

Abstract
We developed a quantum-dot-based fluorescence resonance energy transfer (QD-FRET) nanosensor to visualize the activity of matrix metalloproteinase (MT1-MMP) at cell membrane. A bended peptide with multiple motifs was engineered to position the FRET pair at a close proximity to allow energy transfer, which can be cleaved by active MT1-MMP to result in FRET changes and the exposure of cell penetrating sequence. Via FRET and penetrated QD signals, the nanosensor can profile cancer cells.
Keywords: activatable cell-penetrating peptide, multiplex signals, FRET nanosensor, MT1-MMP, single cell, cancer
Matrix metalloproteinases (MMPs) are promising bio-markers of tumorigenesis that play important roles in extracellular matrix (ECM) remodeling, cancer cell migration, and angiogenesis.1,2 Among the many classes of MMPs, the membrane-anchored MT1-MMP plays a dominant role in regulating cancer and stromal cell trafficking through the ECM of host tissues.3–6 MT1-MMP is the only known MMP capable of degrading and disrupting structural barriers of covalently linked networks composed of type I collagen within the ECM, which is critical for cancer cell invasion in 3D matrices.6,7 Selective inhibition of MT1-MMP blocks tumor growth, invasion and angiogenesis in vivo.8 Therefore, it would be of great utility for cancer research and therapeutic applications to develop a highly efficient and sensitive MT1-MMP biosensor.
In previous publications we developed several FRET biosensors for visualizing MT1-MMP activity at cell surfaces.9,10
The most efficient versions contained an enhanced cyan fluorescent protein (ECFP) as a FRET donor and a yellow fluorescent protein (YPet) as a FRET acceptor, tethered together through a cleavable peptide substrate (AHLR) flanked by flexible linkers.9,10 This AHLR-based biosensor was shown to be highly sensitive and specific for proteolytic cleavage by MT1-MMP, with moderate cleavage by invasion-promoting MMP2 and MMP9, but not protective MMP8,9–12 attributes appropriate for cancer detection. However, as a genetically encoded biosensor, its delivery into the cells remains low in efficiency.13 In this report, we demonstrate the development of a new class of FRET nanosensors based on quantum dots and Cy3 dyes for measurement and imaging of MT1-MMP activity in cancer cells without the need for transfection.
Quantum dots (QDs) are fluorescent nanocrystals with superb optical properties for imaging, including a tunable and narrow emission spectrum,14,15 exceptional brightness,15,16 and high photostability.17,18 These nanoparticles have proved to be versatile probes for applications spanning in vivo imaging,19–21 DNA hybridization,22,23 and drug delivery24,25 and have recently been engineered for improved biocompatibility in living cells and animals.26,27 QDs have also been conjugated with a specific peptide and employed as FRET donors for in vitro sensing of calcium and caspase activity.28,29 However, the application of QD-peptide and FRET donor in single-cell imaging has yet to be extensively explored.
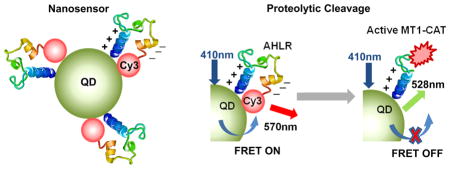
Here, we demonstrate the development of a new general-izable architecture for multifunctional QD-FRET protease nanosensors. FRET is enabled by the usage of a bent peptide sequence stabilized by electrostatic attraction between a sequence of nine cationic arginines (9× arginine) and a sequence of eight anionic glutamates (8× glutamate). When attached to the same backbone, these domains electrostatically bind together to yield a hairpin loop with a relatively weak affinity (Kd = ~6 μM, Figure 1). Thus, we conjugated these bent peptide sequences to a FRET pair, with the N-terminus coupled to a QD FRET donor (QD525) via a hexahistidine sequence and the C-terminus coupled to an acceptor (Cy3) via maleimide–cysteine reactions (Figure 1). The usage of this bent structure to bring close the donor and acceptor at the rest state and enhance FRET efficiency is different from several earlier work of Rao and Medintz’s groups.26,29–31 Then, this peptide was also engineered to contain a cleavage site specific for MT1-MMP to allow monitoring of MT1-MMP activity in live cells via changes in FRET signals.9,10 Upon cleavage by the protease, the weakly associated domains dissociate to separate the donor from acceptor and reduce FRET.9,10
Figure 1.

Schematic illustrations of the design and activation mechanism of the QD-FRET nanosensor. (a) Designed sequence composition of a multifunctional Cy3-peptide. (b) Nanosensor contains a QD coupled to multiple Cy3-peptides bent to a position which allows a high FRET between the QD and Cy3. (c) Activation mechanism of the FRET nanosensor upon the cleavage by MT1-CAT. (d) When the sensors bind to cancer cells with active MT1-MMP, they can be cleaved at the substrate (AHLR), with decreasing FRET and increasing QD emission. Subsequently, cell penetration is triggered by the remaining positively charged peptide sequence. In addition, the nanosensors associate with the surfaces of nonmalignant cells, but they are not efficiently cleaved or and do not efficiently enter the cell.
