at a dose of 60 mg to patients with moderate hypercalcemia (serum calcium 12–14 mg/dL, 3–3.5 mmol/L) with 90 mg dose reserved for individuals with severe disease (mean calcium >14.0 mg/dL, 3.5 mmol/L).54
As a class, bisphosphonates are among the best-studied and safest anti-resorptive therapies. Nonetheless, they are subject to certain side effects and limitations. Fever and bone pain during infusion have been reported in approximately 15% of patients. Osteonecrosis of the maxilla and mandible occurs in 1% of treated individuals with nearly all patients suffering from myeloma or breast cancer, having had a preceding dental procedure, and having been exposed to bisphosphonate therapy for at least 6 months, if not 2–3 years.3,55 Ocular complications such as uveitis or orbital inflammation are infrequent, affecting less than 0.5% of patients. Most importantly, the administration of bisphosphonates has been associated with significant nephrotoxicity, specifically pamidronate-induced collapsing focal segmental glomerulosclerosis and acute tubular necrosis (ATN) with zoledronate. The risk of renal failure is directly related to the drug infusion time and dosage. Multiple case series have documented the development of collapsing focal segmental glomerulosclerosis in cancer patients, mostly myeloma, having received more than 1 year of high-dose pamidronate. ATN has been associated with zoledronic acid with acute kidney injury developing within a few doses of therapy. With both medications, renal impairment has been severe enough to result in permanent dialysis dependence.56 In light of their potential nephrotoxicity, the American Society of Clinical Oncology has issued dosing and monitoring guidelines that stipulate bisphosphonate dose and infusion time be tailored to serum creatinine in patients receiving bisphosphonates for treatment of multiple myeloma; however, no guidelines exist for hypercalcemia of malignancy.57 The package insert recommends pamidronate use in patients with creatinine clearance (CrCl) <30 mL/min only in cases of life-threatening tumor-induced hypercalcemia where the benefit outweighs the potential risk.54 The zoledronic acid package insert stipulates that no dose adjustment is needed for treatment of patients with hypercalcemia of malignancy and serum creatinine values <4.5 mg/dL (400 μmol/L).58
Corticosteroids are the therapy of choice for cases of 1,25-dihydroxyvitamin D (calcitriol) mediated hypercalcemia. Steroids inhibit 1-α-hydroxylase conversion of 25-hydroxyvitamin D (calcidiol) into 1,25-dihydroxyvitamin D (calcitriol) therefore lessening intestinal calcium absorption. In the absence of randomized trials, treatment generally consists of intravenous hydrocortisone 200–400 mg/day for 3–5 days followed by oral prednisone 10–20 mg/day for an additional 7 days.59 In addition to a low-calcium diet, case reports indicate a favorable response to the above regimen with declines in serum calcium of >3.0 mg/dL (0.75 mmol/L) within 1 week of initiating therapy.11 The duration of response is uncertain but felt to be in the order of 1 week during which time malignancy-specific interventions are initiated. Side effects of such treatment include hyperglycemia, and further immunosuppression.
Although withdrawn from the US market in 1995, gallium nitrate is likely as effective an anti-hypercalcemic agent as bisphosphonates inhibiting both osteoclast and PTHrP-mediated elevations in calcium.3 In a double blind trial of 64 patients with hypercalcemia of malignancy randomized to either pamidronate or gallium nitrate, restoration of normocalcemia was achieved more frequently and for a longer duration in patients receiving gallium nitrate than pamidronate.60 Clinically, gallium nitrate is administered by continuous infusion at a dosage of 200 mg/m2 daily for 5 days. Owing to its extended administration time, the advent of even more potent bisphosphonates, and a side effect profile including ATN and hypophosphatemia, gallium nitrate is no longer available within the US, but should be considered in bisphosphonate and denosumab refractory disease.
