

Abstract
Recent research on disparate psychiatric disorders has implicated rare variants in genes involved in global gene regulation and chromatin modification, as well as many common variants located primarily in regulatory regions of the genome. Understanding precisely how these variants contribute to disease will require a deeper appreciation for the mechanisms of gene regulation in the developing and adult human brain. The PsychENCODE project aims to produce a public resource of multidimensional genomic data using tissue- and cell type–specific samples from approximately 1,000 phenotypically well-characterized, high-quality healthy and disease-affected human post-mortem brains, as well as functionally characterize disease-associated regulatory elements and variants in model systems. We are beginning with a focus on autism spectrum disorder, bipolar disorder and schizophrenia, and expect that this knowledge will apply to a wide variety of psychiatric disorders. This paper outlines the motivation and design of PsychENCODE.
Noncoding DNA sequences are believed to comprise over 90% of the human genome1,2 and their roles in the spatial, temporal and quantitative regulation of gene expression in different organisms and tissues have been well recognized for decades3–5. However, a comprehensive catalog of noncoding elements is lacking, and the full extent of contributions of these elements to various biological functions has remained unclear and under-investigated. Recent technological and analytical advances have allowed many large-scale studies, including the Encyclopedia of DNA Elements (ENCODE) Consortium6 and the Roadmap Epigenomics Mapping Consortium7 (REMC), to begin systematic characterization of genomic elements of the human genome and of genome-wide regulatory relationships. These studies substantiated previous findings that many human non-protein-coding sequences may be actively transcribed into noncoding RNAs or serve as functional cis-regulatory elements such as promoters, enhancers and insulators, often in a cell- and tissue-specific way. The functional relevance of noncoding sequences is also supported by linkage studies, genome-wide association studies (GWAS) and targeted and whole-genome sequencing studies, which have repeatedly shown that common genetic variations associated with human diseases cluster in putative regulatory regions4,8. Moreover, several studies, including the Genotype-Tissue Expression (GTEx) Consortium9, have shown that noncoding genetic variation is associated with expression differences across various human tissues. Bridging efforts across various studies using a well-phenotyped, disease-relevant population is therefore needed to provide a mechanistic link between disease-associated genetic variants and disease phenotypes.
Psychiatric disorders such as autism spectrum disorder (ASD), bipolar disorder and schizophrenia are often devastating illnesses, with high personal and societal costs and limited treatment options. They show distinct symptomatology, age of onset and progression, and are highly heritable, with a complex, polygenic risk architecture10,11. The multitude of discrete loci involved makes identifying the molecular and cellular mechanisms underlying a disease problematic and hampers development of therapies. Additionally, while the functional implications of rare variations in coding sequences are usually readily apparent, the effects of the more common noncoding variants can be challenging to intuit8. Therefore, to understand disease mechanisms, it is necessary to perform comprehensive analyses of the regulatory regions, epigenetic modifications and gene expression patterns present across different ages, regions and cell types in both the healthy and the disease-affected human CNS. Unfortunately, so far, neither such a comprehensive data set nor any systematic characterization of the functional genomic elements and noncoding RNAs linked to psychiatric disorders is available. The PsychENCODE project was founded to begin to rectify this deficiency and to facilitate research on psychiatric diseases; a list of participating institutions and groups is available at http://www.psychENCODE.org/.
The necessity and difficulties of studying human brain
Vertebrate and invertebrate model systems offer powerful means of dissecting molecular, cellular and circuit functions at a level often not possible in humans and thus provide a necessary adjunct to study of the human brain. However, the human brain is not simply a larger replicate of the CNS of rodents or other commonly studied experimental species and certain aspects of its development and physiology are not well recapitulated in model organisms. The utility of model organisms is also limited by our evolutionary distance from these organisms. While coding sequences are generally conserved across species, noncoding sequences may be subject to less selective pressure and, as such, regulatory elements, transcriptional regulatory networks and gene expression patterns may differ between species12–15. Moreover, null mutations in human and mouse orthologous genes can result in different phenotypes, including the complete absence of a neural phenotype in one of the two species16. Therefore, psychiatric disorders need to be understood and characterized within the context of the human brain.