Results and Discussion
Design of the QD-FRET MT1-MMP Nanosensor
The engineered QD-FRET MT1-MMP nanosensor is composed of a CdSe/ZnS QD that functions as a FRET donor and multiple Cy3-peptides that function as FRET acceptors (Figure 1). The QDs have a metal-rich surface, allowing spontaneous association with hexa-histidine peptides via oriented self-assembly.32 Each Cy3-peptide consists of a QD binding domain (6× histidine), a positive-charged 9× arginine sequence,33 a 3× RGD (Arg–Gly–Asp) sequence for cell-targeting, the MT1-MMP cleavable sequence AHLR, a negative-charged 8× glutamate sequence, and a Cy3 dye as the FRET acceptor (Figure 1a). The arginine and glutamate sequences are both flanked by flexible linker sequences GGSGGT.10 By this design, the electrostatic interaction between arginine and glutamate bends the peptide-Cy3 module in a hairpin-liked shape, allowing FRET between QD and Cy3 when the peptide-Cy3 module is attached to the QD surface (Figure 1b). The substrate sequence in the nanosensor can be cleaved in vitro by the active catalytic domain of MT1-MMP (MT1-CAT),2,34 thus separating Cy3 from the QD and disrupting FRET (Figure 1c). This decrease of energy transfer between the QD and Cy3 causes an increase in QD emission and decrease in FRET emission (Cy3 emission with QD excitation). As a result, the emission ratio of QD/FRET increases, which can be used to represent the level of MT1-MMP proteolytic activity (Figure 1d). After incubation with cells expressing integrin surface receptors, the QD-FRET nanosensors can be concentrated to the extracellular surface by the binding of RGD ligand sequences to integrins (Figure 1c and d).35,36 For cancer cells with high MT1-MMP activity, the nanosensor will be cleaved at the specific substrate sequence (AHLR) so that the negatively charged Cy3 component can diffuse away from the cell membrane. This exposes the positively charged 9× arginine sequence that also serves as a cell-penetrating peptide to allow entry of the nanosensors into the cell (Figure 1d).33,37 As a result, cells with high MT1-MMP activity are expected to contain internalized nanosensors with high QD/FRET emission ratios, whereas cells with low MT1-MMP activity will exhibit lower QD/FRET emission ratios at the cell membrane (Figure 1d).
The absorption spectrum of Cy3 significantly overlaps with the emission of a 525 nm emitting QD, with the emission peaks of QD and Cy3 well separated (by 45 nm), allowing FRET to occur with the QD serving as a donor and Cy3 an acceptor (Figure 2a). Indeed, our results show that after self-assembly of the QD and Cy3-peptides, the QD emission peak dropped and the Cy3 emission peak at 570 nm increased due to FRET (Figure 2b). The relatively low peak value at 570 nm indicates that Cy3-peptides can also quench the QD while serving as a FRET acceptor (Figure 2b). The FRET pair formation was further confirmed by adding imidazole as a binding competitor to separate the histidine-containing Cy3-peptide from the QD, resulting in rapid recovery of QD emission (Supporting Information Figure S1).
Figure 2.

In vitro calibration of the nanosensor. (a) Normalized absorption and emission spectra of the QD-donor and Cy3-acceptor. (b) Spectra of QD before and after 2 h incubation with the 1 μM Cy3-labeled peptide (QD/peptide = 1:31). The nanosensor was excited at 410 ± 5 nm.
In Vitro MT1-MMP Proteolysis Yields Rapid Nanosensor Response
To quantitatively assess the sensitivity of the QD-FRET nanosensor to MT1-MMP activity, the nanosensors were cleaved in vitro using the active recombinant MT1-CAT. As shown in Figure 3a, when a solution of purified nanosensor (32 nM) was mixed in a 4:1 molar ratio with MT1-CAT (0.7 μM) at 37 °C, an increase of the QD emission accompanied by a drop of FRET emission was observed within minutes. This trend continued up to 120 min after incubation, causing a substantial increase of the QD/FRET emission ratio (Figure 3b). These results confirm that the QD-FRET nanosensors can be efficiently cleaved by MT1-CAT, allowing physical separation between the QD and Cy3-peptide, leading to a decrease of energy transfer and an increase in the QD/FRET emission ratio (Figure 3b).
Figure 3.

QD-FRET nanosensor cleavage assay using multiple concentrations of MT1-CAT in vitro. (a) QD signal recovered dramatically after treatment with 140 nM MT1-CAT for up to 120 min. (b) Time courses of donor (QD)/acceptor (Cy3-FRET) emission ratios (n = 3, error bar: SEM). (c) Representative spectra of nanosensors upon cleavage for 15 min at 37 °C with different concentrations of MT1-CAT. (Inset shows the emission ratios of QD/FRET). (d) Time courses of QD/FRET emission ratios of the nanosensor when incubated with different concentrations of MT1-CAT. Excitation was performed at 410 ± 5 nm and emission was collection at 528 ± 5 nm for the QD and 570 ± 5 nm for the FRET signal.