Since the introduction of bisphosphonate therapy, the need for hemodialysis to control hypercalcemia of malignancy has diminished. However, indications remain and include those individuals with oliguric renal failure whose volume status cannot be managed with diuretics alone. Additional categories include patients with severe symptomatic (ie, coma) elevations in serum calcium despite hydration and high-dose bisphosphonate therapy. In such cases, dialysis is indicated for symptom relief and control of hypercalcemia until chemotherapy has been administered. Renal replacement therapy with low or calcium-free dialysate is highly effective at restoring normocalcemia with reductions of 3–5 mg/dL (0.75–1.25 mmol/L) achieved over a 3–4 hour treatment.61,62 The duration of effect is usually on the order of hours to days. While case reports in the literature demonstrate the safety of calcium-free dialysate baths, our preference is to use low-calcium (<2.0 mmol/L) baths to minimize the risk of cardiac arrhythmia, hypotension, and seizures. Given that humoral hypercalcemia is often accompanied by hypophosphatemia and the lack of phosphorus in standard dialysate solutions, hemodialysis-induced hypophosphatemia should be monitored and corrected for.63
Novel therapies for the treatment of hypercalcemia of malignancy include denosumab and cinacalcet with animal models, suggesting a possible future role for anti-PTHrP antibodies. In 2010, the fully human monoclonal antibody denosumab was approved for the treatment of osteoporosis and the prevention of skeletal-related events in patients with solid tumors. In 2014, it was subsequently approved for bisphosphonate refractory hypercalcemia.64 Denosumab is an RANKL antibody that inhibits osteoclast maturation, activation, and function (Figure 1). In case reports, denosumab demonstrated noteworthy anti-hypercalcemic effects in patients with refractory disease despite high-dose bisphosphonate therapy. In an open label, single arm phase two study, 33 patients with a mean serum calcium of 13.5 mg/dL (3.4 mmol/L) despite intravenous bisphosphonate therapy received 120 mg denosumab subcutaneously weekly for 4 weeks followed by monthly injections. Nearly 65% of study patients experienced a normalization of serum calcium by, on average, day 9 with a median response duration of 104 days.65 Denosumab appears to be well-tolerated apart arthralgias and a risk of jaw osteonecrosis comparable to that of zoledronate.64 In trials evaluating the efficacy of denosumab in preventing skeletal-related events, rates of hypocalcemia were close to 10%, nearly double that of bisphosphonate therapy.66 Severe hypocalcemia has also been reported in prostate cancer patients treated with denosumab for the management of osteo-blastic bone metastases.67 Since it is not metabolized by the kidney, acute kidney injury or chronic kidney disease is not a contraindication to its use. However, denosumab must be used with caution in such patients because the risk of hypocalcemia is augmented in patients with impaired calcium homeostasis characteristic of renal failure.68 In light of the above, the optimal dosing regimen for denosumab in those with impaired renal function remains to be determined. However, it is useful to note that in patients with preexisting hypercalcemia of malignancy treated with denosumab, only 2 of 33 patients developed mild hypocalcemia. The median (range) of calculated CrCl in this group was 76 (13–311) mL/min.65 Some authors advocate the use of oral vitamin D supplementation in patients with low vitamin D levels who are hypercalcemic and are treated with denosumab. However, the use of oral vitamin D supplements should be carefully considered against the risks of exacerbating underlying hypercalcemia.
Approved in 2004 for secondary hyperparathyroidism of renal failure and parathyroid carcinoma, cinacalcet is a calcimimetic that interacts with the calcium sensing receptor on parathyroid cells leading to the downregulation of PTH with an attendant decline in serum calcium levels.69 Also expressed in bone and renal tissue, the calcium sensing receptor is responsible for osteoblast differentiation and bone resorption by osteoclasts.70 Cinacalcet effectively lowers serum calcium levels in inoperable parathyroid cancer patients with high PTH levels.71 To date, there have been no trials of cinacalcet for other types of hypercalcemia of malignancy but we reported a patient with metastatic renal cell carcinoma, a PTH of 10 pg/dL (reference range: 12–88) and a PTHrP of 114 pg/dL (reference range: 14–27) with a serum calcium of 14.2 mg/dL (3.6 mmol/L) despite bisphosphonate and deno-sumab therapy. Titration of cinacalcet to 60 mg/day resulted in a serum calcium of 10.1 mg/dL (2.5 mmol/L) at 10 weeks follow-up with a PTHrP of 159 pg/mL at that time.41,72 As such, cinacalcet may yet have a role in the treatment of hypercalcemia, particularly when provoked by malignancy of the parathyroid gland.
