

Abstract
Fibroblasts are the most abundant cellular components of connective tissue. They possess phenotypical heterogenicity and may be present in the form of smooth muscle cells or myofibroblasts (MFs). MFs are spindle-shaped cells with stress fibres and well-developed fibronexus, and they display α-smooth muscle actin immunohistochemically and smooth-muscle myofilaments ultrastructurally. MFs play a crucial role in physiological and pathological processes. Derived from various sources, they play pivotal roles not only by synthesizing and producing extracellular matrix components, such as other connective tissue cells, but also are involved in force production. In the tissue remodelling phase of wound closure, integrin-mediated interactions between MFs and type I collagen result in scar tissue formation. The tumour stroma in oral cancer actively recruits various cell types into the tumour mass, where they act as different sources of MFs. This article reviews the importance of MFs and its role in pathological processes such as wound healing, odontogenic cysts and tumours, salivary gland tumours, oral preneoplasia, and oral squamous cell carcinoma. Research oriented on blocking the transdifferentiation of fibroblasts into MFs can facilitate the development of noninvasive therapeutic strategies for the treatment of fibrosis and/or cancer.
Keywords: Myofibroblasts, Neoplasm, Fibroblasts, Precancerous lesions, Carcinoma-associated fibroblasts, Precancerous conditions
Core tip: Myofibroblast (MFs) are spindle-shaped cells consisting α-smooth muscle actin myofilament. They have a multicellular origin. MFs of the oral cavity have more contractile ability than dermal fibroblasts in physiologic wound healing. Recently, carcinoma-associated fibroblasts (CAFs) have received considerable attention because of their role in carcinogenesis. Mainly, transforming growth factor-β released from oral cancer cells is responsible for transforming fibroblasts into CAFs, which leads to tumour progression. However, the role of MFs in oral leukoplakia and oral submucous fibrosis is not completely understood. Understanding the implications of therapeutic approaches for the transdifferentiation of fibroblasts into MFs at different stages of carcinogenesis will facilitate in developing a treatment plan.
INTRODUCTION
Oral mucosa comprises stratified squamous epithelium, and its underlying connective tissue harbours various mesenchymal cells such as fibroblasts, endothelial cells, and pericytes[1,2]. These cells and their extracellular matrix (ECM) play pivotal roles in cell differentiation and proliferation during tissue development, wound healing processes, and pathological alterations[2,3].
Fibroblasts, the most abundant cellular components of connective tissue, are phenotypically heterogenous and are present in the form of smooth muscle cells or myofibroblasts (MFs)[4]. Gibbiani was the first to observe fibroblasts under an electron microscope and coined the term “myofibroblast”[5].
MFs may be defined as large spindle-shaped cells with stress fibres and well-developed fibronexus. They display α-smooth muscle actin (α-SMA) immunohistochemically and smooth muscle myofilaments ultrastructurally[6,7]. Therefore, MFs, which are found in normal skin tissue; pulmonary septa; and periodontal ligament[8], are unique[4] and owing to their location, they are called as “juxtaparenchymal cells”[4].
MFs show functional heterogenecity through various mechanisms. During mesenchymal-epithelial interactions, they play a pivotal role in organogenesis and morphogenesis. They secrete the components of the ECM and basement membrane[4]. Their myomechanical function is crucial during wound healing[9]. Their role in the carcinogenic process is well-recognised[10]. This article describes the cascade of events pertaining to the role of MFs in wound healing, oral squamous cell carcinoma (OSCC), odontogenic cysts and tumours, and salivary gland tumours.
DISCUSSION
Genesis and identification of MFs
MFs are multicellular in origin (Figure 1). Local fibroblasts, pericytes, endothelial cells, circulating hematopoietic precursor cells, and fibrocytes are various sources of MFs. They are also derived from bone marrow and are known as bone marrow-derived MFs[11]. The main factor responsible for the transition (activation) of fibroblasts to MFs is transforming growth factor-β1 (TGF-β1)[12]. A previous study demonstrated that connective tissue factors (thrombin and endothelin) and platelet-derived growth factor (PDGF) are also responsible for the differentiation of fibroblasts into MFs[11]. According to Werner et al[13], keratinocytes play an important role by releasing interleukin-1, activin, and TGF-β1 during the differentiation of fibroblasts into promyofibroblasts into MFs. Thus, three local events are required to generate α-SMA-positive differentiated MFs: (1) the accumulation of biologically active TGF-β1, which enhances the assembly of stress fibres and the formation of fibronexus adhesion complexes; (2) the accumulation of an ED-A splice variant of fibronectin, which are specialised ECM proteins; and (3) the mechanical properties of the ECM and cell remodelling activities, which are responsible for high extracellular stress[9-11]. Carthy et al[14] demonstrated that Wnt3a promotes MF-like phenotype formation in cultured fibroblasts. Based on MF gene expression patterns, various studies have revealed that distinct subtypes of fibroblasts exist at different sites of the body[14,15]. Nevertheless, an inactive JunD, which protects the cell against oxidative stress, promotes MF differentiation[16]. Their presence at a site may either be pre-existing, or they may originate denovo from the surrounding subpopulation[15].
Figure 1.