We further investigated the in vitro sensitivity of the QD-FRET nanosensors to increasing concentrations of MT1-CAT. The resulting QD/FRET ratio was proportional to the concentration of MT1-CAT after 15 min incubations (Figure 3c), demonstrating a quantitative measure of MT1-CAT activity based on ratio signals. The time course of changes in QD/FRET emission ratios with different MT1-CAT concentrations confirmed that this proportionality was robust for extended durations (Figure 3d). Therefore, the signals from our FRET nanosensor can be applied to quantify the level of MT1-MMP activity with high sensitivity.
QD-FRET Nanosensor Can Be Activated by Cancer Cells with High MT1-MMP Activity
To further examine the sensitivity and specificity of the nanosensor for detecting MT1-MMP activity in cells, the nanosensors were applied to assess the invasive human breast cancer cell line, MDA-MB-231 (MDA). MDA cells grown on cover glass were incubated with the nanosensors for 3 h and washed to remove any that were unbound. Fluorescence microscopy images were then acquired and analyzed. In parallel experiments, a potent MMP inhibitor GM6001 (GM), which forms a bidentate complex with zinc at the enzyme active site, was applied to MDA cells to selectively inhibit MT1-MMP activity prior to addition of the nano-sensor.38,39 The MDA cells without GM showed high QD/ FRET ratio and high intracellular QD intensity, whereas the cells with GM showed low QD/FRET ratio and low intracellular QD intensity (Figure 4a). These results indicate that active MT1-MMP at the surface of cancer cells is needed for the activation and internalization of the QD-Cy3 nano-sensors to result in high QD/FRET ratio and high QD intensity.
Figure 4.

Quantification of MT1-MMP QD/FRET ratio and QD intensity in MDA-MB-231 breast cancer cells. (a) Representative images of cells and nanosensors with (left) or without (right) GM pretreatment. QD/FRET ratio images (top panels) were overlaid with cell edges (white lines). QD intensity images (middle panels) were overlaid with detected QD clusters (red lines) and cell edges (white lines). DIC images are shown on the bottom row. (b) Pixel-wise histogram of QD/FRET ratio (top) and QD intensity (bottom) within the detected nanosensor clusters of the cell. (c) Comparison of the QD/FRET ratio, QD intensity, and cell sizes between the cells with (n = 20) or without GM pretreatment (n = 22). * indicates statistically significant differences, p < 0.002. Error bar: SEM (d) Comparison of the QD intensity in cells before and after BCG treatment. The intensity values were normalized to those in the same cell before BCG treatment, so that the QD intensity before BCG treatment is 1.
To examine the distribution of nanosensor signals within a single cell, we quantified the distribution of QD intensities and QD/FRET ratios at the level of individual pixels (Figure 4b). In normal MDA cells there was a large population of nanosensor signals with both high QD/FRET ratio and high QD intensity values compared with GM-inhibited cells. This result demonstrates that the nanosensors can be cleaved to show FRET changes. A fraction of pixels in the normal MDA cells were similar to those of GM-inhibited MDA cells in both the QD/FRET ratio and QD intensity, possibly due to nonspecific endocytosis of a small number of intact nanosensors.35
Because the nanosensors predominantly cluster after cellular uptake, possibly through self-aggregation during endocyto-sis,24,40 we quantified the QD/FRET ratio by accumulating all ratio values within the selected region based on an intensity threshold and then dividing by the area of the region to obtain the average QD/FRET ratio of each single cell. The intensity threshold was chosen below the QD-intensity of the nano-sensor-covered regions but slightly above the autofluorescence background based on the histogram distribution. In addition, the normalized QD intensity was obtained by averaging the QD emission intensity above the threshold and normalizing by the cell size to account for size variances. The nanosensors in normal MDA cells showed significantly higher average QD/ FRET ratio and normalized QD intensity than the GM-inhibited cells (Figure 4c). To gain additional insight into the function and internalization of the nanosensor, we also used a GM-washout assay. The GM-inhibited live MDA cells were incubated with the nanosensors for 1 h to allow association with the cell membrane, but little internalization. The QD/ FRET ratio of the nanosensors increased in 5 min after removing GM inhibition to activate cellular MT1-MMP (Supporting Information Figure S2). In contrast, after more than 6 h of incubation, some nanosensors can enter the cell nonspecifically, albeit were no longer cleavable upon GM washout (Supporting Information Figure S2b). Furthermore, we treated the cells with an efficient, membrane-impermeable quencher of QDs, bromocresol green (BCG), which did not cause a statistically significant reduction of the QD intensity Figure 4d). This result indicates that majority of the observed nanosensors were inside the cells and cannot be efficiently quenched by BCG.41 Taken together, the above results support our proposed mechanism that active MT1-MMP at the surface of MDA cells specifically cleaves the QD-Cy3 nanosensor, induces cell penetration, and allows a large population of nanosensors with high QD/FRET ratio to enter the cells, likely via endocytosis.33