A future molecular target for the treatment of hypercalcemia of malignancy involves PTHrP-related antibodies. Using chimeric anti-PTH antibodies, murine models of hypercalcemia suggest infusions of PTHrP antibodies both corrects hypercalcemia and suppresses the release of malignancy associated cytokines such as TNF and multiple interleukins. Moreover, attenuation of these cytokines was accompanied by improvement in appetite, weight, and energy.73
Prognosis
Literature from the early 1990s suggests a median survival of 1–3 months after the diagnosis of hypercalcemia of malignancy.5,74–77 In a retrospective study of 126 patients with cancer-associated hypercalcemia (mean serum calcium 13.2 mg/dL, 3.3 mmol/L) and chemotherapy refractory disease, median survival was 1 month with 75% mortality by 3 months.74 In those whom specific anticancer treatment was available, median survival was 4 months. Another retrospective analysis of nearly 8,000 hypercalcemia of malignancy patients (serum calcium >12 mg/dL, 3.0 mmol/L) from the same time period found a 12-month survival of only 25%.5 No distinction was made between those with chemotherapy sensitive or resistant disease. In a recent study of 138 patients with humoral hypercalcemia of malignancy, the median survival was 52 (21–132) days from the time the PTHrP level was obtained.78 Therefore, while effective therapy exists for controlling serum calcium levels, hypercalcemia of malignancy portends an ominous prognosis, indicating advanced and often refractory cancer. As such, while the aforementioned therapies have markedly decreased the symptoms of hypercalcemia, there is little evidence to suggest that such modalities affect the natural history or mortality rates associated with the underlying malignancy.
Footnotes
Disclosure
Dr Ilya Glezerman has received institutional research support from and had served on advisory board for Amgen Inc. Dr Hillel Sternlicht reports no conflicts of interest in this work.
References
- 1.Horwitz M, Hodak S, Stewart A. Non-parathyroid hypercalcemia. In: Favus M, editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Washington, DC; American Society of Bone and Mineral Research; 2008. pp. 307–312. [Google Scholar]
- 2.Stewart AF. Clinical practice. Hypercalcemia associated with cancer. New Engl J Med. 2005;352(4):373–379. doi: 10.1056/NEJMcp042806. [DOI] [PubMed] [Google Scholar]
- 3.Rosner MH, Dalkin AC. Onco-nephrology: the pathophysiology and treatment of malignancy-associated hypercalcemia. Clin J Am Soc Nephrol. 2012;7(10):1722–1729. doi: 10.2215/CJN.02470312. [DOI] [PubMed] [Google Scholar]
- 4.Burt ME, Brennan MF. Incidence of hypercalcemia and malignant neoplasm. Arch Surg. 1980;115(6):704–707. doi: 10.1001/archsurg.1980.01380060012004. [DOI] [PubMed] [Google Scholar]
- 5.Vassilopoulou-Sellin R, Newman BM, Taylor SH, Guinee VF. Incidence of hypercalcemia in patients with malignancy referred to a comprehensive cancer center. Cancer. 1993;71(4):1309–1312. doi: 10.1002/1097-0142(19930215)71:4<1309::aid-cncr2820710423>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 6.Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21–33. doi: 10.4065/78.1.21. [DOI] [PubMed] [Google Scholar]
