

Abstract
Autism spectrum disorder (ASD) is a neurodevelopmental disorder (NDD) characterized by impairments in social communication and social interaction and the presence of repetitive behaviors and/or restricted interests. ASD has profound etiological and clinical heterogeneity, which has impeded the identification of risk factors and pathophysiological processes underlying the disorder. A constellation of (i) types of genetic variation, (ii) modes of inheritance and (iii) specific genomic loci and genes have all recently been implicated in ASD risk, and these findings are currently being extended with functional analyses in model organisms and genotype–phenotype correlation studies. The overlap of risk loci between ASD and other NDDs raises intriguing questions around the mechanisms of risk. In this review, we will touch upon these aspects of ASD and how they might be addressed.
Introduction
Autism spectrum disorder (ASD) is a neurodevelopmental disorder (NDD) characterized by persistent deficits in social communication and interaction and the presence of repetitive behaviors and/or restricted interests. ASD manifests with broad phenotypic variability and evolves along heterogeneous developmental trajectories (1). Severity of ASD symptoms is variable and inversely correlates with adaptive functioning (2). Language abilities and cognitive function, two domains strongly affecting adaptive behavior (2,3) and predicting later outcome (4), also have a broad range in ASD. For example, ∼30% of individuals with ASD remain minimally verbal throughout life (5) and ∼60% have co-occurring intellectual disability (ID) (3). Other NDDs [e.g. attention deficit hyperactivity disorder (ADHD) and schizophrenia], neurological disorders (e.g. motor deficits, sleep disturbances and epilepsy) and medical conditions (e.g. gastrointestinal problems, congenital anomalies and allergy) are also common in ASD although highly variable (6). Here, we will discuss recent findings on the genetic architecture of ASD and its complex relationship to phenotype.
Etiological heterogeneity
Studies making use of twin pairs (7–11), families (12) and populations (11,13) have provided estimates that over half of risk of developing ASD resides with genetic variation, which explains the elevated recurrence risk of ASD and associated phenotypes observed in families (13).
Diverse forms of genetic variation, differing in frequency (i.e. common, rare and very rare variation), mode of inheritance (i.e. autosomal inherited, X-linked and de novo variation), type of variation [i.e. structural—including aneuploidy, copy number variation (CNV), indel, and single-nucleotide variation] and mode of action (additive, recessive, dominant and hemizygous), all shape risk architecture (11,14,15). Common variation in the form of single-nucleotide polymorphisms (SNPs) has a very weak effect when a given SNP is considered individually (16). As a result, genome-wide association studies carried out to date have been underpowered due to the weak effect of individual SNPs (16) and, hence, insufficient sample size [the largest study has analyzed 2,705 families (16)], and have failed to identify reproducible SNP associations (16–20). However, polygenic risk due to a large number of SNPs is a major determinant of the variance for ASD (11,12,16) (Fig. 1).
Figure 1.
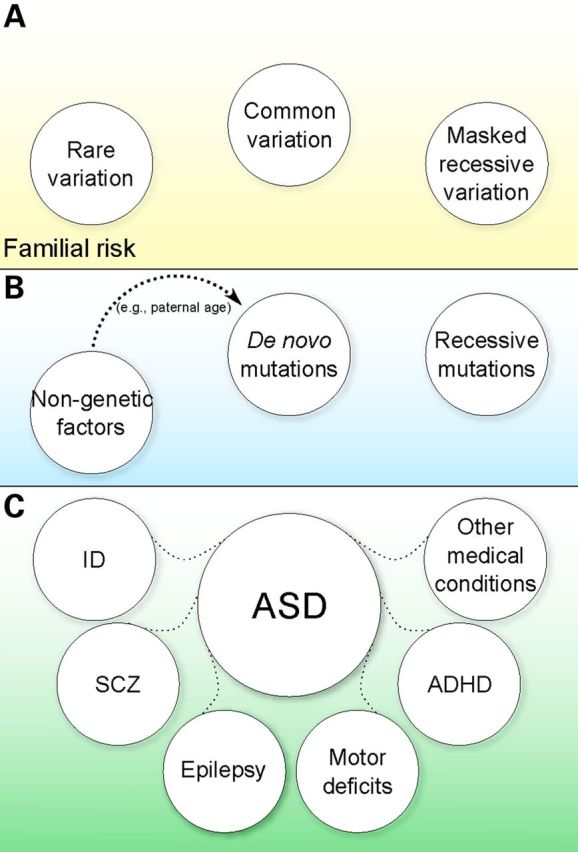
Genotype–phenotype model in ASD. Heritable common and rare variation (A) define familial risk and a liability index. When additional risk variation, for example in the form of rare variation in autosomal recessive or X-linked genes (the latter in males) and/or de novo deleterious variation, occurs in this familial risk environment (B), the liability is increased over a threshold resulting in disease. Non-genetic factors can contribute as well, and some such factors are now beginning to be understood (e.g. paternal age increasing the likelihood of de novo variation). The concerted action of familial background (A) and high-risk events (B) defines the varying clinical manifestations of ASD (C).
Rare genetic risk variation identified to date, in contrast, has a very substantial effect on individual risk. Recent whole-exome sequencing analyses of large ASD cohorts (21–29) have expanded the repertoire of known genes harboring ASD-linked mutations that were previously identifiable only by traditional genetic approaches and targeted sequencing (14,15).
At this point, it is clear that rare microscopically detectable chromosomal rearrangements (14,15), submicroscopic deletions or duplications (CNV or smaller structural variation) (27,29–32), single-nucleotide variation (SNV) and small deletions and duplications (indels) (21–26,33–35) all contribute to risk.
Rare variants can be inherited from unaffected parents (26,28,34,35) or arise as de novo mutations during the meiotic divisions of gametogenesis (21–27,30–32,36,37). Autosomal de novo loss-of-function SNV has been identified that acts in a dominant manner and carries a large burden for individual risk, with odds ratios of 20 or above for genes identified to date (24,26). Transmitted variants that disrupt protein function can also have a strong influence on risk when inherited and acting dominantly (odds ratios ∼3) (26) or if resulting in biallelic disruption and acting in a recessive (34,35) or X-linked (34) manner (odds ratio above 2).