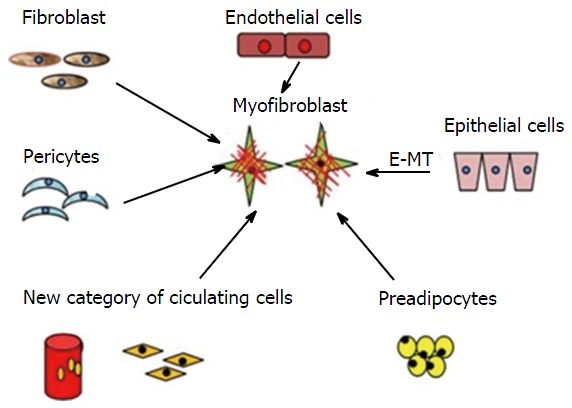
Origin of myofibroblasts-multicellular origin. Multicellular origin of myofibroblasts: Fibroblast, pericytes, endothelial cells, circulating hematopoietic precursor cells and fibrocytes can transform into myofibroblasts. E-MT: Epithelial-mesenchymal transition.
MFs possess several distinguishing morphologic features[17] and are characterised by the highly contractile α-SMA apparatus[9], which is the most significant marker of myofibroblastic cells. They may express smooth muscle myosin heavy chains and desmin[18]. They also express caldesmon, SM22, and tropomyosin. Under the electron microscope, MFs are large cells with abundant rough endoplasmic reticulum and fibronexuses[5], prominent cytoplasmic actin microfilaments (stress fibres), nonmuscle myosin, and vimentin[17], which are connected to each other by adherens and gap junctions[4,9].
Recently, the 4Ig isoform of the protein palladin in stress fibres was proposed as a new marker of MF differentiation[19]. Conversely, another study suggested that interferon-γ reduces α-SMA expression in smooth muscle cells[18].
Role of MFs in wound contraction
The breach of the epithelial layer is followed by changes that occur in the underlying connective tissue resulting in the loss of tissue homeostasis[1]. Normal wound healing is a well-known phenomenon. It involves a sequence of events including inflammation, proliferation, and tissue remodelling[18,20,21]. Wound closure involves connective tissue deposition, epithelization, and contraction[22] (Figure 2).
Figure 2.
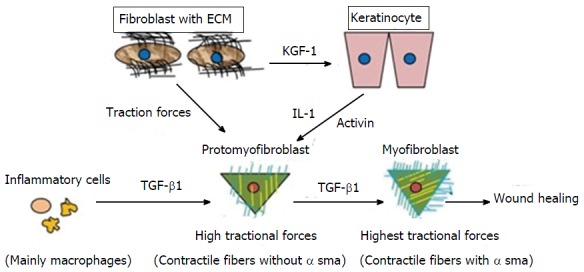
Differentiation of promyofibroblast to myofibroblast. By getting stimulus from various cytokines profibroblasts (promyofibroblasts) transforms into myofibroblasts by expressingα-smooth muscle actin. TGF-β1: Transforming growth factor-β1; ECM: Extracellular matrix; IL-1: Interleukin-1; KGF-1: Keratinocyte growth factor-1.
Wound contraction is mainly carried out by MF, a specialised contractile fibroblast[22]. Initially, small tractional forces exerted by the ECM facilitate the formation of protofibroblasts, which are composed of cytoplasmic actin components and devoid of the contractile apparatus and α-SMA. Protofibroblasts migrate to the wound site by acquiring a migratory phenotype at the fibronectin-fibrin wound interface. At the site, protofibroblasts generate comparably small traction forces. Cytokines, such as PDGF, granulocyte-macrophage colony-stimulating factor (GM-CSF), heparin, integrin[20], and TGF-β, and existing tractional forces stimulate protofibroblast differentiation through α-SMA expression, leading to their transformation into MFs. A study conducted on the role of tenacin in wound contraction demonstrated that increased tenacin expression correlated with MF differentiation[23]. However, interferon-γ, a basic fibroblast growth factor (bFGF), prostaglandin E2, and high cell density inhibit the differentiation of protofibroblasts to MFs[20]. After activation, MFs initiate the synthesis of a new collagen-containing matrix that consists proteoglycans and glycosaminoglycans (highly hydrated molecules)[1,22,24]. They produce tractional forces at the margins for wound contraction, which is known as tractional remodelling[22].
MFs generate forces in two ways. Initially, actin filaments present within the cell form a fibronexus by connecting intracellular actin and extracellular fibronectin fibrils using integrins. Integrins mediate the reorganisation and contraction of collagen matrices with the help of fibroblasts[20]. A study on α1 integrin knockout mice revealed impaired wound healing[20]. The assembly formed by MF with integrin and the actin filament is responsible for the “mechano-transduction system”, which produces a high degree of tractional forces[9]. Later, MFs connect to each other through gap junctions to form a “multicellular contractile unit”. They again exert a force on the ECM by implicating the use of this unit[9,22]. Both mechanisms exert a high level of tractional forces for wound closure.
After complete wound closure and re-epithelization, the number of MFs decreases either by reverting to the quiescent form or by undergoing apoptosis[1,9,25]. In the tissue remodelling phase, integrin-mediated interactions between MFs and collagen I results in scar tissue formation[20].
Wound healing in the oral cavity essentially occurs without scarring and is faster than skin healing[20]. Fibroblasts in the oral mesenchyme possess a unique phenotypical character by constitutively expressing elevated α-SMA levels, along with a higher capacity to contract collagen gel and a higher replicative potential than dermal fibroblasts[17], ultimately leading to a “scar-free” healing process. Factors, such as epidermal growth factor, vascular endothelial growth factor, bFGF, and insulin-like growth factor, present in saliva and crevicular fluid are responsible for wound healing in the oral cavity[20].