- 7.Stewart A, Broadus A. Malignancy Associated Hypercalcemia. In: DeGroot L, Jameson J, editors. Endocrinology 2. 5th ed. Philadelphia: Saunders-Elsevier; 2006. pp. 1555–1565. [Google Scholar]
- 8.Inzucchi SE. Understanding hypercalcemia. Its metabolic basis, signs, and symptoms. Postgrad Med. 2004;115(4):69–70. 3–6. doi: 10.3810/pgm.2004.04.1486. [DOI] [PubMed] [Google Scholar]
- 9.Carnaille B, Oudar C, Pattou F, Combemale F, Rocha J, Proye C. Pancreatitis and primary hyperparathyroidism: forty cases. Aust N Z J Surg. 1998;68(2):117–119. doi: 10.1111/j.1445-2197.1998.tb04719.x. [DOI] [PubMed] [Google Scholar]
- 10.Levi M, Ellis MA, Berl T. Control of renal hemodynamics and glomerular filtration rate in chronic hypercalcemia. Role of prostaglandins, renin-angiotensin system, and calcium. J Clin Invest. 1983;71(6):1624–1632. doi: 10.1172/JCI110918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seymour JF, Gagel RF. Calcitriol: the major humoral mediator of hypercalcemia in Hodgkin’s disease and non-Hodgkin’s lymphomas. Blood. 1993;82(5):1383–1394. [PubMed] [Google Scholar]
- 12.Roodman GD. Mechanisms of bone metastasis. New Engl J Med. 2004;350(16):1655–1664. doi: 10.1056/NEJMra030831. [DOI] [PubMed] [Google Scholar]
- 13.Coleman RE, Seaman JJ. The role of zoledronic acid in cancer: clinical studies in the treatment and prevention of bone metastases. Semin Oncol. 2001;28(2 Suppl 6):11–16. doi: 10.1016/s0093-7754(01)90260-x. [DOI] [PubMed] [Google Scholar]
- 14.Abou-Samra AB, Juppner H, Force T, et al. Expression cloning of a common receptor for parathyroid hormone and parathyroid hormone-related peptide from rat osteoblast-like cells: a single receptor stimulates intracellular accumulation of both cAMP and inositol trisphosphates and increases intracellular free calcium. Proc Natl Acad Sci U S A. 1992;89(7):2732–2736. doi: 10.1073/pnas.89.7.2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guise TA, Yin JJ, Taylor SD, et al. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J Clin Invest. 1996;98(7):1544–1549. doi: 10.1172/JCI118947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mundy GR, Raisz LG, Cooper RA, Schechter GP, Salmon SE. Evidence for the secretion of an osteoclast stimulating factor in myeloma. New Engl J Med. 1974;291(20):1041–1046. doi: 10.1056/NEJM197411142912001. [DOI] [PubMed] [Google Scholar]
- 17.Choi SJ, Cruz JC, Craig F, et al. Macrophage inflammatory protein 1-alpha is a potential osteoclast stimulatory factor in multiple myeloma. Blood. 2000;96(2):671–675. [PubMed] [Google Scholar]
- 18.Case Records of the Massachusetts General Hospital (Case 27461) New Engl J Med. 1941;225(20):789–791. [Google Scholar]
- 19.Gellhorn A, Plimpton CH. Hypercalcemia in malignant disease without evidence of bone destruction. Am J Med. 1956;21(5):750–759. [PubMed] [Google Scholar]
- 20.Lafferty FW. Pseudohyperparathyroidism. Medicine (Baltimore) 1966;45(3):247–260. doi: 10.1097/00005792-196605000-00004. [DOI] [PubMed] [Google Scholar]
- 21.Philbrick WM, Wysolmerski JJ, Galbraith S, et al. Defining the roles of parathyroid hormone-related protein in normal physiology. Physiol Rev. 1996;76(1):127–173. doi: 10.1152/physrev.1996.76.1.127. [DOI] [PubMed] [Google Scholar]