Unfortunately, the human brain is difficult to study for many reasons. The difficulty of obtaining high-quality post-mortem human brain tissue (healthy or otherwise) and the lack of sufficient sample sizes to overcome experimental and individual variability make the application of genomic technologies particularly challenging. The age of onset and progression of major psychiatric disorders also varies, necessitating the study of the temporal dynamics of human brain development and recognition of the developmental context of psychiatric disorders. This problem is further compounded by the extensive molecular, cellular and temporal differences in regional brain architecture17–20. Even within a functionally distinct region, there is usually remarkable cellular heterogeneity, with a myriad of molecularly, morphologically and functionally distinct cell types. We are only beginning to develop methods to systematically isolate specific cell types from brain tissue; these techniques are labor intensive and low throughput, and, unfortunately, cell cultures do not accurately represent brain tissue complexity. The necessity of using archival healthy and disease-affected post-mortem brains, uncertainties in when new tissue specimens will be available and the circumstances of the tissue donor’s death (which can dramatically affect the stability of nucleic acids and proteins) further compound these problems. These and other challenges have hindered wider usage of post-mortem human brain tissues in basic neuroscience, genomics and psychiatric disorder research. Overcoming these challenges requires integration of expertise across disciplines (for example, biobanking, human developmental neurobiology, molecular biology, genetics, genomics and high-throughput analyses, computational and systems biology, biostatistics, evolutionary biology, mouse genetics and stem cell biology), as well as the development of new approaches to generate and make the multidimensional genomic data from healthy and disease-affected human post-mortem brain tissues more readily available to the wider community.
The goals and organization of the PsychENCODE project
Key goals of the PsychENCODE project are to provide an enhanced framework of regulatory genomic elements (promoters, enhancers, silencers and insulators), catalog epigenetic modifications and quantify coding and non-coding RNA and protein expression in tissue-and cell-type-specific samples from healthy (neurotypical) control and disease-affected post-mortem human brains (Fig. 1). These efforts will be complemented with integrative analyses, as well as with functional characterizations of disease-associated genomic elements using human neural cells derived from induced pluripotent cells (iPSCs) or the developing mouse brain. The PsychENCODE Consortium represents, to our knowledge, the single largest integrated collaborative effort in neuroscience and psychiatry to collectively analyze genomic regulatory elements in a large cohort of well-curated human brains. The consortium brings together experts in psychiatric disorders, neurodevelopment, biobanking, genetics, genomics, data integration and innovative large-scale analyses who will together provide critical insights into the complexity of functional genomic elements. In addition, these experts have consolidated approximately 1,000 well-curated, high-quality postmortem brain specimens, both healthy and disease-affected, spanning multiple developmental periods. This intersection of experiences and resources, including funding from individual investigator grants, will allow the PsychENCODE Consortium to contribute to the greater understanding of psychiatric disorder genomics and mechanisms.
Figure 1.
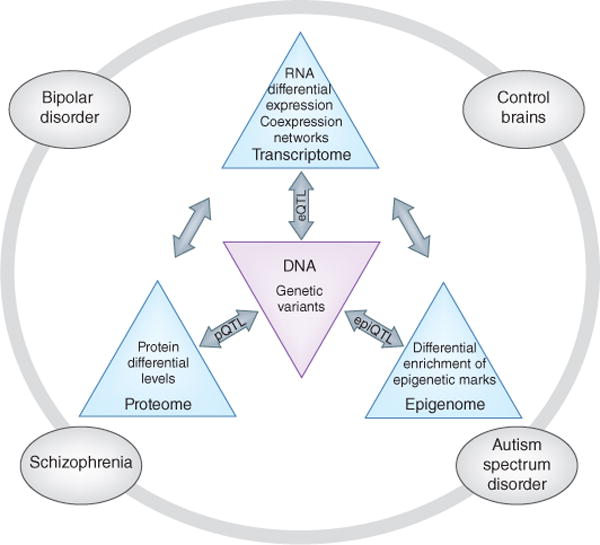
An overview of the PsychENCODE project, showing a schematic of the proposed data types and analyses. The genomic data will be generated from tissues, sorted nuclei and cell culture systems. For each data type, regional, cell-type, developmental, sex-specific and disease-related differences will be identified. Different data modalities will be integrated to identify global mechanisms relevant to disease. Bi-directional flow between genetic information and functional data define eQTLs, epiQTLs and pQTLs, which in turn will assist in prioritizing and refining disease. The specific diseases of interest are shown in outer circle; cross-disorder analysis will also be performed for all data types.