MFs in oral leukoplakia
Leukoplakia is the most common potentially malignant disorder of the oral mucosa[26]. Studies have been conducted on various histopathological grades of oral leukoplakia (OL), but failed to establish a conclusive relationship between OL and MFs. MFs were not found in the stroma under dysplastic epithelium[16]. Myofibroblastic differentiation depends on the following factors: (1) Neoplastic microenvironment, which releases various growth factors[27]; (2) Genetically altered epithelium (carcinomatous epithelium), which is responsible for the inductive effect on the underlying stroma[28]; and (3) Epithelial-mesenchymal interaction (EMI), which plays a role in “epithelial invasion”[16,19].
However, these factors were absent in various grades of epithelial dysplasia[16,19,27,29]. A study conducted by de-Assis et al[29], demonstrated that OL was not associated with MFs because MFs were not found in any of the samples. Therefore, it was hypothesised that myofibroblastic differentiation was entirely dependant on oral carcinoma development and, simultaneously, on the contact of cancer cells with stromal cells achieved by invading the epithelial cells[16].
MFs in oral submucous fibrosis
The most chronic and functionally hampering condition of oral cavity is oral submucous fibrosis (OSMF). OSMF is an abnormal healing process in response to the chronic mechanical and chemical irritation caused by chewing areca nuts[30]. The cellular mediator responsible for fibrosis is MF, which serves as collagen-producing cells when activated[31]. Continuous MF presence stimulates an abnormal repair mechanism, leading to excessive contraction and ECM secretion, subsequently causing fibrosis[30]. It was proposed that in fibrosis, MFs acquire an immune-privileged cell phenotype, which helps them to evade apoptosis and allows their uninterrupted accumulation[31]. A study suggested that OSMF could represent failed wound healing after chronic and sustained injury. Studies have proposed that fibrosis in OSMF could result from a hypersensitivity response caused by arecoline and a juxta-epithelial inflammatory response, which initiates a defective inflammatory response, activates fibroblasts, and culminates in fibrosis[30,32]. In addition, MFs could be used as potential markers for evaluating disease severity because a progressive increase in MFs from the early to the advanced stages was observed[30].
The malignant transformation rate of OSMF ranges from 7% to 13%[33]. Dyavanagoudar[33] hypothesised that precancerous epithelial cells of OSMF acquire multiple genetic mutations in mesenchymal-epithelial crosstalk, and the associated stroma becomes activated and expresses SMA markers. These cells express ECM proteins and growth factors. These factors enhance and support tumour cell survival in an autocrine and paracrine manner, respectively. In contrary, Moutasim et al[34] demonstrated that αvß6 was markedly upregulated in OSMF by oral keratinocytes in an “in vitro” study. The results of the study also revealed that arecoline (the major alkaloid of areca nut) upregulated keratinocyte αvß6 expression, which induced oral fibroblasts to transdifferentiate into MFs and resulted in the upregulation of genes associated with tissue fibrosis (Figure 3). Blocking these specific integrins can develop novel therapies for such fibrotic conditions[35].
Figure 3.
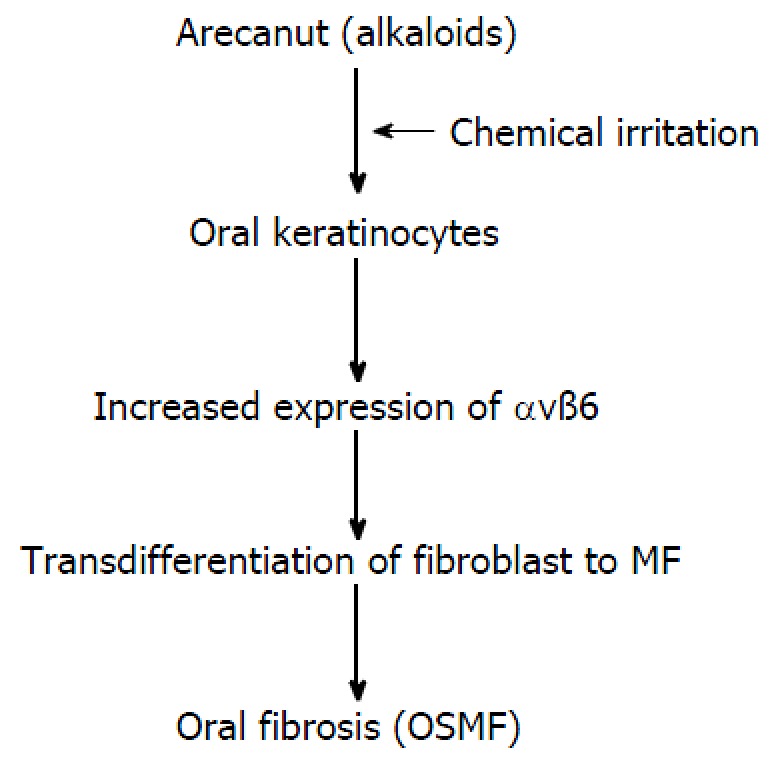
Transdifferentiation of fibroblasts into myofibroblasts. Major alkaloid of areca nuts, up-regulates keratinocyte αvß6 expression which induced transdifferentiation of oral fibroblasts into MF. OSMF: Oral submucous fibrosis.
MFs in OSCC
Stromal changes in wound healing and tumourogenesis depend on epithelial homeostasis[1]. Changes in epithelial homeostasis lead to changes in the underlying connective tissue stroma. This stroma is called reactive or desmoplastic stroma. Reactive stroma plays a significant role in the growth and progression of carcinoma because malignant epithelial cells require support from the surrounding stroma to promote tumorigenic progression[3]. Carcinoma-associated fibroblasts (CAFs) or tumour-associated fibroblasts or MFs are found in considerable quantities in such stroma[36] (Figure 4).