- 22.Burtis WJ, Brady TG, Orloff JJ, et al. Immunochemical characterization of circulating parathyroid hormone-related protein in patients with humoral hypercalcemia of cancer. New Engl J Med. 1990;322(16):1106–1112. doi: 10.1056/NEJM199004193221603. [DOI] [PubMed] [Google Scholar]
- 23.Budayr AA, Nissenson RA, Klein RF, et al. Increased serum levels of a parathyroid hormone-like protein in malignancy-associated hypercalcemia. Ann Intern Med. 1989;111(10):807–812. doi: 10.7326/0003-4819-111-10-807. [DOI] [PubMed] [Google Scholar]
- 24.Orloff JJ, Wu TL, Stewart AF. Parathyroid hormone-like proteins: biochemical responses and receptor interactions. Endocr Rev. 1989;10(4):476–495. doi: 10.1210/edrv-10-4-476. [DOI] [PubMed] [Google Scholar]
- 25.Bender RA, Hansen H. Hypercalcemia in bronchogenic carcinoma. A prospective study of 200 patients. Ann Intern Med. 1974;80(2):205–208. doi: 10.7326/0003-4819-80-2-205. [DOI] [PubMed] [Google Scholar]
- 26.Adami S, Rossini M. Hypercalcemia of malignancy: pathophysiology and treatment. Bone. 1992;13(Suppl 1):S51–S55. doi: 10.1016/s8756-3282(09)80010-0. [DOI] [PubMed] [Google Scholar]
- 27.Nakayama K, Fukumoto S, Takeda S, et al. Differences in bone and vitamin D metabolism between primary hyperparathyroidism and malignancy-associated hypercalcemia. J Clin Endocrinol Metab. 1996;81(2):607–611. doi: 10.1210/jcem.81.2.8636276. [DOI] [PubMed] [Google Scholar]
- 28.Vacher-Coponat H, Opris A, Denizot A, Dussol B, Berland Y. Hypercalcaemia induced by excessive parathyroid hormone secretion in a patient with a neuroendocrine tumour. Nephrol Dial Transplant. 2005;20(12):2832–2835. doi: 10.1093/ndt/gfi065. [DOI] [PubMed] [Google Scholar]
- 29.Hibi M, Hara F, Tomishige H, et al. 1,25-dihydroxyvitamin D-mediated hypercalcemia in ovarian dysgerminoma. Pediatr Hematol Oncol. 2008;25(1):73–78. doi: 10.1080/08880010701774033. [DOI] [PubMed] [Google Scholar]
- 30.Christakos S, Ajibade DV, Dhawan P, Fechner AJ, Mady LJ. Vitamin D: metabolism. Endocrinol Metab Clin North Am. 2010;39(2):243–253. doi: 10.1016/j.ecl.2010.02.002. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hewison M, Kantorovich V, Liker HR, et al. Vitamin D-mediated hypercalcemia in lymphoma: evidence for hormone production by tumor-adjacent macrophages. J Bone Miner Res. 2003;18(3):579–582. doi: 10.1359/jbmr.2003.18.3.579. [DOI] [PubMed] [Google Scholar]
- 32.Nakajima K, Tamai M, Okaniwa S, et al. Humoral hypercalcemia associated with gastric carcinoma secreting parathyroid hormone: a case report and review of the literature. Endocr J. 2013;60(5):557–562. doi: 10.1507/endocrj.ej12-0406. [DOI] [PubMed] [Google Scholar]
- 33.Yoshimoto K, Yamasaki R, Sakai H, et al. Ectopic production of parathyroid hormone by small cell lung cancer in a patient with hypercalcemia. J Clin Endocrinol Metab. 1989;68(5):976–981. doi: 10.1210/jcem-68-5-976. [DOI] [PubMed] [Google Scholar]
- 34.Nussbaum SR, Gaz RD, Arnold A. Hypercalcemia and ectopic secretion of parathyroid hormone by an ovarian carcinoma with rearrangement of the gene for parathyroid hormone. New Engl J Med. 1990;323(19):1324–1328. doi: 10.1056/NEJM199011083231907. [DOI] [PubMed] [Google Scholar]
- 35.Wong K, Tsuda S, Mukai R, Sumida K, Arakaki R. Parathyroid hormone expression in a patient with metastatic nasopharyngeal rhabdomyosarcoma and hypercalcemia. Endocrine. 2005;27(1):83–86. doi: 10.1385/ENDO:27:1:083. [DOI] [PubMed] [Google Scholar]