Beyond genetic factors, there is evidence for a role of non-genetic sources of risk in ASD, although the mechanisms underlying these associations are still unclear. Paternal (38–41) and maternal (41,42) age have been independently associated with ASD risk. In the case of paternal age, one mediating mechanism lies in the increased rate of de novo SNV and CNV with advancing paternal age (22,24,25,43,44). The factors contributing to the maternal age effect, in contrast, have been less explored, but advancing maternal age is a known risk factor for chromosomal aberrations (45).
These findings are consistent with a model whereby inherited variation, both common (11) and rare (26,28), defines a substrate of susceptibility, and additional high-risk genetic (including de novo, recessive and X-linked variation) and/or non-genetic factors then act on this substrate to surpass the liability threshold that results in ASD (Fig. 1). Gaugler et al. (11) provide a clear example by estimating the probability for carriers of de novo mutations (CNV or SNV) to be affected if they were not carriers of these mutations, using a well defined ASD cohort (11). They found that ∼80% of the individuals with a de novo CNV and an ASD diagnosis would not be affected if they did not have the CNV, and 57% of the individuals with a de novo loss-of-function SNV and an ASD diagnosis would not be affected if they did not have the SNV, but common variation, in the form of SNPs, contributes in either case. The model helps in the understanding of the relatively high risk for siblings of individuals diagnosed with ASD (13,46), the increased mutational burden in ASD probands compared with their unaffected siblings (22,28), etiological heterogeneity even within pairs of affected siblings (47,48) and, as we shall discuss further, the variable expressivity of certain loci (49,50) (Fig. 1).
From gene discovery to model organisms and human phenotypes
Simulations based on the observed number of recurrently disrupted genes have estimated that there are some 600–1200 ASD risk genes (21,24,26,51). Studies to identify additional ASD risk are ongoing, but as these genes are distributed along a gradient of effect size, it will become increasingly difficult to identify new genes. To date, in addition to causal and highly penetrant genes and genomic loci identified before whole-exome sequencing (15,30), over 50 high-risk genes have emerged from the large-scale sequencing studies (21–26), most under a model of autosomal dominant variation. Genetic manipulations of these genes in model organisms and subsequent functional analyses are beginning to provide insights into their biology and are highlighting complex phenotypic associations with these genes.
Looking at even just a handful of the top genes reveals an unexpected complexity (Fig. 2, see legend for a detailed discussion). For example, although ADNP is essential for brain development (64) and Adnp+/− mice display a sex-related impairment in learning and memory behaviors (66), the most prominent phenotype of ADNP knockdown in zebrafish is defective erythropoiesis (63).
Figure 2.
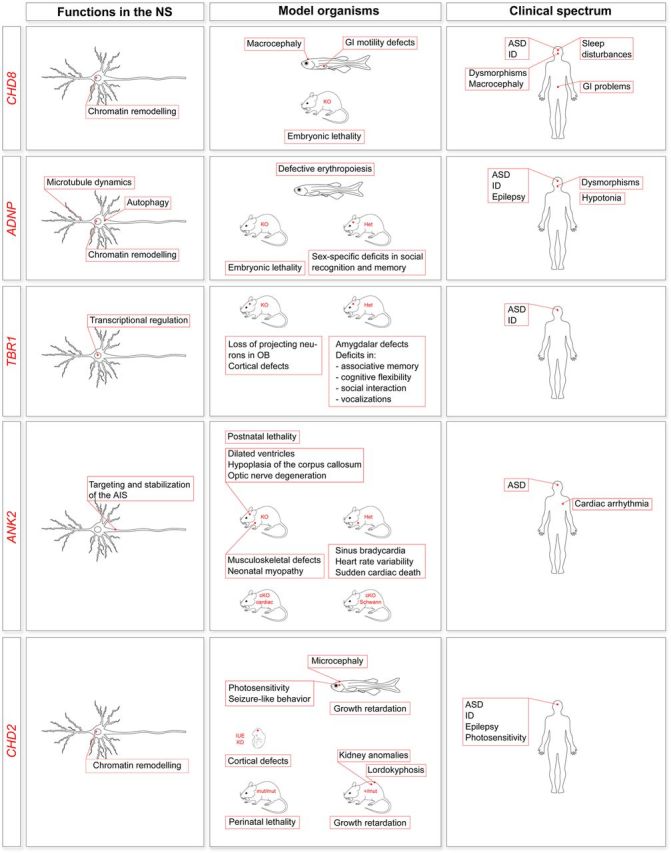
Characterization of newly discovered ASD genes. The figure summarizes, for the indicated genes, what is known about the function in the nervous system, phenotypes in animal models (KD, knockdown; KO, knockout; Het, heterozygotes) and association with human disorders. CHD8 is involved in chromatin remodeling and affects transcriptional regulation (52–54) (a postmitotic neuron is shown, but CHD8 functions in neural progenitor cells as well). In cell models, CHD8 is also implicated in apoptosis (55) (not shown). Both zebrafish (52,56) and mouse models have been generated (55,57). Chd8+/- mice have not been fully characterized. Deep phenotyping of 15 patients with CHD8 mutations has led to the identification of a proposed ASD subtype (56), although for this and many such studies the ascertainment was primarily from samples ascertained on the basis of phenotypes of interest, likely introducing biases. ADNP is involved in chromatin remodeling (58,59), autophagy (60) and microtubule dynamics at synapses through its active peptide (NAP) (61). ADNP is also expressed in glial cells (not shown) (62). ADNP knockdown in zebrafish results in no major brain anomalies but defective erythropoiesis (63). ADNP knockout in mice is embryonic lethal (64), and Adnp heterozygotes show deficits in spatial learning and memory (65) and sex-specific deficits in novel object recognition, social recognition and social memory (66). Deep phenotyping of 10 patients with ADNP mutations has led to the identification of a potential new syndrome (67). TBR1 encodes a transcription factor essential for neural stem cell fate and cerebral cortex development (68–70). TBR1 knockout in mice (71) results in the loss of projection neurons in the olfactory bulbs and olfactory cortex (71) and severe defects of cortical lamination (68,72). Tbr1+/− mice do not show gross lamination defects, but have defective axonal projections in the amygdala and a behavioral phenotype (72). TBR1 has been implicated in ASD (21,22,25,26) and ID (73). ANK2 encodes for a protein regulating the assembly of the submembranous cytoskeleton at the axonal initial segment (AIS) in unmyelinated axons (74) and acting as a glial paranodal scaffolding protein in Schwann cells (75) (not shown). ANK2 knockout in mice reduces survival postnatally (premature death by postnatal day 21) and causes lateral ventricle dilation, hypoplasia of the corpus callosum and pyramidal tracts, degeneration of the optic nerve (76) as well as musculoskeletal defects and neonatal myopathy (77). Ank2+/− mice display a cardiac phenotype (78), but their neurological phenotype has not yet been exhaustively assessed. Two conditional models have also been established: a conditional knockout of the cardiac isoforms (cKO cardiac) not fully characterized yet (79) and a knockout in Schwann cells only (cKO Schwann), with no obvious defects in paranodes of the peripheral nervous system (75). ANK2 has been associated with ASD (22,26,80) and cardiac arrhythmia (78). CHD2 is involved in chromatin remodeling [a postmitotic neuron is shown, but CHD2 functions in neural progenitor cells as well (81)]. CHD2 knockdown in zebrafish causes pericardial edema, microcephaly, body curvature, absent swim bladder, growth retardation, seizure-like behavior (82) and photosensitivity (83). CHD2 knockdown in mice by in utero electroporation causes defective cortical development by affecting neural progenitor cells proliferation (81). Homozygous mice for a CHD2 lacking the DNA-binding domain (mut/mut) have perinatal lethality (84,85). Heterozygosity (+/mut) attenuates lethality, but results in a complex phenotype (84–86). CHD2 has been associated with ASD (22,23), ID (73), epilepsy (82,87) and photosensitivity (83).