Disease focus areas
The PsychENCODE project will focus on three major psychiatric disorders: ASD, bipolar disorder and schizophrenia. Schizophrenia arises during adolescence and adulthood, and diagnosis is based entirely on clinical symptoms, including delusions, hallucinations, anhedonia, social withdrawal, executive function deficits and working memory impairments21,22. Hallmark symptoms of bipolar disorder, which also arises during adolescence and adulthood, include inappropriately elevated mood, rapid speech and decreased need for sleep. These often alternate with episodes of depression, psychotic symptoms similar to those of schizophrenia, and cognitive problems. More than 100 years of family and twin studies have demonstrated substantial heritability for both schizophrenia and bipolar disorder, with estimates of genetic liability ranging from 64 to 67% for schizophrenia and 59 to 62% for bipolar disorder23,24. GWAS have identified common variants in over 100 loci for schizophrenia and 8 for bipolar disorder11,25,26 that are associated with risk. Rates of rare inherited and de novo copy number variations are elevated in schizophrenia (less so in bipolar disorder), with many conferring high risk27. The two diseases also co-occur in families more frequently than expected, and studies suggest there is more than 50% overlap in genetic susceptibility23.
ASD arises during early childhood and is characterized by persistent impairments in reciprocal social communication, accompanied by restrictive, repetitive behaviors, interests or activities28,29. ASD is highly heritable, with rare genetic variations including copy number variations, de novo point mutations, and insertions or deletions in protein-coding regions of the genome30,31 contributing to susceptibility. To date, studies of de novo exonic point mutations have highlighted chromatin remodeling, synaptic structure and function, early embryonic genes and targets of the fragile X mental retardation protein, an RNA-binding protein27,30,31. Recent work has provided strong empirical evidence that single nucleotide polymorphisms (SNPs) and other common variations affect population risk32. Several studies have shown a genetic overlap of de novo variants in genes encoding chromatin modeling proteins or glutamatergic post-synaptic proteins between schizophrenia and ASD11,30,33,34, and common SNPs are shared between schizophrenia and bipolar disorder7. Together, these findings suggest both convergent and distinct underlying pathogenic mechanisms in these and possibly other psychiatric disorders, further strengthening the case for increasing the attention paid to such mechanisms and the need for strategic consolidation of resources.
Human brain regions, developmental periods and cell types
Our investigations will prioritize brain regions and cell types that previous research has suggested contribute to ASD, bipolar disorder and schizophrenia. These include multiple regions of the cerebral neocortex (the dorsolateral prefrontal cortex, anterior cingulate cortex and inferior temporal cortex), hippocampus, amygdala, caudate nucleus, nucleus accumbens and cerebellar cortex21,22,28. Most of the post-mortem tissues will be obtained from brain banks or are already part of the consortium investigators’ tissue collections. To address cellular heterogeneity, PsychENCODE in its first phase will primarily focus on neurons versus non-neuronal cells, using fluorescence-activated nuclear sorting (FANS) to collect molecularly identifiable cell populations from archived and prospectively collected post-mortem human brains35. FANS will be conducted using the NeuN antibody, raised against the pan-neuronal nuclear protein RBFOX3. In addition, exploratory assessment of more neural subtypes for which suitable antibodies against nuclear antigens (which are a prerequisite for FANS) are available and emerging single-cell genomic technologies will also be applied to explore cellular heterogeneity.
To address the developmental changes that may influence disease progression, our studies of the aforementioned brain regions and cell types will also be conducted in the developing human brain, including prenatal and early postnatal brain specimens. Additionally, to complement the use of post-mortem human brain samples, we will conduct parallel investigations of neural cells derived from two model in vitro systems, induced pluripotent stem cells (iPSCs) and cultured neuronal cells derived from olfactory neuroepithelium (CNON cells). iPSC-derived neural cells will be used to model human neurodevelopment in a specific genetic background and to investigate regulatory, epigenetic and transcriptional landscapes during cell differentiation. A previous study has shown that human iPSC-derived forebrain neurons cultured as organoids display transcriptome profiles and cellular phenotypes corresponding to those of the early-fetal human forebrain36, facilitating study of otherwise inaccessible stages of prenatal development. Conversely, primary neural progenitor CNON cells37 are an attractive cell culture system that enable both strict environmental control and a large volume of material to be obtained relatively easily from healthy people and patients. Moreover, CNON cells can be used to study molecular alterations associated with brain disorders, including schizophrenia37. These in vitro cellular systems also allow modeling of genetic variants using genome-editing techniques and comparison with the post-mortem brain–derived findings.