Figure 4.
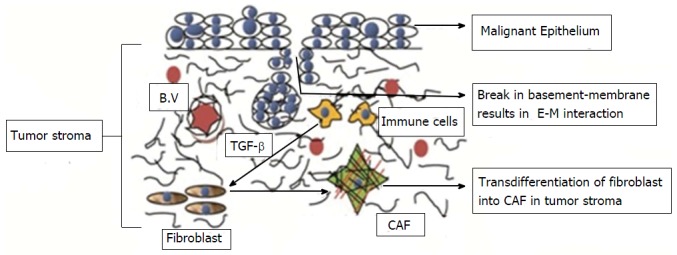
Transdifferentiation of fibroblast into carcinoma associated fibroblast in oral squamous cell carcinoma. Mutual paracrine effect between oral cancer cells and normal fibroblasts is responsible for transdifferentiation of the latter into malignant fibroblasts. CAF: Carcinoma associated fibroblast; TGF-β: Transforming growth factor-β; B.V: Blood Vessels; E-M: Epithelial-mesenchymal.
CAF origin is controversial. A study demonstrated that CAF originate from cancer stem cells, which are a small subpopulation of the tumour stroma[37]. Another study demonstrated that 60% CAF originate from bone marrow-derived mesenchymal cells[36]. Some additional sources may be resident fibroblasts, endothelial cells, pericytes, smooth muscle cells, and preadipocytes. Studies have demonstrated that the mutual paracrine effects between oral cancer cells and normal fibroblasts are responsible for the transdifferentiation of normal fibroblasts into malignant fibroblasts. CAF genesis is also related to an EMI in the stromal population or a possible transdifferentiation of the malignant epithelial cells into MFs during oral carcinogenesis[1,19]. The tumour stroma continues to remodel itself during tumour progression and actively recruits various cell types into the tumour mass where they act as different sources of MFs[3]. TGF-β is the main factor responsible for fibroblastic differentiation leading to activated tumour MFs. It is a main mediator found in the saliva and is expressed by cancer cells. A study demonstrated that the tension exerted in the tumour stroma is also responsible for the transformation of fibroblasts into MFs[1]. Keratinocytes also seem to be responsible for forming tumour-associated fibroblasts[33].
Morphologically tumour fibroblasts differ from normal MFs in their abundant rough endoplasmic reticulum, Golgi apparatus, fibronectin fibrils, and fibronexuses on the cell surface[5]. They also differ in their contractile property, MFs because they can exert more contractile force which is responsible for stiffness in the advanced stages of the neoplasm[1]. MFs secrete several enzymes such as stromelysins and matrix metalloproteinases (MMPs- 1, 2, 3, 9, 13, and 14)[1], which cause ECM degradation. They also release different growth factors such as PDGF, bFGF, keratinocyte growth factor, stem cell factor, epidermal growth factor, GM-CSF, and other cytokines[25]. They also secrete matricellular proteins including CCN2; collagens; tenascins C and FN; and elastins[1]. MFs promote tumour cell migration on tenacin[38]. Hence, MFs play an important role in tumour progression by invading the tumour stroma and consequently remodelling the ECM by forming more desmoplastic responses[25,38]. Most importantly, they secrete fibroblast-associated proteins (FAP). The loss of FAP is associated with inhibition of tumour cell progression and decrease in MF quantity and blood vessel density in the tumour[16].
MFs function as “sentinel cells” by acting as immunoregulatory cells in the stroma, by reducing the physical contact between cancer and immune cells, which is imperative for cancer cell destruction[25].
Several MFs in the tumour stroma are often associated with high-grade malignancies. They promote tumour progression through “neo-angiogenesis” by releasing growth factors, such as fibroblast growth factor-2[3]. A study demonstrated that the most migratory and invasive behaviour of tumour cells correlated with the presence of MFs at the invasive tumour front in OSCC because this precedes the invasive stage of the cancer[39,40]. In addition, abundant MFs in the tumour stroma correlates with a higher tumour incidence, specifically in patients aged below 60 years[19].
MFs are present in the OSCC stroma in two dominant patterns, spindle pattern (MFs are arranged in rows with a few cells around the neoplastic islands) and network pattern (several abundant layers of MFs around the neoplastic islands). The network pattern fibroblasts are exceptionally abundant and occupy almost the entire tumour stroma, whereas the spindle pattern includes spindle cells that are located at the periphery in 1-3 concentric layers[16]. Studies conducted by Seifi et al[39] and Kawashiri et al[41] have demonstrated that the presence of MFs and the arrangement of MFs in the tumour stroma play a role in invasion. They observed that MFs arranged in the network pattern caused tumour invasion and not those arranged in the spindle pattern. They also noted that tumour desmoplasia is associated with aggressive cancer.
Functional similarities and differences between wound healing and tumorigenesis
Morphological similarities exist between the tumour stroma and granulation tissue following wound healing. A wound is disruption of an anatomical structure, such as an epithelial membrane, and healing is the restoration of structure and function[41] (Figure 5).
Figure 5.
