

Summary
Aspirin and naproxen were administered to mice exposed to environmental cigarette smoke (ECS). Both agents inhibited the ECS-related formation of nucleotide alterations. ECS induced pulmonary adenomas and adenocarcinomas, partly Oct-4-positive, which were attenuated by aspirin in female mice.
Abstract
Chemoprevention provides an important strategy for cancer control in passive smokers. Due to the crucial role played by smoke-related chronic inflammation in lung carcinogenesis, of special interest are extensively used pharmacological agents, such as nonsteroidal anti-inflammatory drugs (NSAIDs). We evaluated the ability of aspirin and naproxen, inhibitors of both cyclooxygenase-1 and cyclooxygenase -2, to modulate environmental cigarette smoke (ECS)-induced lung carcinogenesis in A/J mice of both genders. Based on a subchronic toxicity study in 180 postweaning mice, we used 1600mg/kg diet aspirin and 320mg/kg diet naproxen. In the tumor chemoprevention study, using 320 mice, exposure to ECS started soon after birth and administration of NSAIDs started after weaning. At 10 weeks of life, the NSAIDs did not affect the presence of occult blood in feces. As assessed in a subset of 40 mice, bulky DNA adducts and 8-hydroxy-2′-deoxyguanosine levels were considerably increased in ECS-exposed mice and, irrespective of gender, both NSAIDs remarkably inhibited these nucleotide alterations. After exposure for 4 months followed by 5 months in filtered air, ECS induced a significant increase in the yield of surface lung tumors, the 43.7% of which were adenomas and the 56.3% were adenocarcinomas. Oct-4 (octamer-binding transcription factor 4), a marker of cell stemness, was detected in some adenocarcinoma cells. The NAIDs attenuated the yield of lung tumors, but prevention of ECS-induced lung adenomas was statistically significant only in female mice treated with aspirin, which supports a role for estrogens in ECS-related lung carcinogenesis and highlights the antiestrogenic properties of NSAIDs.
Introduction
Environmental cigarette smoke (ECS) or environmental tobacco smoke (ETS) or second-hand smoke (SHS) is a complex mixture that is inhaled by involuntary (or passive) smokers in indoor environments. It is a mixture, at a ratio of ~8:1, of sidestream cigarette smoke (SCS), generated at 900°C at the tip of a lit cigarette, and of that portion of mainstream cigarette smoke (MCS), generated at 1200–1600°C at the proximal extremity of a burning cigarette, which is exhaled by active smokers (1,2). MCS, SCS and ECS contain several thousands compounds (3). Combustion of tobacco leaves generates more than 5000 identified products, including radioactive substances and compounds that virtually belong to any chemical family, 73 of which have been evaluated by IARC to be carcinogenic to humans and/or experimental animals (4). Qualitatively, the chemical composition of SCS and MCS is similar but, due to the lower combustion temperature, there are quantitative differences (5). At equivalent concentrations, SCS is approximately four times more toxic than MCS, and SCS condensates are 2–6 times more tumorigenic than MCS condensates in the mouse skin (6).
There is sufficient evidence for the carcinogenicity to humans of SHS, which is categorized as an IARC Group 1 carcinogen (4). In addition, SHS causes sudden infant syndrome, respiratory and ear infections, and asthma attacks in infants and children, and coronary heart diseases, stroke and lung cancer in adult nonsmokers, being on the whole responsible for more than 41 000 deaths in adults and 400 deaths in infants each year in the USA (7). In Korean women, more lung cases and deaths were attributable to SHS than ever-smoking (8).
A primary objective for preventing SHS-related diseases is to implement antismoke campaigns and to avoid exposure to ECS in indoor environments. Indeed, in the USA, the prevalence of SHS exposure in nonsmokers declined from 52.5% during 1999–2000 to 25.3% during 2011–2012, but 58 million persons are still exposed to SHS, especially among children and certain ethnic and social groups (7). A complementary strategy to control cancer consists of chemoprevention by means of dietary and pharmacological agents. In particular, due to the crucial role played by smoke-related chronic inflammation in lung carcinogenesis (9) and the general role of chronic inflammation as a promoting mechanism (10), the use of anti-inflammatory agents provides a promising approach (11). Of special interest are those drugs that are extensively used in the population, such as nonsteroidal anti-inflammatory drugs (NSAIDs), whose main mechanism is to inhibit cyclooxygenase (COX) activities. While COX-1 is the constitutive isoform that favors the homeostatic maintenance of the gastric mucosa, COX-2 is the inducible, proinflammatory isoform (12).
Among NSAIDs, the salicylate derivative, aspirin or acetylsalicylic acid (AsA), and the propionic acid derivative, naproxen, are dual inhibitors of COX-1 and COX-2. After more than a century of clinical use, AsA remains the most extensively used drug in the world not only for therapeutic purposes but also for the prevention of cardiovascular diseases, and holds promises also in cancer chemoprevention (13). Meta-analyses of clinical trials provided evidence that AsA users have a lower incidence of certain cancers, and especially colorectal cancers (14), but the protective role against lung cancer is uncertain, with either negative findings (15) or modest results (16) or more convincing data (17). A pooled analysis showed a weak, yet significant, attenuation of deaths due to lung cancer, particularly to adenocarcinoma (18). Moreover, AsA exerted protective effects in mice exposed either to 9,10-dimethylbenz(a)anthracene (DMBA) (19) or to 4-(methylnitrosamino)- 1-(3-pyridyl)-1-butanone (NNK) (20). In rodent models, naproxen inhibited colonic adenocarcinoma, oral cancer, and urinary bladder cancer (21–24) but did not protect rats from methylnitrosourea (MNU)-induced mammary cancers (22) and mice from NNK-induced lung tumors (20).
Since no experimental studies are available in animal models involving exposure to cigarette smoke as a complex mixture, the goal of the present study was to evaluate in parallel the ability of NSAIDs to modulate nucleotide modifications and lung tumors in A/J mice. This multitask project involved a series of coordinated studies. The first one was a 6-week subchronic toxicity study aimed at assessing the doses of AsA and naproxen to be used in the subsequent studies. Thereafter, groups of mice were either kept smoke-free or were exposed to ECS since birth and/or treated with oral AsA or naproxen since weaning. At 10 weeks of age, the levels of bulky DNA adducts and oxidative DNA damage were evaluated in the lungs of a subset of mice. The remaining mice were maintained under ECS exposure during the first 4 months of life and then kept in filtered air for an additional 5 months. This protocol is similar to the model developed by Hanspeter Witschi (25), and also used in our laboratory (26), except that exposure to ECS started at birth because our studies have shown that MCS becomes a potent carcinogen when exposure starts at birth (27). In the Witschi’s model, the lung tumorigenicity is due to gas phase components of ECS (28). According to the classical lung tumor assay, surface lung tumors were scored. All of them were confirmed and classified histologically. In addition, the sections from most nodules were tested by immunohistochemistry for the detection of Oct-4 (octamer-binding transcription factor 4), a homeodomain transcription factor of the POU family that is used as a marker for undifferentiated stem cells (29).