Control experiments using nanosensors without the RGD motif confirmed that the highly intracellular QD intensity can only be obtained in cells incubated with sensors harboring RGD motif (Supporting Information Figure S3). Indeed, QDs without the RGD-containing nanosensor coating did not effectively stain the cells and showed no intensity (Supporting Information Figure S3a). To examine the possibility that the MT1-MMP substrate AHLR sequence in the nanosensor can interact with the cell surface and allow nanosensor attachment, we incubated the cells with sensors containing the motifs of ECFP-AHLR-YPet FRET biosensor with or without the RGD motif. The results confirmed that the AHLR sequence was not sufficient to mediate biosensor attachment and only the RGD containing biosensor stained cells with high intensity (Supporting Information Figure S3b). Therefore, these results indicate that the RGD motif in the nanosensor is crucial for the nanosensor-cell coupling, which are consistent with the previously reported RGD-integrin interaction at cellular surface.42,43
Classification of Cell Lines through a Dual-Index Readout of QD-FRET Nanosensors
The FRET nanosensor provides two indices, the QD/FRET ratio for MT1-MMP activity and the QD intensity for cellular penetration of the nanosensor. Using these two metrics, we evaluated the ability of the nanosensors to distinguish cell lines with different invasion potentials (Figure 5). The results show that MDA and HT1080 cells with substantial MT1-MMP expression had significantly higher QD/FRET ratios than the MT1-MMP deficient HeLa cells (Figure 5b),42,43 suggesting that the nanosensor responses can reflect the active MT1-MMP levels among different cell types. By this metric, MDA and HT1080 cells are not distinguishable; however, the MDA cells exhibited significantly more accumulation of QD intensity than HeLa and HT1080 cells, possibly reflecting distinct adhesion processes regulating endocytosis in MDA and HT1080 cells after nanosensor cleavage on the cell surface.44 As a result, the MDA and HT1080 cell populations become distinguishable when the quantified QD/FRET ratio was plotted vs normalized QD intensity for single cells (Figure 5c).
Figure 5.

Different cancer cell lines can be classified by the nanosensor QD/FRET ratio and QD intensity. (a) Representative QD/FRET ratio images of MDA-MB-231 (left), HeLa (middle), and HT1080 (right) cell lines. (b) Comparison of the QD/FRET ratio and QD intensity among MDA-MB-231 (n = 22), HeLa (n = 23), and HT1080 (n = 23). ** indicates statistically significant differences from the other two groups, p < 0.001. Error bar: SEM (c) 2D plots with MDA-MB-231 (blue), HeLa (red), and HT1080 (green) cell lines with the x axis depicting the normalized QD intensity and the y axis depicting the QD/FRET ratio.
We further characterized the capacity of our nanosensors to distinguish cells with specific cleavage and adhesion/uptake potentials. The nanosensors were incubated with three different cell lines: MCF7 cells lacking endogenous MT1-MMP and integrin β3 subunits, MCF7 cells overexpressing exogenous MT1-MMP (MCF-MT1), or MCF7 cells expressing both exogenous integrin β3 subunit and MT1-MMP (MCF-β3-MT1). MCF-MT1 cells are expected to exhibit enhanced cleavage the nanosensors due to higher MT1-MMP activity. Consistent with this prediction, the nanosensors had significantly higher QD/FRET ratios after incubating with the MCF-MT1 cells compared with the original MCF cells (Figure 6a), although the QD intensities were not significantly different (Figure 6b). These results indicate that exogenous MT1-MMP in MCF7 cells can enhance the ability of the cells to cleave the nanosensors but with only minor effect on the cell penetration. Since MCF7 cells express endogenous integrin αv subunits, the integrin receptors in MCF-β3-MT1 cells are expected to form αvβ3 dimers that may facilitate the adhesion of the nanosensors to the extracellular surface as well as enhance penetration of the nanosensors.45 Indeed, coexpressing integrin β3 in the MCF-MT1 cells significantly enhanced the ability of the cells to engulf the nanosensors, and slightly decreased the nanosensor cleavage (Figures 6a–b), possibly reflecting a shortened duration of MT1-MMP and nanosensor interaction for cleavage before endocytosis. These results indicate that our nanosensor can provide a multiplex measure of individual cancer cell potential toward tissue degradation, migration, and invasion.
Figure 6.

Multiplex nanosensor readout of QD/FRET ratio and QD intensity depends on MT1-MMP and integrin expression. Comparison of (a) the QD/FRET ratio and (b) QD intensity among the cell lines MCF7 (MCF, blue, n = 29), MCF7 expressing MT1-MMP only (MCF-MT1, red, n = 25), and MCF7 expressing both integrin β3 subunit and MT1-MMP (MCF-β3-MT1, green, n = 35). * indicates statistically significant difference (p < 0.05) by the Bonferroni multiple comparison test.
