

Abstract
AP endonuclease 1 (APE 1) is a crucial enzyme of the base excision repair pathway (BER) in human cells. APE1 recognizes apurinic/apyrimidinic (AP) sites and makes a nick in the phosphodiester backbone 5′ to them. The conformational dynamics and presteady-state kinetics of wild-type APE1 and its active site mutant, Y171F-P173L-N174K, have been studied. To observe conformational transitions occurring in the APE1 molecule during the catalytic cycle, we detected intrinsic tryptophan fluorescence of the enzyme under single turnover conditions. DNA duplexes containing a natural AP site, its tetrahydrofuran analogue, or a 2′-deoxyguanosine residue in the same position were used as specific substrates or ligands. The stopped-flow experiments have revealed high flexibility of the APE1 molecule and the complexity of the catalytic process. The fluorescent traces indicate that wild-type APE1 undergoes at least four conformational transitions during the processing of abasic sites in DNA. In contrast, nonspecific interactions of APE1 with undamaged DNA can be described by a two-step kinetic scheme. Rate and equilibrium constants were extracted from the stopped-flow and fluorescence titration data for all substrates, ligands, and products. A replacement of three residues at the enzymatic active site including the replacement of tyrosine 171 with phenylalanine in the enzyme active site resulted in a 2 × 104-fold decrease in the reaction rate and reduced binding affinity. Our data indicate the important role of conformational changes in APE1 for substrate recognition and catalysis.
Genomic DNA continuously undergoes the loss of its heterocyclic bases as a result of spontaneous degradation and the action of various metabolic and exogenous factors (1, 2). Apurinic/apyrimidinic (AP1) sites, which arise in the DNA through hydrolysis of N-glycosidic bonds, are highly mutagenic and cytotoxic since they are noninstructive for DNA polymerases and are easily converted to single- and double-strand breaks (3). Repair of such lesions in human cells is accomplished in different ways, the most prominent of which is base excision repair (BER) (4–6). The crucial enzyme of this pathway in human cells is AP endonuclease 1 (APE1), which recognizes AP sites in dsDNA and makes a single nick in the phosphodiester backbone 5′ to the AP site (7–9), generating a hydroxyl 3′ end. In addition to the incision of spontaneously formed AP sites, APE1 operates in BER after the removal of damaged bases by specific DNA glycosylases. DNA polymerase β then inserts the correct dNMP and removes the hanging 2′-deoxyribo-5′-phosphate, and DNA ligase seals the remaining nick (10). APE1 can also cleave DNA 5′ to some base-containing damaged deoxynucleotides in a process termed nucleotide incision repair (11). Finally, besides its role in DNA repair, APE1is an important regulator of the redox state of several transcription factors including AP-1, HIF1α, and p53 (12–15). Because of its multifunctional nature, human APE1 is indispensable to normal embryonic development and cell survival in both mouse (16) and zebrafish (17). It was shown that over-expression of APE1 confers resistance to DNA-damaging agents in several human tumor cell lines (18). Conversely, decreasing APE1 expression using small interfering RNAs or a dominant-negative form of the protein results in hypersensitivity to chemically induced DNA damage in both cell culture and tumor xenograft models (19–22).
It was reported earlier that incision of AP-containing DNA catalyzed by wild-type APE1 is a very rapid process that cannot be described by a first-order reaction scheme (23, 24). Therefore, the reaction may proceed in several steps, and a simple Michaelis–Menten model may be insufficient for a complete kinetic analysis of APE1. In a recent paper (25), the AP endonuclease activity of this enzyme was characterized using equilibrium binding and presteady-state quench-flow kinetic analysis, and it was suggested that the efficiency of APE1 is limited by a slow step of product release.
It is well known that most enzymatic reactions are accompanied by conformational changes in the enzyme molecule. Conformational transitions in proteins can be followed by changes in the intrinsic fluorescence intensity of their tryptophan residues. Fluorescence measurements combined with the stopped-flow technique provide a powerful approach for direct observation of individual events occurring along a reaction pathway (26–28). This presteady-state kinetic method has been successfully employed earlier to define the elementary steps of enzymatic reaction pathways in the millisecond time range (29). Our stopped-flow fluorescence study of Escherichia coli formamidopyrimidine-DNA glycosylase (Fpg) has revealed the details of conformational dynamics of the enzyme–substrate complex during the catalytic reaction (30–32). It was found that the recognition of damaged bases by Fpg involves at least five equilibrium steps and that incorrect substrates are mostly rejected at early stages of the recognition complex formation, supporting the role of indirect readout in lesion recognition. This approach was also applied to address the kinetic mechanism of damaged base excision by human 8-oxoguanine-DNA glycosylase (33, 34).