Studies in evolutionary and developmental biology have shown that some phenotypic variations among species are driven by the evolution of gene regulatory networks, including changes in transcription factors and cis-regulatory elements12–14. The same regulatory networks are also affected in many human diseases. Therefore, complementary analyses of post-mortem brains of rhesus macaque and chimpanzee, two closely related nonhuman primates, will be conducted to identify genomic elements that may be conserved or distinct across species. Better annotation and discovery of regulatory elements in the brains of other species will improve our ability to interpret variations in cis-regulatory elements and the evolutionary conservation of those elements.
Multidimensional genomic data
According to some estimates, the human genome harbors some 400,000 putative enhancers and 70,000 promoters6. To identify active cis-regulatory regions relevant to psychiatric diseases, the consortium will assay for the hallmark features—chromatin accessibility, nucleosome depletion and enrichment for certain post-translational histone modifications— of these regions in relevant brain tissues and cells. Chromatin immunoprecipitation coupled with next-generation sequencing (ChIP-seq) has been extensively applied to map histone modifications and transcription factor binding sites in cell lines6 and in brain tissue7,13,14. Enrichment for histone 3 Lys4 trimethylation (H3K4me3) and histone 3 Lys27 acetylation (H3K27ac) will be assayed to identify putative active promoters and enhancers, respectively, To identify open chromatin regions, we will take advantage of the small amounts of tissue required for the assay of transposase-accessible chromatin (ATAC-seq)38. This assay will be optimized using freshly frozen human brains.
One of the challenges of studying regulatory regions is finding the target genes of those regulatory regions: because higher order chromatin conformations such as chromosomal looping allow transcriptional control across considerable genomic distances and sometimes from locations as remote as another chromosome39, functionally linking cis-regulatory elements such as enhancers to downstream targets can be difficult. Chromosome conformation capture39 (also known as 3C), a standard approach to map chromosomal loops and Hi-C, a high-throughput adaptation of 3C, will be part of PsychENCODE efforts. Enhancer function will be further studied using mouse transgenesis and the self-transcribing active regulatory region sequencing (STARR-seq)40 assay to measure the activity of multiple enhancers simultaneously.
Methylation of DNA, at CpG and non-CpG dinucleotides, will be assayed by whole-genome bisulfite sequencing to obtain genome-wide DNA methylation profiles during development and late adolescence, when most neuropsychiatric illnesses first manifest. Nucleosome occupancy and, methylome sequencing (NOMe-seq)41, a recently developed assay that provides both a nucleosome footprint and a DNA methylation profile for a DNA strand, will also be applied.
The PsychENCODE consortium will quantify both coding and noncoding RNA by RNA-seq and long-read sequencing (Iso-seq). Ribosome footprinting and micro-western arrays coupled with reverse-phase protein arrays (MWA-RPPA) will be used to reveal the portions of RNA that are directly translated into peptides and proteins and to quantify protein levels.
Another important goal of the PsychENCODE consortium is to systematically evaluate variations in major epigenomic features within a well-phenotyped human population. ENCODE and REMC generated reference maps from a variety of control tissues and cell types, but these studies did not focus on the impact of genetic variation on chromatin state and structure. Likewise, GTEx identified expression quantitative trait loci (eQTLs) from a variety of human tissues, but focused primarily on samples from healthy individuals that have not been extensively phenotyped9. PsychENCODE will stand at the interface of these efforts by assembling epigenomic and transcriptomic data sets from approximately 1,000 post-mortem brains from healthy controls or individuals affected with schizophrenia, bipolar disorder or ASD. To this end, genotype data will be available either from genome-wide SNP arrays or by whole genome sequencing.