ANK2 has been consistently associated with ASD (22,26,80) and also with cardiac arrhythmia (OMIM no. 600919) (78). Consistently, neurological (76) and cardiac (77,78) phenotypes have been described in mouse models with a disruption of this gene. As most ANK2 missense mutations in patients with cardiac arrhythmia map to both cardiac and neuronal isoforms (88), ASD and cardiac arrhythmia might be common manifestations of a distinct nosological entity.
Mutations in CHD2 have been detected in individuals with a diagnosis of ASD (22), ID (73) or epilepsy (82,83,87,89), as have many of the known and novel ASD genes, and individuals carrying gene deletions in CHD2 display ID, epilepsy and autistic-like behaviors (90). However, in addition to postnatal growth retardation, zebrafish and mouse models share some unexpected phenotypic traits, including cardiovascular and renal defects, and lordokyphosis (82,84–86). Broad phenotyping of model systems, beyond social and repetitive behaviors, is clearly important (91).
Genotype–phenotype relationships
The scenario emerging from these few examples shows how ASD risk loci and genes identified to date rarely map exclusively to ASD (Figs 1 and 2), raising questions about pleiotropy or other mechanisms of action and rendering genotype–phenotype correlations complex. Chromosomal microarray and sequencing studies have found remarkable genetic overlap between ASD and other neurodevelopmental and neurological conditions, including ID, epilepsy and schizophrenia (92,93). Genetic commonalities might extend beyond NDD. In fact, recent discoveries indicate shared genes and pathways between ASD and congenital heart disease (CHD) (26,93,94).
Using a genotype-first approach, studies reporting the comprehensive clinical characterization of cohorts of patients with shared etiology have shown a high degree of variability in the expressivity of recurrent CNVs and mutations in single genes (95). Among the best examples are 22q11.2 deletions and 16p11.2 deletions. Both deletions are in fact associated, with variable severity, with various neuropsychiatric disorders, including ASD, ID and schizophrenia, among others (50,96). The deletions can also be detected in healthy individuals, an observation consistent with incomplete penetrance (50,96), which is true for most of the genes and loci that have been identified. One intriguing hypothesis is that common variation may be a factor determining the expression of specific traits when a highly penetrant mutation occurs (Fig. 1), and recent findings in both 22q11.2 and 16p11.2 deletion syndromes provide some support of this. A study focussing on the variable expressivity of CHD in carriers of 22q11.2 deletion has identified a common CNV that could act as a modifier on the cardiac phenotype in individuals with the syndrome (49). Also, by analyzing quantitative cognitive, social, motor and anthropometric traits that are highly heritable, a study on 56 individuals with de novo 16p11.2 deletions has shown the importance of family background (which in turn might be mediated by common variation and/or non-genetic factors) on clinical variability and shown the value of using quantitative rather than dichotomous measures to better assess and account for such familial influence (50).
Among the most studied connections is that between ASD and ID. In addition to the high co-occurrence, there is also interdependence between cognitive function and response to social cues. Recent observations in the Simons Simplex Collection (SSC) suggest that ASD risk architecture might differ in individuals with lower and higher IQ, although this must also be examined in additional cohorts. In the SSC, individuals with ASD and IQ < 100 have an excess of de novo loss-of-function mutations in comparison to their higher functioning counterparts (97,98), whereas individuals with ASD but not ID have a higher rate of family history of psychiatric disease and thus a greater familial burden (98). Stratifying patients by IQ, as well as other clinical variables, has been proposed as an approach to reduce phenotypic variability (99–103) and to enhance genetic discoveries. However, in one recent study, grouping by ASD diagnostic category, IQ, ASD severity, insistence on sameness and symptom profiles did not significantly increase genetic homogeneity, at least as defined by SNPs nor does it yield more discoveries for common variation (104,105).
All of these observations may complicate the validity of ASD as a specific construct and support a shared risk between diverse NDDs. To capture this complexity, Ledbetter and co-workers have proposed returning to the term ‘developmental brain dysfunction’ to harmonize and merge classically defined categorical diagnoses, including minimal brain disorders (learning disabilities, language disorders, developmental coordination disorder and ADHD), NDDs (ID, ASD and cerebral palsy) and some neuropsychiatric disorders (schizophrenia and major affective disorders to a certain extent) (50), whereas Gillberg (105,106) has proposed the ESSENCE prospective. Extending these perspectives, cognitive, motor, neurobehavioral and neuroanatomical/neurophysiological traits would ideally be expressed as quantitative measures that account for familial background, and penetrance and expressivity will be considered for a specific trait.