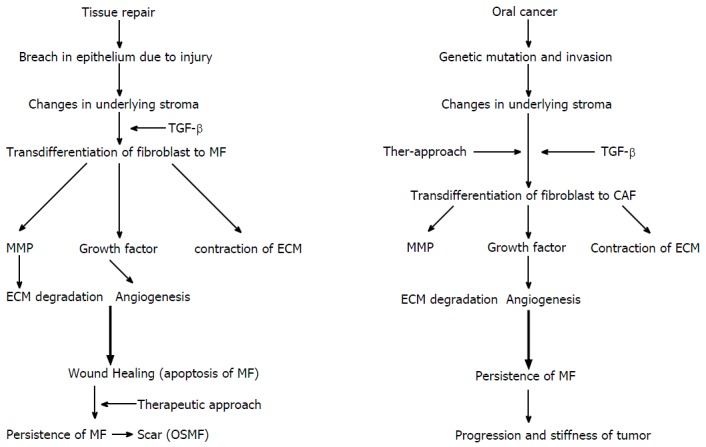
Functional similarities and differences between wound healing and tumerogenesis. Wound healing and tumour progression phenomenon in relation with MF. TGF-β: Transforming growth factor-β; ECM: Extracellular matrix; CAF: Carcinoma-associated fibroblasts; OSMF: Oral submucous fibrosis; MMP: Matrix metalloproteinase; MF: Myofibroblast.
Wound healing and tumour progression are loosely related in terms of MFs[1]. In wound healing, changes in the underlying stroma occur with a breach in the normal lining epithelium. The appearance of a break causes changes in the underlying connective tissue stroma. Inflammatory cells release TGF-β1, which stimulate the differentiation of fibroblast into proto-myofibroblast and finally into MFs[9]. MFs later release MMPs and growth factors, which lead to matrix degradation and new blood vessel formation (angiogenesis), respectively. MFs also cause wound contraction and undergo apoptosis. However, in the last stage of wound healing, a defect in the apoptosis of MF and the persistence of MF results in an hypertrophic scar tissue[42].
A tumorigenic process involves acquiring multiple genetic mutations. In the normal cell ecosystem, a continuous cross-talk between epithelial cells and stroma occurs. Epithelial cells acquiring neoplastic properties result in altered stromal compartment. Neoplastic cells release TGF-β1 and PDGF, which result in the emergence of CAF[3,10,43]. The petite concentration of TGF-β1 increases (femtomolar to picomolar) as the fibroblasts approach the cancer cells and transdifferentiate into MFs[43]. CAF release growth factors and cause angiogenesis similar to wound healing[10]. However, in carcinogenesis, MMPs (1, 2, 9 and 13) liberated by CAF are approximately double than the normal fibroblasts[44]. These factors together promote tumour progression and invasion[43].
Thus, the loss of epithelial homeostasis is a common trigger for the stromal reaction against tumours and for normal wound healing[1]. In both processes, growth factors and MMPs liberated by MFs are responsible for progression.
In wound healing, MFs transiently acquire the phenotype[1], which persists during fibrosis in the tumour environment because of an imbalance between the apoptosis and the proliferation of transformed cells leading to disease progression[40].
MFs in odontogenic cyst and tumour
Smith was the first to question the relationship between MFs and the aggressive behaviour of a neoplasm[45]. Rothouse reviewed the ultrastructural features of ameloblastoma and found that the stromal component of an ameloblastoma is composed of MFs along with associated collagen and basal lamina material[46]. Vered et al[43] studied the number of MFs in a solid ameloblastoma and parakeratinised odontogenic keratocyst to find that the mean number of MFs in the odontogenic lesions was the same as that in OSCC[43]. The number of MFs was high in these lesions, which were responsible for aggressive and invasive behaviour. They explained that the epithelium of parakeratinised odontogenic keratocyst and solid ameloblastoma behave like the OSCC epithelium by releasing more TGF-β1 and simultaneously modulating stromal MFs, making the tumour more aggressive and invasive[43]. This explains the biologic behaviour of these odontogenic lesions. Another study demonstrated the invasive behaviour of a recurrent infiltrative ameloblastoma because of a high number of MFs in stroma[46]. Thus, the presence and the frequency of MFs in the stroma possibly determine the biologic behaviour of the lesions.
Odontogenic myxoma, which is considered to be a slow-growing invasive tumour, showed abundant MFs in its stroma associated with its invasive behaviour. Effiom et al[47] hypothesised that MFs present in the stroma modify the ECM by releasing various cytokines, which are responsible for epithelial invasion.
Various authors have studied the relationship between different cysts and MFs in their stroma. The increased MFs in the stroma directly related to a more aggressive behaviour of the odontogenic cysts[48], radicular cysts[49], dentigerous cysts[49], and keratocystic odontogenic tumours[49]. The results indicated that MFs were present in a decreasing order in the cyst stroma of keratocystic odontogenic tumours, dentigerous cysts, and radicular cysts. However, the results were not statistically significant. Some authors suggest that the presence of MFs in the cyst wall might be part of a homeostatic response to the distension caused by cyst enlargement[50].
MFs in salivary glands
The tumorigenic role of MFs in the salivary gland is controversial. Studies conducted on MFs in the tumour stroma of several salivary gland tumours, such as carcinoma ex pleomorphic adenoma, mucoepidermoid carcinoma (MEC), and adenocarcinoma, revealed the presence of tumour MFs at the tumour front in all the malignant lesions, which further correlated with the invasiveness of the cancer cells[40]. Gupta et al[51] demonstrated that a high MF density in adenoid cystic carcinoma and MEC at tumour front, contributing to the aggressiveness of the lesions, whereas a moderate MF density was observed in polymorphous low grade adenocarcinoma. Conversely, Soma et al[52] demonstrated that MFs inhibit tumour growth because they found more MF presence at the tumour border in benign lesions (pleomorphic adenoma) than that in malignant lesions. Thus, they postulated that the MFs present at the periphery of benign lesions were responsible for containing the tumour, whereas the absence of MFs in malignant tumours was responsible for the progression of the tumour cells owing to no containment of the tumour[52]. Sobral et al[53] reported similar findings. They demonstrated that MF was higher in low grade MEC than the intermediate and high grade suggesting that the inflammatory infiltrate in the tumour stroma causes the cessation of MF differentiation. With the increasing grades of MEC, the inflammatory infiltration increased, and therefore, the number of MF decreased, leading to a poor prognosis[53,54].