Materials and methods
Mice
A total of 260 A/J mice were purchased from Harlan Laboratories (San Pietro al Natisone, Udine, Italy). They include 80 adult mice (20 males and 60 females), to be used for breeding, and 180 post-weaning mice (90 males and 90 females), to be used in the subchronic toxicity study, The mice were housed in MakrolonTM cages on sawdust bedding and maintained on standard rodent chow (Teklad 9607, Harlan Laboratories) and tap water ad libitum. The animal room temperature was 23±2°C, with a relative humidity of 55% and a 12-h day-night cycle. Housing and treatment of mice were in accordance with NIH, European (2010/63/UE Directive), and institutional guidelines.
Subchronic toxicity study
AsA and naproxen, purchased from Sigma-Aldrich (Milan, Italy), were incorporated in the Teklad 9607 diet at 4 doses each, which were selected based on literature data in rodent studies and taking into account the pharmacological doses in humans. Postweaning mice were divided into nine groups, each one composed of 20 mice (10 males and 10 females), as follows. Group A, untreated (sham-exposed) mice; Groups B, C, D, and E, mice receiving AsA at 2000, 1000, 500 and 250mg/kg diet, respectively; Groups F, G, H, I, mice receiving naproxen at 400, 200, 100 and 50mg/kg diet, respectively. The mice were inspected daily for 6 weeks in order to detect possible interim deaths and signs of sufferance and behavioral alterations. Moreover, they were weighed individually at weekly intervals. The maximum tolerated dose is assumed as the highest dose that does not lower the body weight of mice by more than 10%, as compared with untreated mice, and that does not result in behavioral changes.
Exposure of mice to ECS
A whole-body exposure of mice to ECS was achieved by using a smoking machine (model TE-10C, Teague Enterprises, Davis, CA), in which mainstream CS (11%) and sidestream CS (89%) are mixed to generate ECS. Five 3R4F Kentucky reference cigarettes (College of Agriculture, The Reference Cigarette Program, University of Kentucky, Lexington, KY), having a declared content of 9.4mg tar and 0.7mg nicotine and delivering 12mg CO each, were burnt at one time. Cigarettes were smoked using the FTC (Federal Trade Commission) method of puffing for 2 s, once a min, at a volume of 35cm3. Two rounds of exposure were performed daily by burning a total of 120 cigarettes per day. The total particulate matter in the exposure chambers was on an average 95mg/m3 and CO was 610 ppm.
Treatment of neonatal mice
A total of 320 newborn mice were divided into four groups, each one composed of 40 males and 40 females, as follows. Group J, mice kept in filtered air for up to 9 months (sham-exposed mice); Group K, mice exposed to ECS for up to 4 months, starting within 12h after birth (ECS-exposed mice), and then kept in filtered air for an additional 5 months; Group L, ECS-exposed mice receiving a diet supplemented with AsA, at the dose indicated under Results, starting after weaning (∼4 weeks) and continuing for up to 9 months; Group M. ECS-exposed mice receiving for 6 weeks a diet supplemented with naproxen, at the dose indicated under Results, starting after weaning (∼4 weeks) and continuing for up to 9 months. Each experimental group (cases and controls) was composed of 80 mice, thus allowing a statistical power of 90% to detect differences of at least 20% in lung tumor incidence given an alpha error 0.05’.
Collection of biological samples for the study of early biomarkers
At the age of 10 weeks, samples of feces were collected from sham-exposed mice (Group A, 5 males and 5 females) and mice treated with the highest nontoxic doses either of AsA (Group B, 5 males and 5 females) or of naproxen (Group F, 5 males and 5 females), which were housed individually for 24h the day before the end of the subchronic toxicity study.
Ten mice (5 males and 5 females) belonging to Groups B (AsA-treated mice), F (naproxen-treated mice), J (sham-exposed mice), K (ECS-exposed mice), L (ECS-exposed mice treated with AsA) and M (ECS-exposed mice treated with naproxen) were killed by CO2 asphyxiation at the age of 10 weeks, and the lungs were collected individually.
Evaluation of occult blood in feces
The guaiac fecal occult blood test (gFOBT) was used in order to evaluate whether administration of NSAIDs may result in the presence of blood in feces.
Evaluation of bulky DNA adducts and oxidative DNA damage in lung
DNA was extracted from the left lungs of 59 mice, i.e. all mice belonging to the above specified groups, excepting one ECS-exposed female treated with AsA that died at the age of 7 weeks. Homogenization of lung fragments was performed by continuous shaking for 2min at 30 Hz using a tissue lyser (TissueLyser, Qiagen, Gaithersburg, MD) in the presence of a 5-mm stainless steel bead immersed in lysis buffer. Cell debris were removed by centrifugation at 14 000× g at 4°C for 15min. DNA was purified from the supernatant by using a commercially available kit (GenEluteTM Mammalian Genomic DNA Miniprep kit, Sigma, St Louis, MO). Spin columns were washed twice and the trapped DNA was eluted by ultrapure water (100 μl). To avoid DNA oxidation, the extraction procedure was performed under oxygen-free helium atmosphere using an automatic DNA extractor (Genepure 341, Applied Biosystems, Foster City, CA).
Bulky DNA adducts were measured by means of 32P postlabeling analyses, which were performed as described previously (30). Briefly, aliquots of DNA (10 μg) were depolymerized by micrococcal nuclease/spleen phosphodiesterase digestion. Lipophilic bulky adducts were separated from normal nucleotides by butanol enrichment and labeled by T4 nucleotide kinase action using AT-γ-32P (64 μCi, specific activity ≥ 6,000 Ci/mmol, ICN, Irvine, CA) as 32P donor. 32P labeled DNA adducts were purified from the reaction mixture by multidirectional thin layer chromatography on a 10×8cm cellulose sheet coated with the anion exchanger polyethylenimine (Macherey and Nagel, Düren, Germany). Four chromatographic developments (D1, D2, D3 and D4) were performed by rotating the development directions. DNA adducts were detected as radioactive spots on chromatographic sheet by electronic autoradiography performed in a phosphorimager (InstantImager, Packard, Meriden, CT). DNA-free samples were used as negative controls. 7R,8S,9S-trihydroxy-10R-(N2-deoxyguanosyl-3′-phosphate)-7,8,9,10-tetrahydrobenzo(a)pyrene (BPDE-N2-dG) reference standard (National Cancer Institute Chemical Carcinogen Reference Standard Repository, Mid-West Research Institute, Kansas City, MO) was used as a positive control. The results are expressed as DNA adducts/108 nucleotides.