Conclusions
The integration of genetic discoveries and clinical observations in ASD is delineating a scenario whereby common and rare variations combine to produce a diagnosis, and, possibly, common variation (genetic or familial background) defines the specific phenotypes that are manifested. Genotype–phenotype correlation analyses would thus benefit from quantitative analyses of specific traits that take into account the common and rare genetic variation.
The observations on disease co-occurrence in ASD and the emerging evidence of shared risk across NDDs from genetic studies stress the importance of future studies that can determine whether specific gene signatures exist for a given disorder, while also making it clear that aggregating data from multiple NDDs are an efficient means to enhance power for gene discovery.
Finally, deep phenotyping studies of patients sharing highly penetrant mutations, accompanied by functional analyses in model organisms, will also clarify the pathobiological mechanisms of specific genetic loci, lead to the identification of ASD subtypes and novel syndromes and enhance the potential for novel therapeutics.
Conflict of Interest statement. None declared.
Funding
This work was supported by the Seaver Foundation and the National Institute of Mental Health (NIMH grant U01MH100233). S.D.R. is a Seaver Postdoctoral Fellow.
References
- 1.Szatmari P., Georgiades S., Duku E., Bennett T.A., Bryson S., Fombonne E., Mirenda P., Roberts W., Smith I.M., Vaillancourt T., et al. (2015) Developmental trajectories of symptom severity and adaptive functioning in an inception cohort of preschool children with autism spectrum disorder. JAMA Psychiatry, 72, 276–283. [DOI] [PubMed] [Google Scholar]
- 2.Paul R., Loomis R., Chawarska K. (2014) Adaptive behavior in toddlers under two with autism spectrum disorders. J. Autism Dev. Disord., 44, 264–270. [DOI] [PubMed] [Google Scholar]
- 3.Vivanti G., Barbaro J., Hudry K., Dissanayake C., Prior M. (2013) Intellectual development in autism spectrum disorders: new insights from longitudinal studies. Front. Hum. Neurosci., 7, 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Magiati I., Tay X.W., Howlin P. (2014) Cognitive, language, social and behavioural outcomes in adults with autism spectrum disorders: a systematic review of longitudinal follow-up studies in adulthood. Clin. Psychol. Rev., 34, 73–86. [DOI] [PubMed] [Google Scholar]
- 5.Tager-Flusberg H., Kasari C. (2013) Minimally verbal school-aged children with autism spectrum disorder: the neglected end of the spectrum. Autism Res., 6, 468–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aldinger K.A., Lane C.J., Veenstra-VanderWeele J., Levitt P. (2015) Patterns of risk for multiple co-occurring medical conditions replicate across distinct cohorts of children with autism spectrum disorder. Autism Res., 10.1002/aur.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bailey A., Le Couteur A., Gottesman I., Bolton P., Simonoff E., Yuzda E., Rutter M. (1995) Autism as a strongly genetic disorder: evidence from a British twin study. Psychol. Med., 25, 63–77. [DOI] [PubMed] [Google Scholar]
- 8.Ritvo E.R., Freeman B.J., Mason-Brothers A., Mo A., Ritvo A.M. (1985) Concordance for the syndrome of autism in 40 pairs of afflicted twins. Am. J. Psychiatry, 142, 74–77. [DOI] [PubMed] [Google Scholar]
- 9.Steffenburg S., Gillberg C., Hellgren L., Andersson L., Gillberg I.C., Jakobsson G., Bohman M. (1989) A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. J. Child Psychol. Psychiatry, 30, 405–416. [DOI] [PubMed] [Google Scholar]
- 10.Folstein S., Rutter M. (1977) Infantile autism: a genetic study of 21 twin pairs. J. Child Psychol. Psychiatry, 18, 297–321. [DOI] [PubMed] [Google Scholar]
- 11.Gaugler T., Klei L., Sanders S.J., Bodea C.A., Goldberg A.P., Lee A.B., Mahajan M., Manaa D., Pawitan Y., Reichert J., et al. (2014) Most genetic risk for autism resides with common variation. Nat. Genet., 46, 881–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klei L., Sanders S.J., Murtha M.T., Hus V., Lowe J.K., Willsey A.J., Moreno-De-Luca D., Yu T.W., Fombonne E., Geschwind D., et al. (2012) Common genetic variants, acting additively, are a major source of risk for autism. Mol. Autism, 3, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sandin S., Lichtenstein P., Kuja-Halkola R., Larsson H., Hultman C.M., Reichenberg A. (2014) The familial risk of autism. JAMA, 311, 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Devlin B., Scherer S.W. (2012) Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev., 22, 229–237. [DOI] [PubMed] [Google Scholar]
- 15.Betancur C. (2011) Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res., 1380, 42–77. [DOI] [PubMed] [Google Scholar]
- 16.Anney R., Klei L., Pinto D., Almeida J., Bacchelli E., Baird G., Bolshakova N., Bolte S., Bolton P.F., Bourgeron T., et al. (2012) Individual common variants exert weak effects on the risk for autism spectrum disorders. Hum. Mol. Genet., 21, 4781–4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang K., Zhang H., Ma D., Bucan M., Glessner J.T., Abrahams B.S., Salyakina D., Imielinski M., Bradfield J.P., Sleiman P.M., et al. (2009) Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature, 459, 528–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma D., Salyakina D., Jaworski J.M., Konidari I., Whitehead P.L., Andersen A.N., Hoffman J.D., Slifer S.H., Hedges D.J., Cukier H.N., et al. (2009) A genome-wide association study of autism reveals a common novel risk locus at 5p14.1. Ann. Hum. Genet., 73, 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weiss L.A., Arking D.E., Daly M.J., Chakravarti A. (2009) A genome-wide linkage and association scan reveals novel loci for autism. Nature, 461, 802–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anney R., Klei L., Pinto D., Regan R., Conroy J., Magalhaes T.R., Correia