The role of MFs in the pathogenesis of mucocele or chronic sialadenitis was not found. It was postulated that MFs may play a “supportive muscular role” around the cystic wall of the mucous retaining cyst and distended excretory duct[55].
CONCLUSION
MFs are known to contribute to the biological behaviour of various lesions. They actively participate in diseases characterised by tissue fibrosis because of their ability to secrete and degrade ECM components. MFs also play a role in ECM remodelling induced by tumour cells. It, thus, creates a permissive or suitable environment for tumour progression. Therefore, MFs are unique contractile cells that play a role in not only growth, development, and wound healing but also in inflammation, fibrosis, and tumour progression. Benefitting from their advantages in physiologic processes and blocking the processes leading to the causation and progression of a disease are the need of the hour. Additional knowledge and clinical studies involving this unique cell may provide us with an effective target for cancer therapy. Limited studies on oral lesions calls for further research for understanding the molecular mechanisms of MFs in the progression of these lesions.
Footnotes
Conflict-of-interest statement: The author confirm that the manuscript is original and has not been published elsewhere. Nor is it sent to any other journals for consideration. In addition, the authors report no conflict of interest, and the manuscript in its submitted form has been read and approved by all authors.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: April 11, 2015
First decision: June 4, 2015
Article in press: September 8, 2015
P- Reviewer: Gong YW, Gürel P
S- Editor: Ji FF L- Editor: A E- Editor: Li D
References
- 1.Otranto M, Sarrazy V, Bonté F, Hinz B, Gabbiani G, Desmoulière A. The role of the myofibroblast in tumor stroma remodeling. Cell Adh Migr. 2012;6:203–219. doi: 10.4161/cam.20377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lekic PC, Pender N, McCulloch CA. Is fibroblast heterogeneity relevant to the health, diseases, and treatments of periodontal tissues? Crit Rev Oral Biol Med. 1997;8:253–268. doi: 10.1177/10454411970080030201. [DOI] [PubMed] [Google Scholar]
- 3.Polanska UM, Mellody KT, Orimo A. Tumour-promoting stromal myofibroblasts in human carcinomas. In Rebecca G Bagley, editor. Text book of The Tumour Microenvironment, Cancer Drug Discovery and Development. New York Springer Science Business Media. 2010. pp. 325–494. [Google Scholar]
- 4.Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB. Myofibroblasts. I. Paracrine cells important in health and disease. Am J Physiol. 1999;277:C1–C9. doi: 10.1152/ajpcell.1999.277.1.C1. [DOI] [PubMed] [Google Scholar]
- 5.Eyden B. The myofibroblast, electron microscopy and cancer research. Int J Cancer. 2009;125:1743–1745; author reply 1746. doi: 10.1002/ijc.24550. [DOI] [PubMed] [Google Scholar]
- 6.Sappino AP, Schürch W, Gabbiani G. Differentiation repertoire of fibroblastic cells: expression of cytoskeletal proteins as marker of phenotypic modulations. Lab Invest. 1990;63:144–161. [PubMed] [Google Scholar]
- 7.Schmitt-Gräff A, Desmoulière A, Gabbiani G. Heterogeneity of myofibroblast phenotypic features: an example of fibroblastic cell plasticity. Virchows Arch. 1994;425:3–24. doi: 10.1007/BF00193944. [DOI] [PubMed] [Google Scholar]
- 8.Santa Cruz Biotechnology, Inc. Myofibroblasts [Pr 2d3]: Sc-53383. [cited 2014 Sept 18] Available from: http://www.scbt.com/
- 9.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 10.Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432:332–337. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thannickal VJ, Lee DY, White ES, Cui Z, Larios JM, Chacon R, Horowitz JC, Day RM, Thomas PE. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem. 2003;278:12384–12389. doi: 10.1074/jbc.M208544200. [DOI] [PubMed] [Google Scholar]
- 13.Werner S, Krieg T, Smola H. Keratinocyte-fibroblast interactions in wound healing. J Invest Dermatol. 2007;127:998–1008. doi: 10.1038/sj.jid.5700786. [DOI] [PubMed] [Google Scholar]
- 14.Carthy JM, Garmaroudi FS, Luo Z, McManus BM. Wnt3a induces myofibroblast differentiation by upregulating TGF-β signaling through SMAD2 in a β-catenin-dependent manner. PLoS One. 2011;6:e19809. doi: 10.1371/journal.pone.0019809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phan SH. Biology of fibroblasts and myofibroblasts. Proc Am Thorac Soc. 2008;5:334–337. doi: 10.1513/pats.200708-146DR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thode C, Jørgensen TG, Dabelsteen E, Mackenzie I, Dabelsteen S. Significance of myofibroblasts in oral squamous cell carcinoma. J Oral Pathol Med. 2011;40:201–207. doi: 10.1111/j.1600-0714.2010.00999.x. [DOI] [PubMed] [Google Scholar]