- 36.Lafferty FW. Differential diagnosis of hypercalcemia. J Bone Miner Res. 1991;6(Suppl 2):S51–S59. doi: 10.1002/jbmr.5650061413. discussion S61. [DOI] [PubMed] [Google Scholar]
- 37.Reagan P, Pani A, Rosner MH. Approach to diagnosis and treatment of hypercalcemia in a patient with malignancy. Am J Kidney Dis. 2014;63(1):141–147. doi: 10.1053/j.ajkd.2013.06.025. [DOI] [PubMed] [Google Scholar]
- 38.Hutchesson AC, Bundred NJ, Ratcliffe WA. Survival in hypercalcaemic patients with cancer and co-existing primary hyperparathyroidism. Postgrad Med J. 1995;71(831):28–31. doi: 10.1136/pgmj.71.831.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Strodel WE, Thompson NW, Eckhauser FE, Knol JA. Malignancy and concomitant primary hyperparathyroidism. J Surg Oncol. 1988;37(1):10–12. doi: 10.1002/jso.2930370104. [DOI] [PubMed] [Google Scholar]
- 40.Casez J, Pfammatter R, Nguyen Q, Lippuner K, Jaeger P. Diagnostic approach to hypercalcemia: relevance of parathyroid hormone and parathyroid hormone-related protein measurements. Eur J Intern Med. 2001;12(4):344–349. doi: 10.1016/s0953-6205(01)00124-8. [DOI] [PubMed] [Google Scholar]
- 41.Bilezikian JP. Management of acute hypercalcemia. New Engl J Med. 1992;326(18):1196–1203. doi: 10.1056/NEJM199204303261806. [DOI] [PubMed] [Google Scholar]
- 42.Davidson TG. Conventional treatment of hypercalcemia of malignancy. Am J Health Syst Pharm. 2001;58(Suppl 3):S8–S15. doi: 10.1093/ajhp/58.suppl_3.S8. [DOI] [PubMed] [Google Scholar]
- 43.Hosking DJ, Cowley A, Bucknall CA. Rehydration in the treatment of severe hypercalcaemia. Q J Med. 1981;50(200):473–481. [PubMed] [Google Scholar]
- 44.LeGrand SB, Leskuski D, Zama I. Narrative review: furosemide for hypercalcemia: an unproven yet common practice. Ann Intern Med. 2008;149(4):259–263. doi: 10.7326/0003-4819-149-4-200808190-00007. [DOI] [PubMed] [Google Scholar]
- 45.Vaughn CB, Vaitkevicius VK. The effects of calcitonin in hypercalcemia in patients with malignancy. Cancer. 1974;34(4):1268–1271. doi: 10.1002/1097-0142(197410)34:4<1268::aid-cncr2820340437>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 46.Deftos LJ, First BP. Calcitonin as a drug. Ann Intern Med. 1981;95(2):192–197. doi: 10.7326/0003-4819-95-2-192. [DOI] [PubMed] [Google Scholar]
- 47.Purdue BW, Tilakaratne N, Sexton PM. Molecular pharmacology of the calcitonin receptor. Receptors Channels. 2002;8(3–4):243–255. [PubMed] [Google Scholar]
- 48.Carano A, Teitelbaum SL, Konsek JD, Schlesinger PH, Blair HC. Bisphosphonates directly inhibit the bone resorption activity of isolated avian osteoclasts in vitro. J Clin Invest. 1990;85(2):456–461. doi: 10.1172/JCI114459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rogers MJ, Gordon S, Benford HL, et al. Cellular and molecular mechanisms of action of bisphosphonates. Cancer. 2000;88(12 Suppl):2961–2978. doi: 10.1002/1097-0142(20000615)88:12+<2961::aid-cncr12>3.3.co;2-c. [DOI] [PubMed] [Google Scholar]
- 50.Nussbaum SR, Younger J, Vandepol CJ, et al. Single-dose intravenous therapy with pamidronate for the treatment of hypercalcemia of malignancy: comparison of 30-, 60-, and 90-mg dosages. Am J Med. 1993;95(3):297–304. doi: 10.1016/0002-9343(93)90282-t. [DOI] [PubMed] [Google Scholar]