C., Abrahams B.S., Sykes N., Pagnamenta A.T., et al. (2010) A genome-wide scan for common alleles affecting risk for autism. Hum. Mol. Genet., 19, 4072–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neale B.M., Kou Y., Liu L., Ma'ayan A., Samocha K.E., Sabo A., Lin C.F., Stevens C., Wang L.S., Makarov V., et al. (2012) Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature, 485, 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iossifov I., O'Roak B.J., Sanders S.J., Ronemus M., Krumm N., Levy D., Stessman H.A., Witherspoon K.T., Vives L., Patterson K.E., et al. (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature, 515, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iossifov I., Ronemus M., Levy D., Wang Z., Hakker I., Rosenbaum J., Yamrom B., Lee Y.H., Narzisi G., Leotta A., et al. (2012) De novo gene disruptions in children on the autistic spectrum. Neuron, 74, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanders S.J., Murtha M.T., Gupta A.R., Murdoch J.D., Raubeson M.J., Willsey A.J., Ercan-Sencicek A.G., DiLullo N.M., Parikshak N.N., Stein J.L., et al. (2012) De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature, 485, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O'Roak B.J., Vives L., Girirajan S., Karakoc E., Krumm N., Coe B.P., Levy R., Ko A., Lee C., Smith J.D., et al. (2012) Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature, 485, 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Rubeis S., He X., Goldberg A.P., Poultney C.S., Samocha K., Cicek A.E., Kou Y., Liu L., Fromer M., Walker S., et al. (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature, 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krumm N., O'Roak B.J., Karakoc E., Mohajeri K., Nelson B., Vives L., Jacquemont S., Munson J., Bernier R., Eichler E.E. (2013) Transmission disequilibrium of small CNVs in simplex autism. Am. J. Hum. Genet., 93, 595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krumm N., Turner T.N., Baker C., Vives L., Mohajeri K., Witherspoon K., Raja A., Coe B.P., Stessman H.A., He Z.X., et al. (2015) Excess of rare, inherited truncating mutations in autism. Nat. Genet., 47, 582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poultney C.S., Goldberg A.P., Drapeau E., Kou Y., Harony-Nicolas H., Kajiwara Y., De Rubeis S., Durand S., Stevens C., Rehnstrom K., et al. (2013) Identification of small exonic CNV from whole-exome sequence data and application to autism spectrum disorder. Am. J. Hum. Genet., 93, 607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pinto D., Delaby E., Merico D., Barbosa M., Merikangas A., Klei L., Thiruvahindrapuram B., Xu X., Ziman R., Wang Z., et al. (2014) Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet., 94, 677–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sebat J., Lakshmi B., Malhotra D., Troge J., Lese-Martin C., Walsh T., Yamrom B., Yoon S., Krasnitz A., Kendall J., et al. (2007) Strong association of de novo copy number mutations with autism. Science, 316, 445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanders S.J., Ercan-Sencicek A.G., Hus V., Luo R., Murtha M.T., Moreno-De-Luca D., Chu S.H., Moreau M.P., Gupta A.R., Thomson S.A., et al. (2011) Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron, 70, 863–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dong S., Walker M.F., Carriero N.J., DiCola M., Willsey A.J., Ye A.Y., Waqar Z., Gonzalez L.E., Overton J.D., Frahm S., et al. (2014) De novo insertions and deletions of predominantly paternal origin are associated with autism spectrum disorder. Cell Reports, 9, 16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lim E.T., Raychaudhuri S., Sanders S.J., Stevens C., Sabo A., MacArthur D.G., Neale B.M., Kirby A., Ruderfer D.M., Fromer M., et al. (2013) Rare complete knockouts in humans: population distribution and significant role in autism spectrum disorders. Neuron, 77, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu T.W., Chahrour M.H., Coulter M.E., Jiralerspong S., Okamura-Ikeda K., Ataman B., Schmitz-Abe K., Harmin D.A., Adli M., Malik A.N., et al. (2013) Using whole-exome sequencing to identify inherited causes of autism. Neuron, 77, 259–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marshall C.R., Noor A., Vincent J.B., Lionel A.C., Feuk L., Skaug J., Shago M., Moessner R., Pinto D., Ren Y., et al. (2008) Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet., 82, 477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levy D., Ronemus M., Yamrom B., Lee Y.H., Leotta A., Kendall J., Marks S., Lakshmi B., Pai D., Ye K., et al. (2011) Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron, 70, 886–897. [DOI] [PubMed] [Google Scholar]
- 38.Hultman C.M., Sandin S., Levine S.Z., Lichtenstein P., Reichenberg A. (2011) Advancing paternal age and risk of autism: new evidence from a population-based study and a meta-analysis of epidemiological studies. Mol. Psychiatry, 16, 1203–1212. [DOI] [PubMed] [Google Scholar]
- 39.McGrath J.J., Petersen L., Agerbo E., Mors O., Mortensen P.B., Pedersen C.B. (2014) A comprehensive assessment of parental age and psychiatric disorders. JAMA Psychiatry, 71, 301–309. [DOI] [PubMed] [Google Scholar]
- 40.D'Onofrio B.M., Rickert M.E., Frans E., Kuja-Halkola R., Almqvist C., Sjolander A., Larsson H., Lichtenstein P. (2014) Paternal age at childbearing and offspring psychiatric and academic morbidity. JAMA Psychiatry, 71, 432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sandin S., Schendel D., Magnusson P., Hultman C., Suren P., Susser E., Gronborg T., Gissler M., Gunnes N., Gross R., et al. (2015) Autism risk associated with parental age and with increasing difference in age between the parents. Mol. Psychiatry, 10.1038/mp.2015.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sandin S., Hultman C.M., Kolevzon A., Gross R., MacCabe J.H., Reichenberg A. (2012) Advancing maternal age is associated with increasing risk for autism: a review and meta-analysis. J. Am. Acad. Child Adolesc. Psychiatry, 51, 477–486.e471. [DOI] [PubMed] [Google Scholar]