Data analysis
Analysis of the PsychENCODE data sets will be pursued as a joint effort by all individual groups and by the PsychENCODE data analysis core. To enable comparison and integration of the data generated by all groups, the data analysis core will normalize the data to remove batch effects, establish uniform data processing pipelines and build calibration resources for all assays. Established processing pipelines and data quality control metrics for major data types such as RNA-seq, ChIP-seq of transcription factors and histone marks, whole-genome bisulfite sequencing and Hi-C will be adapted for the data generated by the PsychENCODE Consortium; new pipelines will be developed for NOMe-seq and ATAC-seq.
In addition to analyzing individual data sets, an important goal of the PsychENCODE project is to integrate multiple data modalities to identify global mechanisms relevant to disease. Having a large sample size will provide sufficient statistical power to reliably detect different types of genomic elements active in the developing and adult human brain and those that are altered in psychiatric disorders. Data sets measuring transcript abundance will be integrated with maps of regulatory regions, providing putative disease-, tissue-or cell-type-specific relationships between cis-regulatory elements and gene expression. DNA variations that affect chromatin state (epigenetic quantitative trait loci, or epiQTLs), transcript abundance (expression QTL, or eQTLs) and protein expression (pQTLs) will also be identified by integrating genotyping information (Fig. 1). Histone acetylation QTLs (haQTLs) will be identified with high sensitivity from ChIP-seq data by jointly scoring allelic imbalance and peak height variation in a cohort; regulatory variants identified in this manner are strongly enriched for linkage with disease-causing variants42. In all QTL analyses, known confounding factors such as age, sex and technical covariates will be corrected for and unidentified confounding factors will be minimized through principal component analysis or other methods. Because SNP-associated eQTLs in the brain are significantly enriched in GWAS of multiple psychiatric disorders43,44, RNA quantification data will be analyzed in conjunction with genotype data to identify eQTLs associated with brain regions in both normal and disease cohorts. pQTLs will be mapped using the same sets of samples as in the eQTL analyses. We expect that some pQTLs will be traced back to eQTLs. However, many pQTLs are not explained by eQTLs45, but rather by effects of the genetic variation on translational control or regulation of protein stability. Our pQTL analysis will identify such effects.
Many studies have shown that cis-eQTLs, methylation QTLs, DNase I sensitivity QTLs, haQTLs and pQTLs have large effect sizes that can be detected in less than 100 samples9,42,46,47. It is expected that other epiQTLs will have similar number and magnitude of effects. PsychENCODE, which includes data from nearly 1,000 individuals, will allow detection of cis-QTLs and also opens the possibility of detecting trans signals that require larger sample sizes. PsychENCODE will thus have similar statistical power to that of GTEx, which is examining a similar number of individuals for cis and trans eQTLs in a variety of other tissues.
Data integration will be performed initially for the individual diseases. First, the extent and nature of inter-individual differences will be characterized. Second, case-control differences will be investigated. Third, association with risk of disease will be tested and, where possible, these data will be used to refine SNP association by prioritizing individual genes and causal alleles in nominally associated GWAS regions48. For assays common across disorders (for example, RNA-seq and some histone modifications), well-powered sample sizes approaching several hundred controls will be achieved and cross-disorder analysis will be performed.
The initial comparisons will use classical, primarily pairwise, methods followed by systems-level analyses to identify gene regulatory networks. Coexpression networks, such as those based on weighted gene coexpression network analysis, permit construction of networks12,17,18,29–31,34,36 using expression data obtained in brain, in cellular fractions and in cellular models. Identified gene coexpression networks will be refined by incorporating diverse data types, including noncoding RNA expression patterns, splicing and ChIP-seq data to identify those regulatory networks most relevant for psychiatric disease. The comparison of gene coexpression networks between normal and disease-affected brains represents another platform that allows prioritization of gene variants and associated networks as potential candidates for functional validation using mouse models and iPSCs. Comparison of these gene networks with large sample cohorts available through PsychENCODE will also allow population-level analyses.