- 51.Green JR MK, Jaegggi KA. Preclinical pharmacoloyg of CGP 42446 a new potent heterocyclic bisphosphonate compound. J Bone Miner Res. 1994;9:745–751. doi: 10.1002/jbmr.5650090521. [DOI] [PubMed] [Google Scholar]
- 52.Dunford JE, Thompson K, Coxon FP, et al. Structure-activity relationships for inhibition of farnesyl diphosphate synthase in vitro and inhibition of bone resorption in vivo by nitrogen-containing bisphosphonates. J Pharmacol Exp Ther. 2001;296(2):235–242. [PubMed] [Google Scholar]
- 53.Major P, Lortholary A, Hon J, et al. Zoledronic acid is superior to pamidronate in the treatment of hypercalcemia of malignancy: a pooled analysis of two randomized, controlled clinical trials. J Clin Oncol. 2001;19(2):558–567. doi: 10.1200/JCO.2001.19.2.558. [DOI] [PubMed] [Google Scholar]
- 54.Pharmaceutical Partners of Canada Inc. (Pamidronate) [package insert] Richmond Hill, ON: Pharmaceutical Partners of Canada Inc; 2010. [Accessed July 8, 2015]. Available from: http://fresenius-kabi.ca/wp-content/uploads/2015/01/EN_Web_Insert_Pamid_NL.pdf. [Google Scholar]
- 55.Tanvetyanon T, Stiff PJ. Management of the adverse effects associated with intravenous bisphosphonates. Ann Oncol. 2006;17(6):897–907. doi: 10.1093/annonc/mdj105. [DOI] [PubMed] [Google Scholar]
- 56.Perazella MA, Markowitz GS. Bisphosphonate nephrotoxicity. Kidney Int. 2008;74(11):1385–1393. doi: 10.1038/ki.2008.356. [DOI] [PubMed] [Google Scholar]
- 57.Kyle RA, Yee GC, Somerfield MR, et al. American Society of Clinical Oncology 2007 clinical practice guideline update on the role of bisphosphonates in multiple myeloma. J Clin Oncol. 2007;25(17):2464–2472. doi: 10.1200/JCO.2007.12.1269. [DOI] [PubMed] [Google Scholar]
- 58.Zometa [Accessed July 8, 2015]. Available from: https://www.pharma.us.novartis.com/product/pi/pdf/Zometa.pdf.
- 59.Santarpia L, Koch CA, Sarlis NJ. Hypercalcemia in cancer patients: pathobiology and management. Horm Metab Res. 2010;42(3):153–164. doi: 10.1055/s-0029-1241821. [DOI] [PubMed] [Google Scholar]
- 60.Cvitkovic F, Armand JP, Tubiana-Hulin M, Rossi JF, Warrell RP., Jr Randomized, double-blind, phase II trial of gallium nitrate compared with pamidronate for acute control of cancer-related hypercalcemia. Cancer J. 2006;12(1):47–53. doi: 10.1097/00130404-200601000-00009. [DOI] [PubMed] [Google Scholar]
- 61.Kaiser W, Biesenbach G, Kramar R, Zazgornik J. Calcium free hemodialysis: an effective therapy in hypercalcemic crisis – report of 4 cases. Intensive Care Med. 1989;15(7):471–474. doi: 10.1007/BF00255605. [DOI] [PubMed] [Google Scholar]
- 62.Koo WS, Jeon DS, Ahn SJ, Kim YS, Yoon YS, Bang BK. Calcium-free hemodialysis for the management of hypercalcemia. Nephron. 1996;72(3):424–428. doi: 10.1159/000188907. [DOI] [PubMed] [Google Scholar]
- 63.Leehey DJ, Ing TS. Correction of hypercalcemia and hypophosphatemia by hemodialysis using a conventional, calcium-containing dialysis solution enriched with phosphorus. Am J Kidney Dis. 1997;29(2):288–290. doi: 10.1016/s0272-6386(97)90044-5. [DOI] [PubMed] [Google Scholar]