There is a possibility of cross-talk between the adhesion and cleavage signals when the functional receptors are excessively expressed. We reasoned that the excessive expression of integrin in MCF-β3-MT1 may have enhanced the nanosensor adhesion and endocytosis (Figure 6b), which shortened the nanosensor interaction with MT1-MMP on the plasma membrane. This may negate the effect of a higher level of MT1-MMP in MCF-β3-MT1 cells, which led to a similar level of QD/FRET ratio in MCF-β3-MT1 and original MCF7 cells (Figure 6a). It is, however, clear that the two indices of QD intensity and QD/FRET ratio related to adhesion and cleavage, respectively, are quite separable in different cell types when only the endogenous receptors and their expression levels are involved (Figure 5b–c). As such, we reasoned that the relationship between the adhesion and cleavage signals is complex and nonlinear and, hence, requires a multiplex readout for assessing the invasive potential of cancer cells.
Conclusion
In this study, we have demonstrated that our QD-FRET nanosensor with an engineered bent peptide can serve as a potent and multiplex analysis tool to visualize MT1-MMP activity and integrin adhesion in single cells. Our in vitro studies showed that this sensor can be precisely cleaved by catalytically active MT1-CAT domain in a concentration-dependent manner. Single cell imaging further revealed that the nanosensor not only detects cancer cells expressing MT1-MMP through peptide cleavage but also measures integrin receptor expression through RGD-binding. This multiplex detection capability of the nanosensor should allow the profiling of different cancer cells in their potentials for tissue degradation, migration, and invasion.
Experimental Details
Peptide Construction and Dye Conjugation
The FRET nanosensor peptide was constructed by fusing several functional motifs in a specific order. The quantum dots binding site (6× histidine) was coded by pRsetB vector at the N-terminus of the linker peptide; the cell-penetrating peptide (9× arginine) and its blocking peptide (8× glutamate) were derived from reported cell-penetrating peptides;33 the cell-targeting peptide (3× RGD motifs) was derived from α5β1 integrin binding motif of fibronectin; the MT1-MMP cleavable peptide CRPAHLRDSG was reported in a previous publication9 with a C-terminal cysteine for Cy3 labeling. These motifs were flanked by GGSGGT linker peptides. The oligonucleotide constructs were synthesized and inserted into a pRsetB vector using the BamH I/BglII restriction site with a stop codon. The construct was confirmed by sequencing. The synthesized plasmid was expressed in BL21 competent E. coli for protein production and then purification by nickel chelation chromatography extraction described in our previous publication.46
For Cy3 conjugation, the purified protein was dissolved at 1 mg/mL in degassed PBS buffer and incubated at room temperature for 30 min. Then, 100 molar excess of Tris(2-carboxethyl) phosphine (TCEP) was added and incubated with nitrogen gas at room temperature for 10 min. After that, 50 μL of anhydrous dimethylformamide was added to one pack of Cy3-maleimide (GE Healthcare) and added to the TCEP-reduced protein. The final solution was further incubated with in nitrogen at room temperature for 2 h with additional mixing every 30 min, then left for reaction overnight (~10 h) at 2–8 °C. The conjugated peptide was purified using Ni-NTA beads following standard protein purification procedures. The final concentration of conjugated Cy3-peptide construct was determined with an UV–visible spectrophotometer (Fisher Scientific) using a Cy3 extinction coefficient of 150 000 M−1 cm−1 at 550 nm.
QD Nanosensor Assembly and Biocompatibility
The QD-FRET nanosensor was fabricated by incubating 32 nM of 525 nm emitting ITK carboxyl quantum dots (Invitrogen) with 1 μM Cy3 labeled linker peptide (amount of peptide-Cy3 is chosen to saturate QDs and was determined experimentally as shown in Supporting Information Figure S4) for 2 h at 4 °C. The mixture was diluted 10 times with a proteolysis assay buffer (50 mM HEPES, 10 mM CaCl2, 0.5 mM MgCl2, 50 μM ZnCl2, and 0.01% Brij-35, pH 6.8), the excess dye-labeled peptide was removed with a centrifugal 50 kDa MW cutoff filter (Millipore) at 4000 rpm for 5 min, and then restored to the original concentration with the proteolysis assay buffer.