Finally, the PsychENCODE project will avail itself of data from other consortia, including BrainSpan (http://www.brainspan.org/), CommonMind (http://www.commonmind.org/), ENCODE (http://www.encodeproject.org/), REMC (http://www.roadmapepigenomics.org/), GTEx (http://www.gtexportal.org/home/) and the Psychiatric Genomics Consortium (PGC; http://www.med.unc.edu/pgc/). To allow comparison of these data sets, parameters in the processing pipelines of different projects will be evaluated and calibrated by the data analysis core. In addition, we will compare the functional genomic elements identified across these projects, with reference to the specific tissues and cell types studied by these consortia. Major differences in identified genomic elements, if any are detected, will be further examined to determine whether they are due to the underlying raw data or analytical approaches. After building the calibration resources, PsychENCODE data will be compared against data from tissues other than brain to identify transcripts and enhancers specific to brain regions and cell types, and such information will be used to deconvolve cellular diversity and variability in post-mortem tissues.
Functional characterization of disease-associated elements
The functional contribution of regulatory elements to the development, evolution, function and pathogenesis of the brain is well recognized, but still under-studied by neuroscientists13,14,49. We will assess consequences of noncoding disease-associated variants on DNA-binding protein occupancy, epigenetic modifications and gene expression in the context of neurodevelopment by engineering select variants into mice and/or isogenic human iPSCs using CRISPR-based genome editing tools. Variants will be prioritized on a number of criteria, including the strength of association between any given variant and disease, the likelihood that the variant perturbs cis-regulatory elements or genes known to be differentially expressed in disorders or active in the developing human brain, the proximity of the variant to previously identified disease-related genes or loci, the presence of binding sites for relevant disease-related transcription factors within the putative regulatory element, the relative impact of the variant on high-throughput reporter gene activity and the evolutionary conservation of the putative regulatory element.
iPSCs provide an in vitro system on a controlled genetic background that has been used to study molecular and cellular alterations associated with psychiatric disorders36,50. Multidimensional genomic maps complementary to those generated by studies of the post-mortem human brain will be generated using human iPSCs differentiated into cortical organoids containing neural progenitors, excitatory projection neurons and inhibitory interneurons, thus validating this system as a model to study human brain development. Then iPSC-derived organoids carrying control or disease variants and their constituent cell types isolated by FANS will be analyzed at different time points during differentiation and the resulting data compared to that generated from post-mortem tissue. Additionally, to account for sex-related differences and off-target effects, iPSCs derived from both male and female donors will be used and each mutation will be made in two independent isogenic iPSC clones.
The mouse model provides a physiologically and developmentally relevant in vivo system. It will be used primarily for disease variants that occur in genomic regions that are highly conserved between species and that have already been validated using iPSCs. Comparisons between the results generated from human iPSCs and mouse models will demonstrate the extent to which disease variants converge on common molecular and cellular mechanisms across development, species and cell types. Additionally, the development of these mouse models will provide the basis for future studies on neural circuits and behavior.
Data accessibility
The PsychENCODE Consortium will share information among consortium members and the broader research community through a website (http://www.psychENCODE.org/) and a knowledge portal (http://www.synapse.org/pec). The website will provide descriptive information about each project, news about the consortium and up-to-date information on tissue banks, protocols and sample sizes. The knowledge portal, developed by the PsychENCODE data coordination core at Sage Bionetworks, is designed to provide a centralized environment for accessing data, protocols and analytical output to enable collaboration among and beyond consortium members.
Data will be released to the broader research community at six-month intervals beginning in January 2016. Access to data derived from human samples will be shared using a controlled access mechanism that complies with regulatory requirements and governance policies regarding protection of personal information. Tissue samples from the investigators’ collections, when available, will also be obtainable upon request using similar guidelines. CNON cells, fibroblasts and iPSCs will be made available through the NIMH Repository and Genomics Resource (NIMH-RGR; http://www.nimhgenetics.org/). PsychENCODE investigators will ensure that data can be visualized through genome browsers such as the UCSC Genome Browser and/or IGV.