- 17.Lygoe KA, Wall I, Stephens P, Lewis MP. Role of vitronectin and fibronectin receptors in oral mucosal and dermal myofibroblast differentiation. Biol Cell. 2007;99:601–614. doi: 10.1042/BC20070008. [DOI] [PubMed] [Google Scholar]
- 18.Gabbiani G. Evolution and clinical implications of the myofibroblast concept. Cardiovasc Res. 1998;38:545–548. doi: 10.1016/s0008-6363(98)00065-0. [DOI] [PubMed] [Google Scholar]
- 19.Lúcio PS, Cavalcanti AL, Alves PM, Godoy GP, Nonaka CF. Myofibroblasts and their relationship with oral squamous cell carcinoma. Braz J Otorhinolaryngol. 2013;79:112–118. doi: 10.5935/1808-8694.20130019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beurden HV. Characterization of fibroblast phenotypes in intra-oral wound healing. Thesis Radboud University Nijmegen Medical Centre, Nijmegen, The Netherlands, 2005. [cited. 2014. p. Aug 15]. Available from: http://hdl.handle.net/2066/51360. [Google Scholar]
- 21.Peters T, Sindrilaru A, Hinz B, Hinrichs R, Menke A, Al-Azzeh EA, Holzwarth K, Oreshkova T, Wang H, Kess D, et al. Wound-healing defect of CD18(-/-) mice due to a decrease in TGF-beta1 and myofibroblast differentiation. EMBO J. 2005;24:3400–3410. doi: 10.1038/sj.emboj.7600809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grinnell F. Mini-Review on the cellular mechanisms of disease, fibroblasts, myofibroblasts, and wound contraction. J Cell Biol. 1994;124:401–404. doi: 10.1083/jcb.124.4.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Häkkinen L, Hildebrand HC, Berndt A, Kosmehl H, Larjava H. Immunolocalization of tenascin-C, alpha9 integrin subunit, and alphavbeta6 integrin during wound healing in human oral mucosa. J Histochem Cytochem. 2000;48:985–998. doi: 10.1177/002215540004800712. [DOI] [PubMed] [Google Scholar]
- 24.Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127:526–537. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- 25.Desmoulière A, Guyot C, Gabbiani G. The stroma reaction myofibroblast: a key player in the control of tumor cell behavior. Int J Dev Biol. 2004;48:509–517. doi: 10.1387/ijdb.041802ad. [DOI] [PubMed] [Google Scholar]
- 26.Sciubba JJ. Oral leukoplakia. Crit Rev Oral Biol Med. 1995;6:147–160. doi: 10.1177/10454411950060020401. [DOI] [PubMed] [Google Scholar]
- 27.Chaudhary M, Gadbail AR, Vidhale G, Mankar Gadbail MP, Gondivkar SM, Gawande M, Patil S. Comparison of myofibroblasts expression in oral squamous cell carcinoma, verrucous carcinoma, high risk epithelial dysplasia, low risk epithelial dysplasia and normal oral mucosa. Head Neck Pathol. 2012;6:305–313. doi: 10.1007/s12105-012-0335-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Etemad-Moghadam S, Khalili M, Tirgary F, Alaeddini M. Evaluation of myofibroblasts in oral epithelial dysplasia and squamous cell carcinoma. J Oral Pathol Med. 2009;38:639–643. doi: 10.1111/j.1600-0714.2009.00768.x. [DOI] [PubMed] [Google Scholar]
- 29.de-Assis EM, Pimenta LG, Costa-e-Silva E, Souza PE, Horta MC. Stromal myofibroblasts in oral leukoplakia and oral squamous cell carcinoma. Med Oral Patol Oral Cir Bucal. 2012;17:e733–e738. doi: 10.4317/medoral.17834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Angadi PV, Kale AD, Hallikerimath S. Evaluation of myofibroblasts in oral submucous fibrosis: correlation with disease severity. J Oral Pathol Med. 2011;40:208–213. doi: 10.1111/j.1600-0714.2010.00995.x. [DOI] [PubMed] [Google Scholar]
- 31.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dabelsteen S, Christensen S, Gron B, Bardow A, Dabelsteen E. HGF is released from buccal fibroblasts after smokeless tobacco stimulation. Oral Oncol. 2005;41:509–514. doi: 10.1016/j.oraloncology.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 33.Dyavanagoudar SN. Oral Submucous Fibrosis: Review on etiopathogenesis. J Cancer Sci Ther. 2009;1:72–77. [Google Scholar]
- 34.Moutasim KA, Jenei V, Sapienza K, Marsh D, Weinreb PH, Violette SM, Lewis MP, Marshall JF, Fortune F, Tilakaratne WM, et al. Betel-derived alkaloid up-regulates keratinocyte alphavbeta6 integrin expression and promotes oral submucous fibrosis. J Pathol. 2011;223:366–377. doi: 10.1002/path.2786. [DOI] [PubMed] [Google Scholar]