- 43.Kong A., Frigge M.L., Masson G., Besenbacher S., Sulem P., Magnusson G., Gudjonsson S.A., Sigurdsson A., Jonasdottir A., Jonasdottir A., et al. (2012) Rate of de novo mutations and the importance of father's age to disease risk. Nature, 488, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hehir-Kwa J.Y., Rodriguez-Santiago B., Vissers L.E., de Leeuw N., Pfundt R., Buitelaar J.K., Perez-Jurado L.A., Veltman J.A. (2011) De novo copy number variants associated with intellectual disability have a paternal origin and age bias. J. Med. Genet., 48, 776–778. [DOI] [PubMed] [Google Scholar]
- 45.Jones K.T. (2008) Meiosis in oocytes: predisposition to aneuploidy and its increased incidence with age. Hum. Reprod. Update, 14, 143–158. [DOI] [PubMed] [Google Scholar]
- 46.Gronborg T.K., Schendel D.E., Parner E.T. (2013) Recurrence of autism spectrum disorders in full- and half-siblings and trends over time: a population-based cohort study. JAMA Pediatrics, 167, 947–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yuen R.K., Thiruvahindrapuram B., Merico D., Walker S., Tammimies K., Hoang N., Chrysler C., Nalpathamkalam T., Pellecchia G., Liu Y., et al. (2015) Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med., 21, 185–191. [DOI] [PubMed] [Google Scholar]
- 48.Autism Genome Project, C. Szatmari P., Paterson A.D., Zwaigenbaum L., Roberts W., Brian J., Liu X.Q., Vincent J.B., Skaug J.L., Thompson A.P., et al. (2007) Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet., 39, 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mlynarski E.E., Sheridan M.B., Xie M., Guo T., Racedo S.E., McDonald-McGinn D.M., Gai X., Chow E.W., Vorstman J., Swillen A., et al. (2015) Copy-number variation of the glucose transporter gene SLC2A3 and congenital heart defects in the 22q11.2 deletion syndrome. Am. J. Hum. Genet., 96, 753–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moreno-De-Luca A., Evans D.W., Boomer K.B., Hanson E., Bernier R., Goin-Kochel R.P., Myers S.M., Challman T.D., Moreno-De-Luca D., Slane M.M., et al. (2015) The role of parental cognitive, behavioral, and motor profiles in clinical variability in individuals with chromosome 16p11.2 deletions. JAMA Psychiatry, 72, 119–126. [DOI] [PubMed] [Google Scholar]
- 51.He X., Sanders S.J., Liu L., De Rubeis S., Lim E.T., Sutcliffe J.S., Schellenberg G.D., Gibbs R.A., Daly M.J., Buxbaum J.D., et al. (2013) Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet., 9, e1003671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sugathan A., Biagioli M., Golzio C., Erdin S., Blumenthal I., Manavalan P., Ragavendran A., Brand H., Lucente D., Miles J., et al. (2014) CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc. Natl Acad. Sci. USA, 111, E4468–E4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cotney J., Muhle R.A., Sanders S.J., Liu L., Willsey A.J., Niu W., Liu W., Klei L., Lei J., Yin J., et al. (2015) The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat. Commun., 6, 6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilkinson B., Grepo N., Thompson B.L., Kim J., Wang K., Evgrafov O.V., Lu W., Knowles J.A., Campbell D.B. (2015) The autism-associated gene chromodomain helicase DNA-binding protein 8 (CHD8) regulates noncoding RNAs and autism-related genes. Transl. Psychiatry, 5, e568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nishiyama M., Oshikawa K., Tsukada Y., Nakagawa T., Iemura S., Natsume T., Fan Y., Kikuchi A., Skoultchi A.I., Nakayama K.I. (2009) CHD8 suppresses p53-mediated apoptosis through histone H1 recruitment during early embryogenesis. Nat. Cell Biol., 11, 172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bernier R., Golzio C., Xiong B., Stessman H.A., Coe B.P., Penn O., Witherspoon K., Gerdts J., Baker C., Vulto-van Silfhout A.T., et al. (2014) Disruptive CHD8 mutations define a subtype of autism early in development. Cell, 158, 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nishiyama M., Nakayama K., Tsunematsu R., Tsukiyama T., Kikuchi A., Nakayama K.I. (2004) Early embryonic death in mice lacking the beta-catenin-binding protein Duplin. Mol. Cell. Biol., 24, 8386–8394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mandel S., Gozes I. (2007) Activity-dependent neuroprotective protein constitutes a novel element in the SWI/SNF chromatin remodeling complex. J. Biol. Chem., 282, 34448–34456. [DOI] [PubMed] [Google Scholar]
- 59.Mandel S., Rechavi G., Gozes I. (2007) Activity-dependent neuroprotective protein (ADNP) differentially interacts with chromatin to regulate genes essential for embryogenesis. Dev. Biol., 303, 814–824. [DOI] [PubMed] [Google Scholar]
- 60.Merenlender-Wagner A., Malishkevich A., Shemer Z., Udawela M., Gibbons A., Scarr E., Dean B., Levine J., Agam G., Gozes I. (2015) Autophagy has a key role in the pathophysiology of schizophrenia. Mol. Psychiatry, 20, 126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oz S., Kapitansky O., Ivashco-Pachima Y., Malishkevich A., Giladi E., Skalka N., Rosin-Arbesfeld R., Mittelman L., Segev O., Hirsch J.A., et al. (2014) The NAP motif of activity-dependent neuroprotective protein (ADNP) regulates dendritic spines through microtubule end binding proteins. Mol. Psychiatry, 19, 1115–1124. [DOI] [PubMed] [Google Scholar]
- 62.Furman S., Steingart R.A., Mandel S., Hauser J.M., Brenneman D.E., Gozes I. (2004) Subcellular localization and secretion of activity-dependent neuroprotective protein in astrocytes. Neuron. Glia Biol., 1, 193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dresner E., Malishkevich A., Arviv C., Leibman Barak S., Alon S., Ofir R., Gothilf Y., Gozes I. (2012) Novel evolutionary-conserved role for the activity-dependent neuroprotective protein (ADNP) family that is important for erythropoiesis. J. Biol. Chem., 287, 40173–40185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pinhasov A., Mandel S., Torchinsky A., Giladi E., Pittel Z., Goldsweig A.M., Servoss S.J., Brenneman D.E., Gozes I. (2003) Activity-dependent neuroprotective protein: a novel gene essential for brain formation. Brain Res. Dev. Brain Res., 144, 83–90. [DOI] [PubMed] [Google Scholar]