8-Hydroxy-2′-deoxyguanosine (8-oxo-dGuo) was measured by 32P postlabeling as described previously (31). Briefly, DNA (3 μg) was depolymerized and normal nucleotides were removed by trifluoroacetic acid incubation. 8-oxo-dGuo was 32P postlabeled by polynucleotide kinase and AT[γ-32P] reaction. The remaining 32P-labeled normal nucleotides were digested by nuclease P1. 32P-labeled 8-oxo-dGuo was purified by monodirectional thin layer chromatography on polyethyleneimine-coated cellulose sheets in formic acid. Quantification by electronic autoradiography was carried out as described for the detection of bulky DNA adducts. DNA-free samples were used as negative controls. An 8-oxodGuo reference standard (National Cancer Institute Chemical Carcinogen Reference Standard Repository) was used as a positive control. The results are expressed as 8-oxo-dGuo/105 normal nucleotides.
Cancer chemoprevention study in mice exposed to ECS
At 9 months of age, all surviving mice from Groups J (sham-exposed mice), K (mice exposed to ECS for 4 months, starting at birth and thereafter kept in filtered air for an additional 5 months), L (ECS-exposed mice treated with AsA after weaning) and M (ECS-exposed mice treated with naproxen after weaning) were killed by CO2 asphyxiation. Since all 280 mice were not born on the same day and taking into account that the yield of spontaneous tumors in A/J mice increases considerably with time (32), we respected exactly the time of killing according to the date of birth. The lungs were collected, immersed in formalin and, according to the classical lung tumor assay in A/J mice (32), surface tumors were scored with the aid of a stereomicroscope. All suspected nodules were embedded in paraffin, stained with hematoxylin and eosin, and subjected to standard histopathological analysis. A subset of nodules (see Results) was analyzed by immunohistochemistry for the detection of Oct-4 in hematoxylin-stained sections. An Oct-4 polyclonal antibody (BioVision Incorporated, Milpitas, CA) was used. The antibody reacts with 45kDa Oct-4 (also designated Oct-3A) and, to a lower extent, with 33kDa Oct-3B from human, mouse, and rat samples.
Statistical analyses
Comparisons between groups regarding survival of mice, incidence of histopathological lesions, and other frequency end-points (e.g. occult blood in feces) were made by χ2 analysis. Body weights, bulky DNA adducts, 8-oxo-dGuo levels, and multiplicity of lung tumors were expressed as means ± SE of the data generated within the mice composing each experimental group. Comparisons between groups regarding body weights, bulky DNA adducts, and 8-oxo-dGuo levels were made by parametric ANOVA and Student’s t-test for unpaired data. Since tumor multiplicity data had not a normal distribution, due to the high prevalence of tumor-free mice, the differences between groups were assayed by using the Kruskal–Wallis rank sum test, and pairwise comparisons between groups were carried out by Wilcoxon rank sums tests. The estimated P values were adjusted for multiple comparisons by using the Bonferroni post hoc test. The correlation between bulky DNA adducts and 8-oxo-dGuo levels was evaluated by Pearson product-moment correlation coefficient.
Results
Subchronic toxicity of AsA and naproxen
All the 180 postweanling mice used in the subchronic toxicity study survived after 6 weeks, with the exception of one sham-exposed female. No signs of morbidity or visible alterations in appearance and behavior of mice were detected throughout the study, irrespective of administration of the NSAIDs. The body weights after 0, 1, 2, 3, 4, 5 and 6 weeks (means ± SE) were 14.5±0.33, 18.8±0.36, 20.0±0.42, 20.7±0.47, 21.7±0.47, 21.8±0.49 and 23.8±0.66g in males, and 14.0±0.30, 16.1±0.30, 16.6±0.32, 17.6±0.47, 17.7±0.38, 17.9±0.37 and 20.0±0.32g in females, respectively. At any dose, AsA and naproxen did not affect the body weight gains of mice by more than 10%, compared to untreated controls. Therefore, it was decided to use in the chemoprevention study the 80% of the maximum tested dose that had not produced any apparent adverse effect in mice, i.e. 1600mg/kg diet for aspirin and 320mg/kg diet for naproxen.
Fecal blood in feces
The guaiac fecal occult blood test (gFOBT), which provides qualitative data, showed that 3 of 10 mice treated with AsA at 2000mg/kg diet (1/5 males and 2/5 females) and 2 of 10 mice treated with naproxen at 400mg/kg diet (1/5 males and 1/5 females) were positive at this test, compared to 1 of 10 mice used as controls (0/5 males and 1/5 females). These differences were not statistically significant. Therefore, even at the highest dose tested, neither AsA nor naproxen caused a significant bleeding of gastric mucosa in A/J mice treated for 6 weeks after weaning.
Modulation of ECS-induced DNA adducts and oxidative DNA damage
Spectrophotometric analyses showed a very good quality of the DNA extracted from all 59 processed samples, the average 260/280 ratio being consistently >1.7, proving an adequate DNA purity and integrity, and the 260/230 ratio being consistently around 2, which rules out any contaminations by proteins. 32P-Postlabeling analyses for bulky DNA adducts showed that exposure of mice to ECS results in the formation of a diagonal radioactive zone (data not shown), which is typical of exposures to complex mixtures. As shown in Table 1, DNA adduct levels increased 7.9 times in ECS-exposed male mice and 9.8 times in ECS-exposed female mice as compared with sham-exposed mice of the same gender. Neither AsA nor naproxen affected the baseline levels of DNA adducts in sham-exposed mice, whereas both NSAIDs significantly inhibited the formation of DNA adducts in ECS-exposed mice, with a more than 2-fold decrease of adduct levels in both males and females.
Table 1.