Conclusions
The main goals of PsychENCODE are to build comprehensive spatio-temporal reference maps of genomic elements active in healthy and disease-affected brains, to assess population epigenetic variations of human brains and to functionally characterize disease-associated variants. The PsychENCODE Consortium brings together distinct projects that will apply comprehensive, unbiased, complementary and non-overlapping approaches to gain insight into the roles of noncoding elements in developing and adult, healthy and disease-affected human brains on a population scale. Although some existing consortia (REMC, GTEx and BrainSpan) have included studies of the healthy human brain, the PsychENCODE project is unlike these efforts in that it will integrate disease-associated variants, changes in gene and protein expression, noncoding DNA elements and many disease phenotypes. Furthermore, PsychENCODE will be the first consortium to compare neuronal and non-neuronal transcriptomes and epigenomes from post-mortem brains of donors with psychiatric phenotypes with similar data sets obtained from tissue homogenates, as a starting point for exploring cell-type-specific regulation of genome organization and function across the timespan and disease states. It will bring together a large collection of well-curated healthy and disease-affected human brains over the course of development and adulthood. The large sample sizes will generate population-level genomic and epigenomic data for both control and disease-affected brains. The PsychENCODE project will also complement these approaches with studies in human neural cell culture systems and in developing nonhuman primate brains. The consortium’s cross-species and cross-model studies will reveal genomic risk factors that may be central to human neuropathology in a comprehensive fashion beyond the purview of any individual laboratory. We expect that the genome-scale data sets provided by the PsychENCODE project will facilitate the identification and functional characterization of genomic elements active in the developing human brain, provide a new foundation on which to build and test hypothesis derived from the data and greatly enhance our understanding of the mechanisms underlying complex psychiatric disorders.
Acknowledgments
The authors would like to acknowledge and thank the tissue donors and their families. We also thank all consortium members for discussions and feedback on this document. The PsychENCODE consortium projects are funded by the US National Institute of Mental Health.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Contributor Information
Geetha Senthil, Email: geetha.senthil2@nih.gov.
Thomas Lehner, Email: tlehner@mail.nih.gov.
Pamela Sklar, Email: pamela.sklar@mssm.edu.
Nenad Sestan, Email: nenad.sestan@yale.edu.
References
- 1.Lander ES, et al. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 2.Venter JC, et al. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 3.Levine M, Davidson EH. Proc Natl Acad Sci USA. 2005;102:4936–4942. doi: 10.1073/pnas.0408031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee TI, Young RA. Cell. 2013;152:1237–1251. doi: 10.1016/j.cell.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Visel A, Rubin EM, Pennacchio LA. Nature. 2009;461:199–205. doi: 10.1038/nature08451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Encode Project Consortium. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roadmap Epigenomics Consortium et al. Nature. 2015;518:317–330. [Google Scholar]
- 8.Ward LD, Kellis M. Nat Biotechnol. 2012;30:1095–1106. doi: 10.1038/nbt.2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.GTEx Consortium. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sullivan PF, Daly MJ, O’Donovan M. Nat Rev Genet. 2012;13:537–551. doi: 10.1038/nrg3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cross-Disorder Group of the Psychiatric Genomics Consortium et al. Nat Genet. 2013;45:984–994. doi: 10.1038/ng.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson MB, et al. Neuron. 2009;62:494–509. doi: 10.1016/j.neuron.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nord AS, Pattabiraman K, Visel A, Rubenstein JL. Neuron. 2015;85:27–47. doi: 10.1016/j.neuron.2014.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shibata M, Gulden FO, Sestan N. Trends Genet. 2015;31:77–87. doi: 10.1016/j.tig.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Konopka G, et al. Neuron. 2012;75:601–617. doi: 10.1016/j.neuron.2012.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liao BY, Zhang J. Proc Natl Acad Sci USA. 2008;105:6987–6992. doi: 10.1073/pnas.0800387105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller JA, et al. Nature. 2014;508:199–206. doi: 10.1038/nature13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang HJ, et al. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson MB, et al. Nat Neurosci. 2015;18:637–646. doi: 10.1038/nn.3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Darmanis S, et al. Proc Natl Acad Sci USA. 2015;112:7285–7290. doi: 10.1073/pnas.1507125112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meyer-Lindenberg A, Weinberger DR. Nat Rev Neurosci. 2006;7:818–827. doi: 10.1038/nrn1993. [DOI] [PubMed] [Google Scholar]