- 65.Vulih-Shultzman I., Pinhasov A., Mandel S., Grigoriadis N., Touloumi O., Pittel Z., Gozes I. (2007) Activity-dependent neuroprotective protein snippet NAP reduces tau hyperphosphorylation and enhances learning in a novel transgenic mouse model. J. Pharmacol. Exp. Ther., 323, 438–449. [DOI] [PubMed] [Google Scholar]
- 66.Malishkevich A., Amram N., Hacohen-Kleiman G., Magen I., Giladi E., Gozes I. (2015) Activity-dependent neuroprotective protein (ADNP) exhibits striking sexual dichotomy impacting on autistic and Alzheimer's pathologies. Transl. Psychiatry, 5, e501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Helsmoortel C., Vulto-van Silfhout A.T., Coe B.P., Vandeweyer G., Rooms L., van den Ende J., Schuurs-Hoeijmakers J.H., Marcelis C.L., Willemsen M.H., Vissers L.E., et al. (2014) A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat. Genet., 46, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hevner R.F., Shi L., Justice N., Hsueh Y., Sheng M., Smiga S., Bulfone A., Goffinet A.M., Campagnoni A.T., Rubenstein J.L. (2001) Tbr1 regulates differentiation of the preplate and layer 6. Neuron, 29, 353–366. [DOI] [PubMed] [Google Scholar]
- 69.Bedogni F., Hodge R.D., Elsen G.E., Nelson B.R., Daza R.A., Beyer R.P., Bammler T.K., Rubenstein J.L., Hevner R.F. (2010) Tbr1 regulates regional and laminar identity of postmitotic neurons in developing neocortex. Proc. Natl Acad. Sci. USA, 107, 13129–13134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Englund C., Fink A., Lau C., Pham D., Daza R.A., Bulfone A., Kowalczyk T., Hevner R.F. (2005) Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J. Neurosci., 25, 247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bulfone A., Wang F., Hevner R., Anderson S., Cutforth T., Chen S., Meneses J., Pedersen R., Axel R., Rubenstein J.L. (1998) An olfactory sensory map develops in the absence of normal projection neurons or GABAergic interneurons. Neuron, 21, 1273–1282. [DOI] [PubMed] [Google Scholar]
- 72.Huang T.N., Chuang H.C., Chou W.H., Chen C.Y., Wang H.F., Chou S.J., Hsueh Y.P. (2014) Tbr1 haploinsufficiency impairs amygdalar axonal projections and results in cognitive abnormality. Nat. Neurosci., 17, 240–247. [DOI] [PubMed] [Google Scholar]
- 73.Hamdan F.F., Srour M., Capo-Chichi J.M., Daoud H., Nassif C., Patry L., Massicotte C., Ambalavanan A., Spiegelman D., Diallo O., et al. (2014) De novo mutations in moderate or severe intellectual disability. PLoS Genet., 10, e1004772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Galiano M.R., Jha S., Ho T.S., Zhang C., Ogawa Y., Chang K.J., Stankewich M.C., Mohler P.J., Rasband M.N. (2012) A distal axonal cytoskeleton forms an intra-axonal boundary that controls axon initial segment assembly. Cell, 149, 1125–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chang K.J., Zollinger D.R., Susuki K., Sherman D.L., Makara M.A., Brophy P.J., Cooper E.C., Bennett V., Mohler P.J., Rasband M.N. (2014) Glial ankyrins facilitate paranodal axoglial junction assembly. Nat. Neurosci., 17, 1673–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Scotland P., Zhou D., Benveniste H., Bennett V. (1998) Nervous system defects of AnkyrinB (−/−) mice suggest functional overlap between the cell adhesion molecule L1 and 440-kD AnkyrinB in premyelinated axons. J. Cell Biol., 143, 1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tuvia S., Buhusi M., Davis L., Reedy M., Bennett V. (1999) Ankyrin-B is required for intracellular sorting of structurally diverse Ca2+ homeostasis proteins. J. Cell Biol., 147, 995–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mohler P.J., Schott J.J., Gramolini A.O., Dilly K.W., Guatimosim S., duBell W.H., Song L.S., Haurogne K., Kyndt F., Ali M.E., et al. (2003) Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature, 421, 634–639. [DOI] [PubMed] [Google Scholar]
- 79.Smith S.A., Sturm A.C., Curran J., Kline C.F., Little S.C., Bonilla I.M., Long V.P., Makara M., Polina I., Hughes L.D., et al. (2015) Dysfunction in the betaII spectrin-dependent cytoskeleton underlies human arrhythmia. Circulation, 131, 695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Willsey A.J., Sanders S.J., Li M., Dong S., Tebbenkamp A.T., Muhle R.A., Reilly S.K., Lin L., Fertuzinhos S., Miller J.A., et al. (2013) Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell, 155, 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shen T., Ji F., Yuan Z., Jiao J. (2015) CHD2 is required for embryonic neurogenesis in the developing cerebral cortex. Stem Cells, 33, 1794–1806. [DOI] [PubMed] [Google Scholar]
- 82.Suls A., Jaehn J.A., Kecskes A., Weber Y., Weckhuysen S., Craiu D.C., Siekierska A., Djemie T., Afrikanova T., Gormley P., et al. (2013) De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am. J. Hum. Genet., 93, 967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Galizia E.C., Myers C.T., Leu C., de Kovel C.G., Afrikanova T., Cordero-Maldonado M.L., Martins T.G., Jacmin M., Drury S., Krishna Chinthapalli V., et al. (2015) CHD2 variants are a risk factor for photosensitivity in epilepsy. Brain, 138, 1198–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Marfella C.G., Ohkawa Y., Coles A.H., Garlick D.S., Jones S.N., Imbalzano A.N. (2006) Mutation of the SNF2 family member Chd2 affects mouse development and survival. J. Cell. Physiol., 209, 162–171. [DOI] [PubMed] [Google Scholar]