Bulky DNA adducts and oxidative DNA damage in the lung of A/J mice, as related to exposure to ECS and/or treatment with either AsA or naproxen
| Treatment | Gender | DNA adducts/108 nucleotides (means ± SE) |
8-oxo-dGuo/105 nucleotides (means ± SE) |
|---|---|---|---|
| Sham | M | 2.0±0.16 | 1.7±0.16 |
| F | 1.7±0.18 | 1.5±0.16 | |
| M + F | 1.9±0.12 | 1.6±1.09 | |
| AsA | M | 1.5±0.32 | 1.4±0.12 |
| F | 1.5±0.21 | 1.3±0.10 | |
| M + F | 1.5±0.18 | 1.4±0.08 | |
| Naproxen | M | 1.9±0.15 | 1.5±0.16 |
| F | 1.9±0.20 | 1.3±0.09 | |
| M + F | 1.9±0.12 | 1.4±0.09 | |
| ECS | M | 15.8±0.95a | 5.8±0.28b |
| F | 16.6±0.84a | 5.8±0.37b | |
| M + F | 16.2±0.61a | 5.8±0.22b | |
| ECS + AsA | M | 7.3±0.59a,d | 3.2±0.33b,d |
| F | 8.0±1.06a,d | 3.7±0.68b,c | |
| M + F | 7.5±0.99a,d | 3.4±0.34b,d | |
| ECS + Naproxen | M | 6.5±0.53a,d | 3.1±0.52b,c |
| F | 7.4±1.09a,d | 4.0±0.43b,d | |
| M + F | 7.0±0.59a,d | 3.6±0.35b,d |
The lungs of 10 mice per experimental group (five males and five females) were examined at the age of 10 weeks.
Statistical analysis: a P < 0.01 and b P < 0.001, as compared with the corresponding Sham; c P < 0.01 and d P < 0.001, as compared with the corresponding ECS.
Likewise, exposure of mice to ECS caused a significant oxidative DNA damage in the lung, as shown by a 3.4- and 3.9-fold increase of 8-oxo-dGuo levels in males and females, respectively (Table 1). Neither AsA nor naproxen significantly affected 8-oxo-dGuo levels, as compared with sham-exposed mice. Again, both NSAIDs were able to significantly attenuate the ECS-related induction of oxidative DNA damage, with a decrease ranging between 1.5- and 1-9-fold as compared with ECS-exposed mice in the absence of chemopreventive agents.
On the whole, there was a high correlation between DNA adduct levels and 8-oxo-dGuo levels in the 59 examined samples (r = 0.919; P < 0.001).
Survival and body weights of the mice used in the chemoprevention study
After killing of 40 mice for evaluating intermediate biomarkers, each one of the four experimental groups (sham-exposed mice, ECS, ECS + AsA and ECS + Naproxen) was composed of 70 mice (35 males and 35 females), for a total of 280 mice. After 9 months, all of them were still alive, excepting two sham-exposed females and 1 ECS-exposed female treated with AsA. Figure 1 shows the body weights of mice measured at monthly intervals from months 1 to 9. ECS-exposed females and even more ECS-exposed males had a significantly decreased body weight compared to sham-exposed mice throughout the 9 months of observation. This effect tended to be less evident in ECS-exposed females treated with either AsA or naproxen after 4 months onwards, when exposure to ECS was discontinued.
Figure 1.
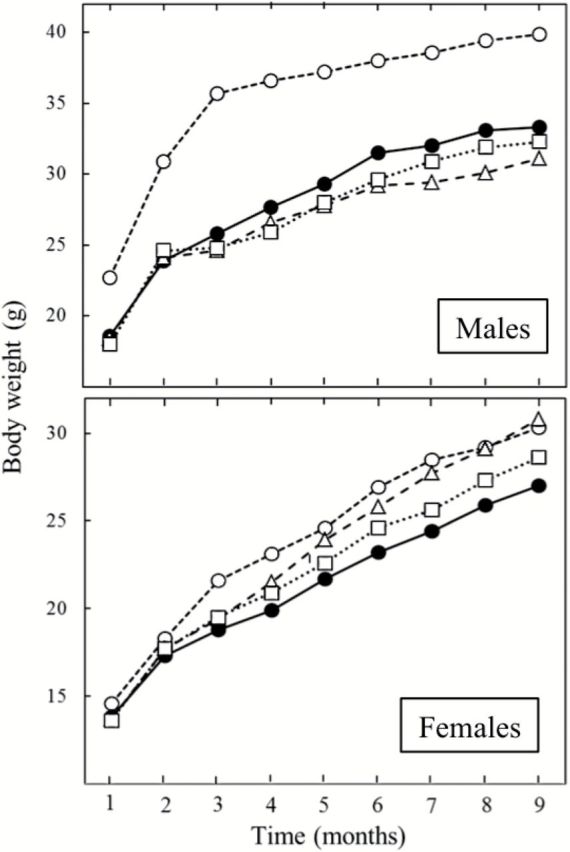
Body weights (means ± SE), measured at monthly intervals during the first 9 months of life, of male and female A/J mice, either sham-exposed (empty circles) or exposed to ECS during the first 4 months (full circles) or ECS-exposed and treated with AsA (triangles) or naproxen (squares) from weaning until the end of the experiment.
Modulation of lung tumors by AsA and naproxen
A total of 72 nodules were detected with the aid of a stereomicroscope on the lung surface of the 277 A/J mice surviving after 9 months. All of them were subjected to standard histopathological analysis. Excepting one hyperplastic and inflammatory nodule, the remaining 71 nodules were diagnosed either as adenomas (31 nodules) or adenocarcinomas (40 nodules). Figure 2 shows both incidence and multiplicity of surface lung tumors in the four experimental groups, divided by gender and exemplifies the appearance of adenomas and adenocarcinomas. Exposure of mice to ECS increased the yield of lung tumors. In particular, adenomas were absent in female sham-exposed mice. Compared to sham-exposed mice, the increase in the incidence of adenomas in ECS-exposed mice was 2-fold in males and 5.7-fold in combined genders. The increase in the incidence of adenocarcinomas was 2.5-fold in males, 1.9-fold in females and 2.3-fold in combined genders. The increase in the incidence of total tumors (adenomas plus adenocarcinomas) was 2.3-fold in males, 5.7-fold in females and 3.2-fold in combined genders. The increase in the multiplicity of adenomas in ECS-exposed mice was 2-fold in males and 5.0-fold in combined genders. The increase in the multiplicity of adenocarcinomas was 2.3-fold in males, 2.0-fold in females and 2.5-fold in combined genders. The increase in the multiplicity of total tumors (adenomas plus adenocarcinomas) was 2.5-fold in males, 5.0-fold in females and 3.3-fold in combined genders. Although the above increases were biologically relevant in all groups of ECS-exposed mice, they attained the statistical significance threshold (P < 0.05) only in the case either of adenomas or of total tumors in females and combined genders.