The QD nanosensors were composed of core/shell CdSe/ ZnS nanocrystals coated with amphiphilic polymers that have previously been used extensively due to high stability of fluorescence.24 Under certain conditions, cadmium-based QDs can leach toxic divalent cadmium ions;24 however, these QDs were coated with a cadmium-free inorganic shell and an amphiphilic polymer that has been demonstrated to allow robust protection from aqueous etchants and to provide long-term stability without nanocrystal degradation,47 which is verified by the lack of a spectral shift from the QD peak during the course of the experiments. During the short duration of these experiments (3 h QD exposure), no significant degradation or acute toxicity is expected to occur.47
MT1-CAT Enzyme Cleavage Assays
The MT1-MMP catalytic domain (MT1-CAT) was expressed and purified as described34,48 and stored at −80 °C prior to use. The cleavage assays were carried out in 96-well polystyrene plate (Greiner). MT1-CAT with different concentrations was added into the freshly prepared nanosensors in the proteolysis assay buffer. The reactions were carried out for 2 h at 37 °C in a fluorescence plate reader (TECAN Infinite M1000 Pro).
Intracellular Imaging
All cells were cultured following ATCC instructions. The glass bottom dishes (Cell E and G) for cell imaging were coated with 20 μM fibronectin solution at 37 °C for 5 h before cells were seeded. Cells were transferred to the coated class bottom dish one night before experiments with culture media containing 1% BSA. The medium was then replaced with a freshly prepared FRET nanosensor solution mixed 1:1 with DMEM culture medium (with 1% BSA). The dishes were further incubated in a 37 °C incubator for 3 h with additional mixing every 30 min to avoid nanosensor aggregation. After incubation, cells were quickly washed with PBS three times to remove QD nanosensors from the medium, then fixed with 4% paraformaldehyde solution for 10 min. The cells were then washed with PBS and covered with cold PBS before imaging. The selected cells had similar sizes. Bromocresol green (BCG, Sigma-Aldrich) is a membrane-impermeable dye that efficiently quenches QDs, so we used it to distinguish internal QDs from external QDs.41 Briefly, after the cells were washed to remove QD nanosensors from the medium, a high concentration of BCG (200 μM) was added so that the QDs outside the cells were quenched without affecting the intracellular QDs. The cells were imaged before and after the addition of BCG to compare the QD intensity (Figure 4d).
Quantitative Analysis and Statistics
Quantitative cell images were obtained with a Nikon Eclipse Ti inverted microscope with a cooled charge-coupled device camera using MetaFluor 6.2 software (Universal Imaging). The emission ratio and intensity values were quantified by MATLAB (MathWorks) with our Fluocell package49 and Excel (Micro-soft). The cell boundaries were drawn manually to identify QD nanosensors located inside the cell bodies. The average QD intensity ±3 times STD was set to identify statistical outliers and remove aberrant cells. For statistical analysis, two-tailed Student’s t test with unequal variance was used except for Figure 6. In Figure 6, we used the Bonferroni multiple comparison test of means at 95% confidence interval, which is provided by the multcompare function in the MATLAB statistics toolbox.50
Supplementary Material
Acknowledgments
This work is supported by grants from NIH HL098472, HL109142, HL121365 (Yingxiao W.), NSF CBET1360341, DMS1361421 (Yingxiao W. and S. L.), UC San Diego, Beckman Laser Institute, Inc. (Yingxiao W.), and NIH R00CA153914 (A.M.S.). This research was also supported by NSF Singapore through the Singapore MIT Alliance for Research and Technology’s BioSystems and Micromechanics Inter-Disciplinary Research programme, and the National Cancer Institute 1R33CA174550 (R.K. and C.O.). The funding agencies had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Author Contributions
S.L., Y.Q., and Y.W. designed research; E.Y.C., C.J.O, Y.W., and L.L. performed research; E.Y.C., S.L., and Y.W. analyzed data; E.Y.C., A.M.S., A.Y.S., R.K., S.L., Y.Q., and Y.W. wrote the paper.
Notes
The authors declare the following competing financial interest(s): Y.W. and S.L. own equity in Cell E&G LLC..
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nano-lett.5b01047.