- 85.Kulkarni S., Nagarajan P., Wall J., Donovan D.J., Donell R.L., Ligon A.H., Venkatachalam S., Quade B.J. (2008) Disruption of chromodomain helicase DNA binding protein 2 (CHD2) causes scoliosis. Am. J. Med. Genet. A, 146A, 1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Marfella C.G., Henninger N., LeBlanc S.E., Krishnan N., Garlick D.S., Holzman L.B., Imbalzano A.N. (2008) A mutation in the mouse Chd2 chromatin remodeling enzyme results in a complex renal phenotype. Kidney Blood Press. Res., 31, 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Epi4K Consortium; Epilepsy Phenome/Genome Project, Allen A.S., Berkovic S.F., Cossette P., Delanty N., Dlugos D., Eichler E.E., Epstein M.P., Glauser T., et al. (2013) De novo mutations in epileptic encephalopathies. Nature, 501, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cunha S.R., Mohler P.J. (2006) Cardiac ankyrins: essential components for development and maintenance of excitable membrane domains in heart. Cardiovasc. Res., 71, 22–29. [DOI] [PubMed] [Google Scholar]
- 89.Carvill G.L., Heavin S.B., Yendle S.C., McMahon J.M., O'Roak B.J., Cook J., Khan A., Dorschner M.O., Weaver M., Calvert S., et al. (2013) Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat. Genet., 45, 825–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chenier S., Yoon G., Argiropoulos B., Lauzon J., Laframboise R., Ahn J.W., Ogilvie C.M., Lionel A.C., Marshall C.R., Vaags A.K., et al. (2014) CHD2 haploinsufficiency is associated with developmental delay, intellectual disability, epilepsy and neurobehavioural problems. J. Neurodev. Disord., 6, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Buxbaum J.D., Betancur C., Bozdagi O., Dorr N.P., Elder G.A., Hof P.R. (2012) Optimizing the phenotyping of rodent ASD models: enrichment analysis of mouse and human neurobiological phenotypes associated with high-risk autism genes identifies morphological, electrophysiological, neurological, and behavioral features. Mol. Autism, 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li J., Cai T., Jiang Y., Chen H., He X., Chen C., Li X., Shao Q., Ran X., Li Z., et al. (2015) Genes with de novo mutations are shared by four neuropsychiatric disorders discovered from NPdenovo database. Mol. Psychiatry, 10.1038/mp.2015.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Buxbaum J.D. (2015) DSM-5 and psychiatric genetics—round hole, meet square peg. Biol. Psychiatry, 77, 766–768. [DOI] [PubMed] [Google Scholar]
- 94.Zaidi S., Choi M., Wakimoto H., Ma L., Jiang J., Overton J.D., Romano-Adesman A., Bjornson R.D., Breitbart R.E., Brown K.K., et al. (2013) De novo mutations in histone-modifying genes in congenital heart disease. Nature, 498, 220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Moreno-De-Luca A., Myers S.M., Challman T.D., Moreno-De-Luca D., Evans D.W., Ledbetter D.H. (2013) Developmental brain dysfunction: revival and expansion of old concepts based on new genetic evidence. Lancet Neurol., 12, 406–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bassett A.S., McDonald-McGinn D.M., Devriendt K., Digilio M.C., Goldenberg P., Habel A., Marino B., Oskarsdottir S., Philip N., Sullivan K., et al. (2011) Practical guidelines for managing patients with 22q11.2 deletion syndrome. J. Pediatr., 159, 332–339 e331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Samocha K.E., Robinson E.B., Sanders S.J., Stevens C., Sabo A., McGrath L.M., Kosmicki J.A., Rehnström K., Mallick S., Kirby A., et al. (2014) A framework for the interpretation of de novo mutation in human disease. Nat. Genet., 46, 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Robinson E.B., Samocha K.E., Kosmicki J.A., McGrath L., Neale B.M., Perlis R.H., Daly M.J. (2014) Autism spectrum disorder severity reflects the average contribution of de novo and familial influences. Proc. Natl Acad. Sci. USA, 111, 15161–15165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Veatch O.J., Veenstra-Vanderweele J., Potter M., Pericak-Vance M.A., Haines J.L. (2014) Genetically meaningful phenotypic subgroups in autism spectrum disorders. Genes Brain Behav., 13, 276–285. [DOI] [PubMed] [Google Scholar]
- 100.Hus V., Pickles A., Cook E.H., Jr, Risi S., Lord C. (2007) Using the autism diagnostic interview—revised to increase phenotypic homogeneity in genetic studies of autism. Biol. Psychiatry, 61, 438–448. [DOI] [PubMed] [Google Scholar]
- 101.Bishop S.L., Hus V., Duncan A., Huerta M., Gotham K., Pickles A., Kreiger A., Buja A., Lund S., Lord C. (2013) Subcategories of restricted and repetitive behaviors in children with autism spectrum disorders. J. Autism Dev. Disord., 43, 1287–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu X.Q., Paterson A.D., Szatmari P; Autism Genome Project Consortium. (2008) Genome-wide linkage analyses of quantitative and categorical autism subphenotypes. Biol. Psychiatry, 64, 561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Vieland V.J., Hallmayer J., Huang Y., Pagnamenta A.T., Pinto D., Khan H., Monaco A.P., Paterson A.D., Scherer S.W., Sutcliffe J.S., et al. (2011) Novel method for combined linkage and genome-wide association analysis finds evidence of distinct genetic architecture for two subtypes of autism. J. Neurodev. Disord., 3, 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chaste P., Klei L., Sanders S.J., Hus V., Murtha M.T., Lowe J.K., Willsey A.J., Moreno-De-Luca D., Yu T.W., Fombonne E., et al. (2015) A genome-wide association study of autism using the Simons Simplex Collection: does reducing phenotypic heterogeneity in autism increase genetic homogeneity? Biol. Psychiatry, 77, 775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Buxbaum J.D. (2015) DSM-5 and psychiatric genetics—round hole, meet square peg. Biol. Psychiatry, 77, 766–768. [DOI] [PubMed] [Google Scholar]
- 106.Gillberg C. (2010) The ESSENCE in child psychiatry: early symptomatic syndromes eliciting neurodevelopmental clinical examinations. Res. Dev. Disabil., 31, 1543–1551. [DOI] [PubMed] [Google Scholar]