- 35.Hinz B, Gabbiani G. Fibrosis: recent advances in myofibroblast biology and new therapeutic perspectives. F1000 Biol Rep. 2010;2:78. doi: 10.3410/B2-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mishra PJ, Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, Ganesan S, Glod JW, Banerjee D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008;68:4331–4339. doi: 10.1158/0008-5472.CAN-08-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen C, Wei Y, Hummel M, Hoffmann TK, Gross M, Kaufmann AM, Albers AE. Evidence for epithelial-mesenchymal transition in cancer stem cells of head and neck squamous cell carcinoma. PLoS One. 2011;6:e16466. doi: 10.1371/journal.pone.0016466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lewis MP, Lygoe KA, Nystrom ML, Anderson WP, Speight PM, Marshall JF, Thomas GJ. Tumour-derived TGF-beta1 modulates myofibroblast differentiation and promotes HGF/SF-dependent invasion of squamous carcinoma cells. Br J Cancer. 2004;90:822–832. doi: 10.1038/sj.bjc.6601611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seifi S, Shafaei S, Shafigh E, Sahabi SM, Ghasemi H. Myofibroblast stromal presence and distribution in squamous epithelial carcinomas, oral dysplasia and hyperkeratosis. Asian Pac J Cancer Prev. 2010;11:359–364. [PubMed] [Google Scholar]
- 40.de Araújo VC, Furuse C, Cury PR, Altemani A, Alves VA, de Araújo NS. Desmoplasia in different degrees of invasion of carcinoma ex-pleomorphic adenoma. Head Neck Pathol. 2007;1:112–117. doi: 10.1007/s12105-007-0028-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kawashiri S, Tanaka A, Noguchi N, Hase T, Nakaya H, Ohara T, Kato K, Yamamoto E. Significance of stromal desmoplasia and myofibroblast appearance at the invasive front in squamous cell carcinoma of the oral cavity. Head Neck. 2009;31:1346–1353. doi: 10.1002/hed.21097. [DOI] [PubMed] [Google Scholar]
- 42.Moulin V, Larochelle S, Langlois C, Thibault I, Lopez-Vallé CA, Roy M. Normal skin wound and hypertrophic scar myofibroblasts have differential responses to apoptotic inductors. J Cell Physiol. 2004;198:350–358. doi: 10.1002/jcp.10415. [DOI] [PubMed] [Google Scholar]
- 43.Vered M, Shohat I, Buchner A, Dayan D. Myofibroblasts in stroma of odontogenic cysts and tumors can contribute to variations in the biological behavior of lesions. Oral Oncol. 2005;41:1028–1033. doi: 10.1016/j.oraloncology.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 44.Sobral LM, Zecchin KG, Nascimento de Aquino S, Lopes MA, Graner E, Coletta RD. Isolation and characterization of myofibroblast cell lines from oral squamous cell carcinoma. Oncol Rep. 2011;25:1013–1020. doi: 10.3892/or.2011.1161. [DOI] [PubMed] [Google Scholar]
- 45.Smith SM, Bartov SA. Ameloblastoma with myofibroblasts: first report. J Oral Pathol. 1986;15:284–286. doi: 10.1111/j.1600-0714.1986.tb00625.x. [DOI] [PubMed] [Google Scholar]
- 46.Rothouse LS, Majack RA, Fay JT. An ameloblastoma with myofibroblasts and intracellular septate junctions. Cancer. 1980;45:2858–2863. doi: 10.1002/1097-0142(19800601)45:11<2858::aid-cncr2820451123>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 47.Effiom OA, Adewole RA, Odukoya O. Clinicopathological characteristics of odontogenic myxoma in Nigerians. West Afr J Med. 2011;30:255–261. [PubMed] [Google Scholar]
- 48.Mashhadiabbas F, Atarbashi Moghadam S, Moshref M, Elahi M. Immunohistochemical detection and ultrastructure of myofibroblasts in the stroma of odontogenic cysts and ameloblastoma. Iran Red Crescent Med J. 2010;12:453–457. [Google Scholar]
- 49.Nadalin MR, Fregnani ER, Silva-Sousa YT, da Cruz Perez DE. Presence of myofibroblasts and matrix metalloproteinase 2 in radicular cysts, dentigerous cysts, and keratocystic odontogenic tumors: a comparative immunohistochemical study. J Endod. 2012;38:1363–1367. doi: 10.1016/j.joen.2012.05.020. [DOI] [PubMed] [Google Scholar]
- 50.Lombardi T, Morgan PR. Immunohistochemical characterisation of odontogenic cysts with mesenchymal and myofilament markers. J Oral Pathol Med. 1995;24:170–176. doi: 10.1111/j.1600-0714.1995.tb01160.x. [DOI] [PubMed] [Google Scholar]
- 51.Gupta V, Ramani P, Chandrasekar A clinico-pathological and immunohistochemical study of salivary gland tumours: A 5 year Indian experience. Int J Oral Maxillofac Pathol. 2012;3:15–22. [Google Scholar]
- 52.Soma L, LiVolsi VA, Baloch ZW. Dendritic interstitial and myofibroblastic cells at the border of salivary gland tumors. Arch Pathol Lab Med. 2001;125:232–236. doi: 10.5858/2001-125-0232-DIAMCA. [DOI] [PubMed] [Google Scholar]
- 53.Sobral AP, Loducca SV, Nunes FD, de Araújo NS, Kowalski LP, de Araújo VC. Relationship between major and minor salivary gland mucoepidermoid carcinoma malignancy grading and presence of stromal myofibroblasts: immunohistochemical study. J Oral Pathol Med. 2004;33:335–339. doi: 10.1111/j.1600-0714.2004.00111.x. [DOI] [PubMed] [Google Scholar]
- 54.Vered M, Nasrallah W, Buchner A, Dayan D. Stromal myofibroblasts in central giant cell granuloma of the jaws cannot distinguish between non-aggressive and aggressive lesions. J Oral Pathol Med. 2007;36:495–500. doi: 10.1111/j.1600-0714.2007.00541.x. [DOI] [PubMed] [Google Scholar]
- 55.Epivatianos A, Iordanidis F, Andreadis D, Markopoulos A, Samara A. Myofibroblasts in mucoceles and chronic sialadenitis of minor salivary glands. Hippokratia. 2011;15:382–383. [PMC free article] [PubMed] [Google Scholar]