Supplementary figures and supplementary figure legends (PDF)
References
- 1.Kessenbrock K, Plaks V, Werb Z. Cell. 2010;141:52. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Strongin AY. Biochim Biophys Acta, Mol Cell Res. 2010;1803:133. doi: 10.1016/j.bbamcr.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wolf K, Friedl P. Clin Exp Metastasis. 2009;26:289. doi: 10.1007/s10585-008-9190-2. [DOI] [PubMed] [Google Scholar]
- 4.Itoh Y, Seiki M. J Cell Physiol. 2006;206:1. doi: 10.1002/jcp.20431. [DOI] [PubMed] [Google Scholar]
- 5.Li XY, Ota I, Yana I, Sabeh F, Weiss SJ. Mol Biol Cell. 2008;19:3221. doi: 10.1091/mbc.E08-01-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sabeh F, Shimizu-Hirota R, Weiss SJ. J Cell Biol. 2009;185:11. doi: 10.1083/jcb.200807195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rowe RG, Li XY, Hu Y, Saunders TL, Virtanen I, Garcia de Herreros A, Becker KF, Ingvarsen S, Engelholm LH, Bommer GT, Fearon ER, Weiss SJ. J Cell Biol. 2009;184:399. doi: 10.1083/jcb.200810113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Devy L, Huang L, Naa L, Yanamandra N, Pieters H, Frans N, Chang E, Tao Q, Vanhove M, Lejeune A, van Gool R, Sexton DJ, Kuang G, Rank D, Hogan S, Pazmany C, Ma YL, Schoonbroodt S, Nixon AE, Ladner RC, Hoet R, Henderikx P, TenHoor C, Rabbani SA, Valentino ML, Wood CR, Dransfield DT. Cancer Res. 2009;69:1517. doi: 10.1158/0008-5472.CAN-08-3255. [DOI] [PubMed] [Google Scholar]
- 9.Ouyang M, Huang H, Shaner NC, Remacle AG, Shiryaev SA, Strongin AY, Tsien RY, Wang Y. Cancer Res. 2010;70:2204. doi: 10.1158/0008-5472.CAN-09-3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ouyang M, Lu S, Kim T, Chen CE, Seong J, Leckband DE, Wang F, Reynolds AB, Schwartz MA, Wang Y. Nat Commun. 2013;4:1589. doi: 10.1038/ncomms2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin M, Matrisian L. Cancer Metastasis Rev. 2007;26:717. doi: 10.1007/s10555-007-9089-4. [DOI] [PubMed] [Google Scholar]
- 12.Balbin M, Fueyo A, Tester AM, Pendas AM, Pitiot AS, Astudillo A, Overall CM, Shapiro SD, Lopez-Otin C. Nat Genet. 2003;35:252. doi: 10.1038/ng1249. [DOI] [PubMed] [Google Scholar]
- 13.Felgner PL, Gadek TR, Holm M, Roman R, Chan HW, Wenz M, Northrop JP, Ringold GM, Danielsen M. Proc Natl Acad Sci U S A. 1987;84:7413. doi: 10.1073/pnas.84.21.7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Michalet X, Pinaud FF, Bentolila LA, Tsay JM, Doose S, Li JJ, Sundaresan G, Wu AM, Gambhir SS, Weiss S. Science. 2005;307:538–544. doi: 10.1126/science.1104274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alivisatos AP, Gu W, Larabell C. Annu Rev Biomed Eng. 2005;7:55. doi: 10.1146/annurev.bioeng.7.060804.100432. [DOI] [PubMed] [Google Scholar]
- 16.Resch-Genger U, Grabolle M, Cavaliere-Jaricot S, Nitschke R, Nann T. Nat Methods. 2008;5:763. doi: 10.1038/nmeth.1248. [DOI] [PubMed] [Google Scholar]
- 17.Reiss P, Bleuse J, Pron A. Nano Lett. 2002;2:781. [Google Scholar]
- 18.Sukhanova A, Devy J, Venteo L, Kaplan H, Artemyev M, Oleinikov V, Klinov D, Pluot M, Cohen JH, Nabiev I. Anal Biochem. 2004;324:60. doi: 10.1016/j.ab.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 19.Dubertret B, Skourides P, Norris DJ, Noireaux V, Brivanlou AH, Libchaber A. Science. 2002;298:1759. doi: 10.1126/science.1077194. [DOI] [PubMed] [Google Scholar]
- 20.Ballou B, Lagerholm BC, Ernst LA, Bruchez MP, Waggoner AS. Bioconjugate Chem. 2004;15:79. doi: 10.1021/bc034153y. [DOI] [PubMed] [Google Scholar]
- 21.Akerman ME, Chan WC, Laakkonen P, Bhatia SN, Ruoslahti E. Proc Natl Acad Sci U S A. 2002;99:12617. doi: 10.1073/pnas.152463399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang C-y, Hu J. Anal Chem. 2010;82:1921. doi: 10.1021/ac9026675. [DOI] [PubMed] [Google Scholar]
- 23.Spillmann CM, Ancona MG, Buckhout-White S, Algar WR, Stewart MH, Susumu K, Huston AL, Goldman ER, Medintz IL. ACS Nano. 2013;7:7101. doi: 10.1021/nn402468t. [DOI] [PubMed] [Google Scholar]
- 24.Smith AM, Duan H, Mohs AM, Nie S. Adv Drug Delivery Rev. 2008;60:1226. doi: 10.1016/j.addr.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pawar VK, Singh Y, Meher JG, Gupta S, Chourasia MK. J Controlled Release. 2014;183:51. doi: 10.1016/j.jconrel.2014.03.030. [DOI] [PubMed] [Google Scholar]