Figure 2.
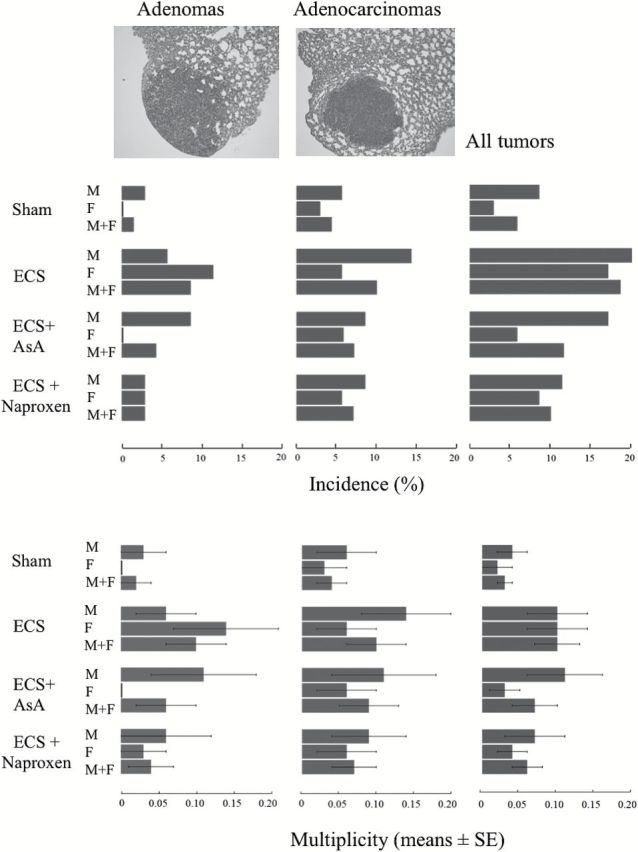
Examples of histopathological appearance of adenomas and adenocarcinomas detected on the lung surface, and their incidence and multiplicity in 9-month old A/J mice, either males (M) or females (F). The mice were either sham-exposed (35M and 33 F) or exposed to ECS during the first 4 months, starting at birth (35M and 35 F), or ECS-exposed and treated with either AsA (35M and 34 F) or naproxen (35M and 35 F) from weaning until the end of the experiment.
AsA attenuated both incidence and multiplicity of lung tumors. In particular, no adenoma was detectable in ECS-exposed female mice treated with AsA. Compared with ECS-exposed mice, the incidence of adenomas was not reduced by AsA in males and was decreased 2.0-fold in combined genders. The incidence of adenocarcinomas in ECS-exposed mice was decreased by AsA 1.7-fold in males, was unchanged in females and was decreased 1.4-fold in combined genders. The incidence of ECS-induced total tumors was decreased by AsA 1.2-fold in males, 2.9-fold in females and 1.6-fold in combined genders. Prevention of ECS-induced lung adenomas by AsA was statistically significant in female mice in terms of incidence (P < 0.05). The decrease by AsA of adenoma multiplicity in female mice (from 0.14±0.07 to 0) was statistically significant when evaluated by Kruskal–Wallis rank sum test followed by Wilcoxon rank sum tests but significance was lost after correction with Bonferroni’s post hoc test.
Likewise, naproxen attenuated both incidence and multiplicity of lung tumors. In particular, compared with ECS-exposed mice, the incidence of adenomas was decreased by naproxen 2.0-fold in males, 3.9-fold in females and 3.0-fold in combined genders, whereas the incidence of adenocarcinomas was unchanged in females, and was decreased 1.7-fold in males and 1.4-fold in combined genders. The incidence of ECS-induced total tumors was decreased by naproxen 1.8-fold in males, 2.0-fold in females and 1.9-fold in combined genders. However, none of the difference between ECS-exposed mice treated with naproxen and ECS-exposed mice in the absence of NSAIDs reached the statistical significance threshold.
Oct-4 in tumors and surrounding tissues
A subset of 25 nodule sections, including 1 hyperplastic and inflammatory nodule, 3 adenomas and 21 adenocarcinomas, were examined by immunohistochemistry for the presence of Oct-4 protein. Excepting 4 adenocarcinomas and the hyperplastic and inflammatory nodule, which were from sham-exposed mice, all remaining lesions were from ECS-exposed mice. Some of the analyzed samples were positive for Oct-4, but with different patterns. In fact, in the hyperplastic and inflammatory nodule the brownish color was localized in the cytoplasm (Figure 3A). In the adenomas there was no detectable Oct-4, whereas in four adenocarcinomas (19.0%) the brownish color was localized in the nucleus (Figure 3B). It is noteworthy that no Oct-4 staining was detected in the tissue surrounding the nodules, which served as negative control in each sample. Due to the low number of available samples, it was not possible to evaluate the influence either of gender or NSAIDs administration on Oct-4 positivity in ECS-exposed mice.
Figure 3.
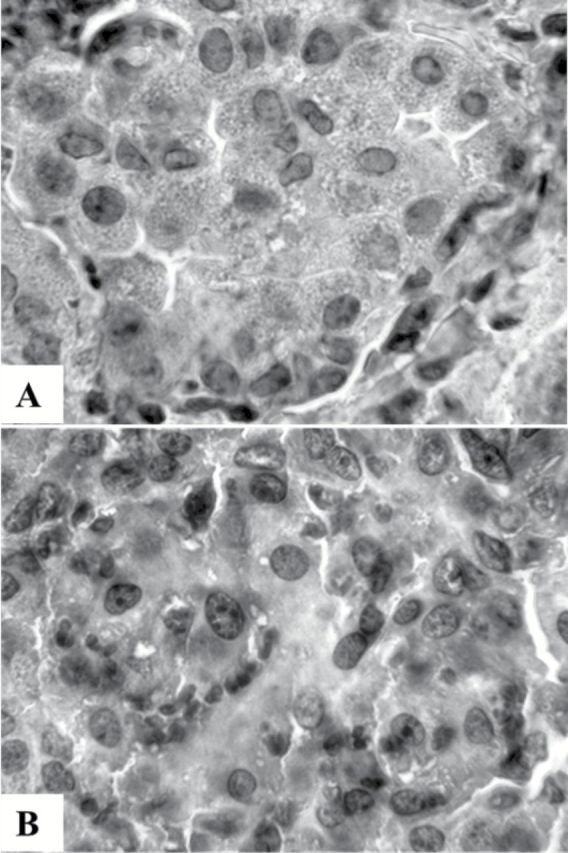
Examples of immunohistochemical detection of Oct-4 in sections of a lung hyperplastic and inflammatory nodule (A) and of an adenocarcinoma (B). See text for details. Original magnification 400×.