- 22.Insel TR. Nature. 2010;468:187–193. doi: 10.1038/nature09552. [DOI] [PubMed] [Google Scholar]
- 23.Lichtenstein P, et al. Lancet. 2009;373:234–239. doi: 10.1016/S0140-6736(09)60072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wray NR, Gottesman II. Front Genet. 2012;3:118. doi: 10.3389/fgene.2012.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Psychiatric GWAS Consortium Bipolar Disorder Working Group. Nat Genet. 2011;43:977–983. doi: 10.1038/ng.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Nature. 2014;511:421–427. [Google Scholar]
- 27.Malhotra D, Sebat J. Cell. 2012;148:1223–1241. doi: 10.1016/j.cell.2012.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amaral DG, Schumann CM, Nordahl CW. Trends Neurosci. 2008;31:137–145. doi: 10.1016/j.tins.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 29.Geschwind DH, State MW. Lancet Neurol. 2015;14:1109–1120. doi: 10.1016/S1474-4422(15)00044-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Rubeis S, et al. Nature. 2014;515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iossifov I, et al. Nature. 2014;515:216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gaugler T, et al. Nat Genet. 2014;46:881–885. doi: 10.1038/ng.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCarthy SE, et al. Mol Psychiatry. 2014;19:652–658. doi: 10.1038/mp.2014.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fromer M, et al. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang Y, Matevossian A, Huang HS, Straubhaar J, Akbarian S. BMC Neurosci. 2008;9:42. doi: 10.1186/1471-2202-9-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mariani J, et al. Cell. 2015;162:375–390. doi: 10.1016/j.cell.2015.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evgrafov OV, et al. Psychiatr Genet. 2011;21:217–228. doi: 10.1097/YPG.0b013e328341a2f0. [DOI] [PubMed] [Google Scholar]
- 38.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. Curr Protoc Mol Biol. 2015;109:21.29. doi: 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dekker J, Marti-Renom MA, Mirny LA. Nat Rev Genet. 2013;14:390–403. doi: 10.1038/nrg3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vockley CM, et al. Genome Res. 2015;25:1206–1214. doi: 10.1101/gr.190090.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kelly TK, et al. Genome Res. 2012;22:2497–2506. doi: 10.1101/gr.143008.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.del Rosario RC, et al. Nat Methods. 2015;12:458–464. doi: 10.1038/nmeth.3326. [DOI] [PubMed] [Google Scholar]
- 43.Gamazon ER, et al. Mol Psychiatry. 2013;18:340–346. doi: 10.1038/mp.2011.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Richards AL, et al. Mol Psychiatry. 2012;17:193–201. doi: 10.1038/mp.2011.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hause RJ, et al. Am J Hum Genet. 2014;95:194–208. doi: 10.1016/j.ajhg.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Degner JF, et al. Nature. 2012;482:390–394. doi: 10.1038/nature10808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bell JT, et al. Genome Biol. 2011;12:R10. doi: 10.1186/gb-2011-12-1-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He X, et al. Am J Hum Genet. 2013;92:667–680. doi: 10.1016/j.ajhg.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.West AE, Greenberg ME. Cold Spring Harb Perspect Biol. 2011;3:a005744. doi: 10.1101/cshperspect.a005744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brennand KJ, Gage FH. Dis Model Mech. 2012;5:26–32. doi: 10.1242/dmm.008268. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The PsychENCODE Consortium will share information among consortium members and the broader research community through a website (http://www.psychENCODE.org/) and a knowledge portal (http://www.synapse.org/pec). The website will provide descriptive information about each project, news about the consortium and up-to-date information on tissue banks, protocols and sample sizes. The knowledge portal, developed by the PsychENCODE data coordination core at Sage Bionetworks, is designed to provide a centralized environment for accessing data, protocols and analytical output to enable collaboration among and beyond consortium members.
Data will be released to the broader research community at six-month intervals beginning in January 2016. Access to data derived from human samples will be shared using a controlled access mechanism that complies with regulatory requirements and governance policies regarding protection of personal information. Tissue samples from the investigators’ collections, when available, will also be obtainable upon request using similar guidelines. CNON cells, fibroblasts and iPSCs will be made available through the NIMH Repository and Genomics Resource (NIMH-RGR; http://www.nimhgenetics.org/). PsychENCODE investigators will ensure that data can be visualized through genome browsers such as the UCSC Genome Browser and/or IGV.