- 26.Ma N, Marshall AF, Gambhir SS, Rao J. Small. 2010;6:1520. doi: 10.1002/smll.200902409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schipper ML, Iyer G, Koh AL, Cheng Z, Ebenstein Y, Aharoni A, Keren S, Bentolila LA, Li J, Rao J, Chen X, Banin U, Wu AM, Sinclair R, Weiss S, Gambhir SS. Small. 2009;5:126. doi: 10.1002/smll.200800003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Medintz IL, Clapp AR, Brunel FM, Tiefenbrunn T, Uyeda HT, Chang EL, Deschamps JR, Dawson PE, Mattoussi H. Nat Mater. 2006;5:581. doi: 10.1038/nmat1676. [DOI] [PubMed] [Google Scholar]
- 29.Prasuhn DE, Feltz A, Blanco-Canosa JB, Susumu K, Stewart MH, Mei BC, Yakovlev AV, Loukov C, Mallet JM, Oheim M, Dawson PE, Medintz IL. ACS Nano. 2010;4:5487. doi: 10.1021/nn1016132. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, So MK, Rao J. Nano Lett. 2006;6:1988. doi: 10.1021/nl0611586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Algar WR, Ancona MG, Malanoski AP, Susumu K, Medintz IL. ACS Nano. 2012:11044. doi: 10.1021/nn304736j. [DOI] [PubMed] [Google Scholar]
- 32.Sapsford KE, Pons T, Medintz IL, Higashiya S, Brunel FM, Dawson PE, Mattoussi H. J Phys Chem C. 2007;111:11528. [Google Scholar]
- 33.Jiang T, Olson ES, Nguyen QT, Roy M, Jennings PA, Tsien RY. Proc Natl Acad Sci U S A. 2004;101:17867. doi: 10.1073/pnas.0408191101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Golubkov VS, Chekanov AV, Shiryaev SA, Aleshin AE, Ratnikov BI, Gawlik K, Radichev I, Motamedchaboki K, Smith JW, Strongin AY. J Biol Chem. 2007;282:36283. doi: 10.1074/jbc.M706290200. [DOI] [PubMed] [Google Scholar]
- 35.Xiong JP, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman SL, Arnaout MA. Science. 2002;296:151. doi: 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- 36.Ruoslahti E. Annu Rev Cell Dev Biol. 1996;12:697. doi: 10.1146/annurev.cellbio.12.1.697. [DOI] [PubMed] [Google Scholar]
- 37.Olson ES, Jiang T, Aguilera TA, Nguyen QT, Ellies LG, Scadeng M, Tsien RY. Proc Natl Acad Sci U S A. 2010;107:4311. doi: 10.1073/pnas.0910283107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grobelny D, Poncz L, Galardy RE. Biochemistry. 1992;31:7152. doi: 10.1021/bi00146a017. [DOI] [PubMed] [Google Scholar]
- 39.Yamamoto M, Tsujishita H, Hori N, Ohishi Y, Inoue S, Ikeda S, Okada Y. J Med Chem. 1998;41:1209. doi: 10.1021/jm970404a. [DOI] [PubMed] [Google Scholar]
- 40.Derfus AM, Chan WCW, Bhatia SN. Adv Mater. 2004;16:961. [Google Scholar]
- 41.Valentine CD, Verkman AS, Haggie PM. Traffic. 2012;13:25. doi: 10.1111/j.1600-0854.2011.01287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhai Y, Hotary KB, Nan B, Bosch FX, Muñoz N, Weiss SJ, Cho KR. Cancer Res. 2005;65:6543. doi: 10.1158/0008-5472.CAN-05-0231. [DOI] [PubMed] [Google Scholar]
- 43.Brekhman V, Neufeld G. BMC Cancer. 2009;9:415. doi: 10.1186/1471-2407-9-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cho JA, Osenkowski P, Zhao H, Kim S, Toth M, Cole K, Aboukameel A, Saliganan A, Schuger L, Bonfil RD, Fridman R. J Biol Chem. 2008;283:17391. doi: 10.1074/jbc.M708943200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deryugina EI, Ratnikov B, Monosov E, Postnova TI, DiScipio R, Smith JW, Strongin AY. Exp Cell Res. 2001;263:209. doi: 10.1006/excr.2000.5118. [DOI] [PubMed] [Google Scholar]
- 46.Wang Y, Botvinick EL, Zhao Y, Berns MW, Usami S, Tsien RY, Chien S. Nature. 2005;434:1040. doi: 10.1038/nature03469. [DOI] [PubMed] [Google Scholar]
- 47.Smith AM, Duan H, Rhyner MN, Ruan G, Nie S. Phys Chem Chem Phys. 2006;8:3895. doi: 10.1039/b606572b. [DOI] [PubMed] [Google Scholar]
- 48.Kridel SJ, Sawai H, Ratnikov BI, Chen EI, Li W, Godzik A, Strongin AY, Smith JW. J Biol Chem. 2002;277:23788. doi: 10.1074/jbc.M111574200. [DOI] [PubMed] [Google Scholar]
- 49.Lu S, Kim TJ, Chen CE, Ouyang M, Seong J, Liao X, Wang Y. PLoS One. 2011;6:e21293. doi: 10.1371/journal.pone.0021293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu S, Ouyang M, Seong J, Zhang J, Chien S, Wang Y. PLoS Comput Biol. 2008;4:e1000127. doi: 10.1371/journal.pcbi.1000127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.