Discussion
The results of the present study showed that exposure of mice to ECS caused the formation of evident nucleotide modifications in the short term and induced a weak tumorigenic response in the lung in the medium term. The ECS doses used in the present experiment, accounting for an average total particulate matter of 96.5mg/m3 air, were within the ranges (70–150mg/m3 air) reported in 28 studies using the same smoking machine available for the present study (33). These doses are high but not unrealistic for human exposures. In fact, the Scientific Review Panel of the California Environmental Protection Agency has reported that the respirable particulate matter in certain entertainment venues is estimated to range from less than 15mg/m3 where smoking is prohibited up to 350mg/m3 where smoking is allowed. In the home environment, peaks up to 300mg/m3 have been found, and inside vehicles concentrations are estimated to range from about 90mg/m3 to well over 1000mg/m3 (3).
The 70-day exposure period, after which bulky DNA adducts and 8-oxo-dGuo were evaluated, covered the immediate post-natal time, during which the sudden transition from the maternal-mediated respiration to the autonomous pulmonary respiration of neonatal mice causes paraphysiological genomic and postgenomic alterations in the lung (34), followed by the lactation period until completion of weaning and then adolescence. It is known that children are especially sensitive to the respiratory effects of exposure to SHS (3), and mice exposed until weaning are more susceptible than their dams to molecular, biochemical and cytogenetic alterations induced by ECS in the respiratory tract, mainly due to oxidative stress and the resulting DNA damage and epigenetic alterations (35,36).
The tumorigenic response, in terms of surface lung tumors as proposed in the classical mouse tumor assay (32), was weak yet appreciable in A/J mice exposed to ECS. It was approximately of the same order of magnitude as the one detected by exposing adult A/J mice to ECS according to the model proposed by Hanspeter Witschi (25,26,37). Probably, one of the reasons accounting for the low yield of lung tumors in this model is that, according to the classical lung tumor assay with chemical carcinogenesis (32), only surface lung tumors are enumerated, which may underestimate the lung tumor yield in the whole organ (37).
Therefore, exposure early in life, which was found to be particularly advantageous in mice exposed to MCS (27), does not appear to increase the lung tumor yield in ECS-exposed mice. This probably depends on the young age of mice (9 months) at the time of sacrifice, since the incidence of lung tumors, both spontaneous and induced by chemical carcinogens, considerably increases with age (32). However, an important difference is that, in the Witschi’s model in adult A/J mice, only the 20% of the lung tumors are adenocarcinomas, a figure that is not increased by ECS (37), whereas in A/J mice exposed early in life the relative proportion of adenocarcinomas (56.3%) was higher than that of adenomas (43.7%), and the incidence of ECS-induced adenocarcinomas was more than twice that of spontaneous adenocarcinomas, although the difference did not reach the statistical significance threshold. The finding that exposure to ECS mainly induces lung adenocarcinomas in mice is of particular interest because this type of tumor is particularly frequent in passive smokers, as shown for instance in Japanese women exposed to ECS both at home and in the workplace (38).
We previously demonstrated that stem cell antigen-1 (Sca-1) is upregulated in the endothelial cells of the pulmonary vasculature of post-weaning CD-1 mice exposed to ECS since birth (39). In the present study, a proportion of the detected adenocarcinomas were positive for Oct-4, which suggests the presence of stem cells within the tumor mass, contrasting with the lack of detectable protein in the surrounding healthy tissue. In particular, Oct-4 was localized in the nucleus of adenocarcinomatous cells, whereas in a hyperplastic and inflammatory nodule Oct-4 was localized in the cytoplasm. In fact, the antibody used reacts both with both Oct-4A, which is localized in the nucleus as a transcription factor related to its pluripotent properties, and Oct-4B, which is localized in the cytoplasm and is related to stress response (40,41).
AsA and naproxen were given with the diet starting after weaning until the end of the experiment. Therefore, this protocol mimics an intervention in a passive smoker that, after a while, avoids further exposure to ECS. The doses of NSAIDs were selected based on a preliminary subchronic toxicity study, and they did not cause any significant bleeding of the gastric mucosa. These doses are 3–4 times higher than those used in humans for therapeutic purposes, but it should be taken into account that, due to metabolic reasons, mice eat as often as they can, and accordingly the intake of drugs with the diet is highly fractionated during the day.
Both AsA and naproxen were able to attenuate the early formation of bulky DNA adducts and oxidative DNA damage in A/J mice exposed to ECS, which is consistent with the finding that COX-2 is able to mediate the formation of etheno adducts via lipid peroxidation in cultured cells (42). Moreover, naproxen significantly attenuated the systemic genotoxic damage induced by MCS in female H mice (43). In addition, AsA inhibited the formation of DNA adducts both in the urinary bladder and kidney of mice treated with the nephrotoxic mycotoxin ochratoxin A (44), which has been shown to behave as a tumor promoter (45), and in a human bladder tumor cell line treated with 2-aminofluorene (46). Furthermore, AsA potently inhibited the induction of oxidative DNA strand-breaks in plasmid DNA, most likely through scavenging of hydroxyl radicals (47). All these studies are consistent with the conclusion that NSAIDs are able to attenuate oxidative DNA damage, and our results provide the first evidence that this mechanism also occurs in the lung of mice exposed to ECS early in life.
AsA significantly suppressed the formation of ECS-induced adenomas in female A/J mice. Naproxen decreased ECS-induced adenomas in female mice almost 4-fold, but this effect did not reach the statistical significance threshold. Interestingly, a selective inhibition of lung adenomas by both NSAIDs was also observed in female Swiss H mice exposed to MCS (43). Thus, at least in mice, the protective effects of NSAIDs towards smoke-induced lung tumors are gender-specific, and they are due to epigenetic mechanisms rather than to antigenotoxic mechanisms because inhibition of ECS-induced DNA adducts by both AsA and naproxen was of the same order of magnitude in males and females. These findings suggest a role of estrogens in smoke-related pulmonary carcinogenesis, as also supported by metabolic studies (48,49). On the other hand, they are consistent with the known antiestrogenic properties of NSAIDs, COX-2 overexpression being a critical component in aromatase-catalyzed estrogen biosynthesis (50). Therefore, the NSAIDs appear to mainly interfere with the tumor promotion process triggered by CS in mouse lung. It should be noted that inhibition of nucleotide alterations and inhibition of lung tumors do not necessarily correlate, also because tumors probably originate from stem cells, whereas nucleotide alterations may affect other pulmonary cell types as well (10).