- 64.Castellano D, Sepulveda JM, Garcia-Escobar I, Rodriguez-Antolin A, Sundlov A, Cortes-Funes H. The role of RANK-ligand inhibition in cancer: the story of denosumab. Oncologist. 2011;16(2):136–145. doi: 10.1634/theoncologist.2010-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hu MI, Glezerman IG, Leboulleux S, et al. Denosumab for treatment of hypercalcemia of malignancy. J Clin Endocrinol Metab. 2014;99(9):3144–3152. doi: 10.1210/jc.2014-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lipton A, Fizazi K, Stopeck AT, et al. Superiority of denosumab to zoledronic acid for prevention of skeletal-related events: a combined analysis of 3 pivotal, randomised, phase 3 trials. Eur J Cancer. 2012;48(16):3082–3092. doi: 10.1016/j.ejca.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 67.Autio KA, Farooki A, Glezerman IG, et al. Severe hypocalcemia associated with denosumab in metastatic castration-resistant prostate cancer: risk factors and precautions for treating physicians. Clin Genitourin Cancer. 2015;13(4):e305–e309. doi: 10.1016/j.clgc.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cicci JD, Buie L, Bates J, van Deventer H. Denosumab for the management of hypercalcemia of malignancy in patients with multiple myeloma and renal dysfunction. Clin Lymphoma Myeloma Leuk. 2014;14(6):e207–e211. doi: 10.1016/j.clml.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 69.Messa P, Alfieri C, Brezzi B. Clinical utilization of cinacalcet in hypercalcemic conditions. Expert Opin Drug Metab Toxicol. 2011;7(4):517–528. doi: 10.1517/17425255.2011.562196. [DOI] [PubMed] [Google Scholar]
- 70.Brown EM, Lian JB. New insights in bone biology: unmasking skeletal effects of the extracellular calcium-sensing receptor. Sci Signal. 2008;1(35):pe40. doi: 10.1126/scisignal.135pe40. [DOI] [PubMed] [Google Scholar]
- 71.Silverberg SJ, Rubin MR, Faiman C, et al. Cinacalcet hydrochloride reduces the serum calcium concentration in inoperable parathyroid carcinoma. J Clin Endocrinol Metab. 2007;92(10):3803–3808. doi: 10.1210/jc.2007-0585. [DOI] [PubMed] [Google Scholar]
- 72.Campbell SR, Flombaum CD, Glezerman IG. Use of cinacalcet for treatment of hypercalcemia of malignancy refractory to conventional therapies [Abstract] J Am Soc Nephrol. 2013;24:614A. [Google Scholar]
- 73.Ogata E. Parathyroid hormone-related protein as a potential target of therapy for cancer-associated morbidity. Cancer. 2000;88(12 Suppl):2909–2911. doi: 10.1002/1097-0142(20000615)88:12+<2909::aid-cncr5>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 74.Ralston SH, Gallacher SJ, Patel U, Campbell J, Boyle IT. Cancer-associated hypercalcemia: morbidity and mortality. Clinical experience in 126 treated patients. Ann Intern Med. 1990;112(7):499–504. doi: 10.7326/0003-4819-112-7-499. [DOI] [PubMed] [Google Scholar]
- 75.Mundy GR, Martin TJ. The hypercalcemia of malignancy: pathogenesis and management. Metabolism. 1982;31(12):1247–1277. doi: 10.1016/0026-0495(82)90012-9. [DOI] [PubMed] [Google Scholar]
- 76.Fisken RA, Heath DA, Bold AM. Hypercalcaemia – a hospital survey. Q J Med. 1980;49(196):405–418. [PubMed] [Google Scholar]
- 77.Blomqvist CP. Malignant hypercalcemia – a hospital survey. Acta Med Scand. 1986;220(5):455–463. doi: 10.1111/j.0954-6820.1986.tb02795.x. [DOI] [PubMed] [Google Scholar]
- 78.Donovan PJ, Achong N, Griffin K, Galligan J, Pretorius CJ, McLeod DS. PTHrP-mediated hypercalcemia: causes and survival in 138 patients. J Clin Endocrinol Metab. 2015;100(5):2024–2029. doi: 10.1210/jc.2014-4250. [DOI] [PubMed] [Google Scholar]