In parallel with the present study, we are evaluating the modulation of microRNA expression in the lung of the same mice. Preliminary results highlight not only the well-known ability of NSAIDs to regulate COX activity but also their involvement in a variety of cell functions (A. Izzotti et al., in preparation). Tobacco smoke is known to trigger a cascade of events involving both genotoxic and epigenetic mechanisms, such as induction of intracellular signaling and modulation of gap junctional intercellular communications, in the multistage process of lung carcinogenesis (5,35,51). On the whole, our results show that AsA and naproxen can interfere with cigarette smoke pulmonary carcinogenesis both via antigenotoxic mechanisms, as documented by inhibition of DNA adducts and oxidative DNA damage in the lung, at a time when no histopathological alterations are detectable in the lung yet, and via epigenetic mechanisms, as documented by modulation of microRNA expression and by the antiestrogenic activity resulting in inhibition of lung tumors in females.
Funding
United States National Cancer Institute (Contract #HHSN- 261201200015I).
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- AsA
aspirin or acetylsalicylic acid
- COX
cyclooxygenase
- ECS
environmental cigarette smoke
- ETS
environmental tobacco smoke
- MCS
mainstream cigarette smoke
- NSAID
nonsteroidal anti-inflammatory drug
- SHS
second-hand smoke
- SCS
sidestream cigarette smoke
References
- 1. Hang B. (2010) Formation and repair of tobacco carcinogen-derived bulky DNA adducts. J. Nucleic Acids, 2010, 709521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. De Flora S., et al. (2014) Rationale and approaches to the prevention of smoking-related diseases: overview of recent studies on chemoprevention of smoking-induced tumors in rodent models. J. Environ. Sci. Health. C Environ. Carcinog. Ecotoxicol. Rev., 32, 105–120. [DOI] [PubMed] [Google Scholar]
- 3. California Environmental Protection Agency, 2005. Proposed Identification of Environmental Tobacco Smoke as a Toxic Air contaminant, State of California. www.arb.ca.gov/regact/ets2006/app3partb.pdf.
- 4. International Agency for Research on Cancer (2004) Tobacco smoke and involuntary smoking. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 83 IARC, Lyon, France. [PMC free article] [PubMed] [Google Scholar]
- 5. Besaratinia A., et al. (2008) Second-hand smoke and human lung cancer. Lancet. Oncol., 9, 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schick S., et al. (2005) Philip Morris toxicological experiments with fresh sidestream smoke: more toxic than mainstream smoke. Tob. Control, 14, 396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Homa DM., et al. (2015) Vital signs: disparities in nonsmokers’ exposure to secondhand smoke—United States, 1999–2012. MMWR Morb. Mortal. WklyRep., 64, 103–108. [PMC free article] [PubMed] [Google Scholar]
- 8. Park S., et al. (2014) Attributable fraction of tobacco smoking on cancer using population-based nationwide cancer incidence and mortality data in Korea. BMC Cancer, 14, 406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takahashi H., et al. (2010) Tobacco smoke promotes lung tumorigenesis by triggering IKKbeta- and JNK1-dependent inflammation. Cancer Cell, 17, 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Trosko J.E., et al. (2006) Adult stem cell theory of the multi-stage, multi-mechanism theory of carcinogenesis: role of inflammation on the promotion of initiated stem cells. Contrib. Microbiol., 13, 45–65. [DOI] [PubMed] [Google Scholar]
- 11. Smith C.J., et al. (2006) Perspectives on pulmonary inflammation and lung cancer risk in cigarette smokers. Inhal. Toxicol., 18, 667–677. [DOI] [PubMed] [Google Scholar]
- 12. Mazhar D., et al. (2005) COX and cancer. QJM, 98, 711–718. [DOI] [PubMed] [Google Scholar]
- 13. Cuzick J., et al. (2009) Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. Lancet Oncol., 10, 501–507. [DOI] [PubMed] [Google Scholar]
- 14. Bosetti C., et al. (2009) Aspirin and cancer risk: a summary review to 2007. Recent Results Cancer Res., 181, 231–251. [DOI] [PubMed] [Google Scholar]
- 15. Oh S.W., et al. (2011) Aspirin use and risk for lung cancer: a meta-analysis. Ann. Oncol., 22, 2456–2465. [DOI] [PubMed] [Google Scholar]
- 16. McCormack V.A., et al. (2011) Aspirin and NSAID use and lung cancer risk: a pooled analysis in the International Lung Cancer Consortium (ILCCO). Cancer Causes Control, 22, 1709–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu J., et al. (2012) Meta-analysis on the association between nonsteroidal anti-inflammatory drug use and lung cancer risk. Clin. Lung Cancer, 13, 44–51. [DOI] [PubMed] [Google Scholar]
- 18. Rothwell P.M., et al. (2011) Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet, 377, 31–41. [DOI] [PubMed] [Google Scholar]
- 19. Saini R.K., et al. (2009) Chemopreventive effect of nonsteroidal anti-inflammatory drugs on 9,10-dimethylbenz[a]anthracene-induced lung carcinogenesis in mice. Oncol. Res., 17, 505–518. [DOI] [PubMed] [Google Scholar]
- 20. Jalbert G., et al. (1992) Effects of NSAIDs on NNK-induced pulmonary and gastric tumorigenesis in A/J mice. Cancer Lett., 66, 21–28. [DOI] [PubMed] [Google Scholar]
- 21. Lubet R.A., et al. et al. (2010) Screening agents for preventive efficacy in a bladder cancer model: study design, end points, and gefitinib and naproxen efficacy. J. Urol., 183, 1598–1603. [DOI] [PubMed] [Google Scholar]
- 22. Steele V.E., et al. et al. (2009) Chemopreventive efficacy of naproxen and nitric oxide-naproxen in rodent models of colon, urinary bladder, and mammary cancers. Cancer Prev. Res. (Phila)., 2, 951–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McCormick D.L., et al. et al. (2010) Overexpression of cyclooxygenase-2 in rat oral cancers and prevention of oral carcinogenesis in rats by selective and nonselective COX inhibitors. Cancer Prev. Res. (Phila)., 3, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Suh N., et al. et al. (2011) Combination of atorvastatin with sulindac or naproxen profoundly inhibits colonic adenocarcinomas by suppressing the p65/β-catenin/cyclin D1 signaling pathway in rats. Cancer Prev. Res. (Phila)., 4, 1895–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Witschi H., et al. et al. (1997) The carcinogenicity of environmental tobacco smoke. Carcinogenesis, 18, 575–586. [DOI] [PubMed] [Google Scholar]
- 26. D’Agostini F., et al. et al. (2001) Pilot studies evaluating the lung tumor yield in cigarette smoke-exposed mice. Int. J. Oncol., 18, 607–615. [DOI] [PubMed] [Google Scholar]
- 27. Balansky R., et al. et al. (2012) Differential carcinogenesis of cigarette smoke in mice exposed either transplacentally, early in life or in adulthood. Int. J. Cancer, 130, 1001–1010. [DOI] [PubMed] [Google Scholar]
- 28. Witschi H., et al. et al. (1997) The carcinogenic potential of the gas phase of environmental tobacco smoke. Carcinogenesis, 18, 2035–2042. [DOI] [PubMed] [Google Scholar]
- 29. Tai M.H., et al. et al. (2005) Oct4 expression in adult human stem cells: evidence in support of the stem cell theory of carcinogenesis. Carcinogenesis, 26, 495–502. [DOI] [PubMed] [Google Scholar]
- 30. Izzotti A., et al. et al. (2001) Modulation of biomarkers by chemopreventive agents in smoke-exposed rats. Cancer Res., 61, 2472–2479. [PubMed] [Google Scholar]
- 31. Izzotti A., et al. et al. (1998) In vitro inhibition by N-acetylcysteine of oxidative DNA modifications detected by 32P postlabeling. Free Radic. Res., 28, 165–178. [DOI] [PubMed] [Google Scholar]
- 32. Shimkin M.B., et al. et al. (1975) Lung tumors in mice: application to carcinogenesis bioassay. Adv. Cancer Res., 21, 1–58. [DOI] [PubMed] [Google Scholar]
- 33. Leberl M., et al. et al. (2013) Tobacco smoke induced COPD/emphysema in the animal model - are we all on the same page? Front Physiol., 4, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Izzotti A., et al. et al. (2003) Birth-related genomic and transcriptional changes in mouse lung. Modulation by transplacental N-acetylcysteine. Mutat. Res., 544, 441–449. [DOI] [PubMed] [Google Scholar]
- 35. Micale R.T., et al. et al. (2013) Oxidative stress in the lung of mice exposed to cigarette smoke either early in life or in adulthood. Arch. Toxicol., 87, 915–918. [DOI] [PubMed] [Google Scholar]
- 36. Upham B.L., et al. et al. (2009) Oxidative-dependent integration of signal transduction with intercellular gap junctional communication in the control of gene expression. Antioxid. Redox Signal., 11, 297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Witschi H. (2005) The complexities of an apparently simple lung tumor model: The A/J mouse. Exp. Toxicol. Pathol., 57 ( suppl. 1, 171–181. [DOI] [PubMed] [Google Scholar]
- 38. Kurahashi N., et al. et al. (2008) Passive smoking and lung cancer in Japanese non-smoking women: a prospective study. Int. J. Cancer, 122, 653–657. [DOI] [PubMed] [Google Scholar]
- 39. Izzotti A., et al. et al. (2009) Upregulation of stem cell antigen-1 in the lung of neonatal mice exposed to environmental cigarette smoke. Oncol. Rep., 22, 469–474. [DOI] [PubMed] [Google Scholar]
- 40. Lee J., et al. et al. (2006) The human OCT-4 isoforms differ in their ability to confer self-renewal. J. Biol. Chem., 281, 33554–33565. [DOI] [PubMed] [Google Scholar]
- 41. Wang X., et al. et al. (2009) Alternative translation of OCT4 by an internal ribosome entry site and its novel function in stress response. Stem Cells, 27, 1265–1275. [DOI] [PubMed] [Google Scholar]
- 42. Lee S.H., et al. et al. (2005) Cyclooxygenase-2-mediated DNA damage. J. Biol. Chem., 280, 28337–28346. [DOI] [PubMed] [Google Scholar]
- 43. Balansky R, et al. (2015) Selective inhibition by aspirin and naproxen of mainstream cigarette smoke-induced genotoxicity and lung tumors in female mice. Arch. Toxicol. [DOI] [PubMed] [Google Scholar]
- 44. Horvath A., et al. et al. (2002) Determination of the epigenetic effects of ochratoxin in a human kidney and a rat liver epithelial cell line. Toxicon, 40, 273–282. [DOI] [PubMed] [Google Scholar]
- 45. Obrecht-Pflumio S., et al. et al. (1996) Protection by indomethacin and aspirin against genotoxicity of ochratoxin A, particularly in the urinary bladder and kidney. Arch. Toxicol., 70, 244–248. [DOI] [PubMed] [Google Scholar]
- 46. Yeh C.C., et al. et al. (1999) Effects of aspirin on arylamine N -acetyltransferase activity and DNA adducts in human bladder tumour cells. J. Appl. Toxicol., 19, 389–394. [DOI] [PubMed] [Google Scholar]
- 47. Hsu C.S., et al. et al. (2002) Aspirin potently inhibits oxidative DNA strand breaks: implications for cancer chemoprevention. Biochem. Biophys. Res. Commun., 293, 705–709. [DOI] [PubMed] [Google Scholar]
- 48. Meireles S.I., et al. et al. (2010) Early changes in gene expression induced by tobacco smoke: Evidence for the importance of estrogen within lung tissue. Cancer Prev. Res. (Phila)., 3, 707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Peng J., et al. et al. (2013) Estrogen metabolism within the lung and its modulation by tobacco smoke. Carcinogenesis, 34, 909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Terry M.B., et al. et al. (2004) Association of frequency and duration of aspirin use and hormone receptor status with breast cancer risk. JAMA, 291, 2433–2440. [DOI] [PubMed] [Google Scholar]
- 51. Upham B., et al. et al. (2008) Inhibition of intercellular signaling, a tumor promotion event, by a cigarette abundant PAH, depends on phosphatidylcholine-specific phospholipase C. Cancer Sci., 99, 696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]