According to its crystal structure, the APE1 molecule contains seven Trp residues, including Trp280 and Trp267 located in the vicinity of the active site (8, 35). Changes in their fluorescence could be sufficient to detect transient intermediates of the enzyme–substrate complexes during the catalytic cycle. Recently, we have studied the presteady-state kinetics of APE1 in the reactions of nucleotide incision repair and base excision repair pathways using a 30-nt AP-DNA substrate (36). Under BER conditions, we have observed a high reaction rate constant (kcatAP = 12 s−1) and the existence of a stable enzyme–product complex (Kp = 4.8 × 10−7M), which precluded the detection of individual transient complexes in the catalytic cycle.
To obtain more detailed information about the APE1 kinetic mechanism, we have designed short double-stranded oligonucleotides containing a natural AP site or its tetrahydrofuran analogue (F) opposite C in the complementary strand (Figure 1). In the undamaged ligand, a deoxyguanosine (G) residue substituted for AP or F. Under single-turnover conditions, we were able to detect multiple conformational transitions in the APE1 molecule during the presteady-state phase of the process, demonstrating a significant role of conformational changes in the enzyme for recognition and catalysis.
Figure 1.
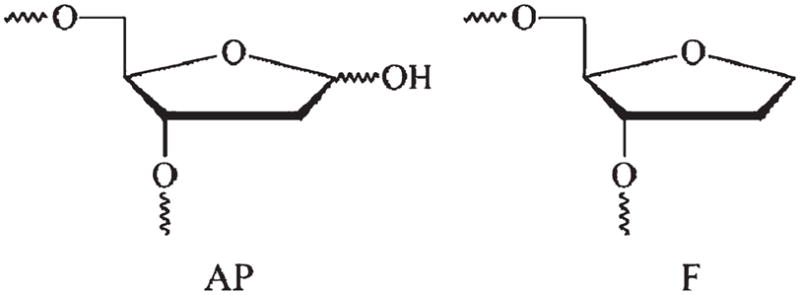
Chemical structures of the AP site (AP) and tetrahydrofuran abasic site analogue (F).
Tyr171, Pro173, and Asn174 are located within the APE1 active site and have been shown to be involved in Mg2+ coordination (35, 37). Furthermore, in several recent publications, the importance of conserved Tyr171 for AP site recognition, binding, and catalysis was reported (38–40). The substitution of a Phe residue for Tyr171 significantly decreases the catalytic efficiency of APE1. In our work, we have also addressed the question as to what stage of the APE1 enzymatic mechanism may be affected by this substitution. Together with other kinetic and structural studies, our data can help to gain a deeper insight of the mechanism of recognition and catalysis by APE1.
EXPERIMENTAL PROCEDURES
Oligonucleotides
Oligodeoxyribonucleotides (ODNs; Table 1) were synthesized on an ASM-700 synthesizer (BIOSSET Ltd., Novosibirsk, Russia) using phosphoramidites purchased from Glen Research (Sterling, VA, USA) and purified by anion exchange high-performance liquid chromatography (HPLC) on a Nucleosil 100–10 N(CH3)2 column followed by reverse-phase HPLC on a Nucleosil 100–10 C18 column (both columns from Macherey-Nagel, Düren, Germany). The purity of the ODNs was assessed by 20% denaturing polyacrylamide gel electrophoresis (PAGE) after staining with Stains-All (Sigma-Aldrich, St. Louis, MO, USA). The concentrations of the ODNs were determined from their absorbance at 260 nm. The AP-containing ODN was prepared as described in ref 41 by incubation of the deoxyuridine-containing ODN (0.1 mmol) for 14 h at 37 °C with 15 U of uracil-DNA glycosylase in 150 μL of 20 mM Tris-HCl (pH 8.0), 1 mM EDTA, 1 mM dithiothreitol (DTT), and 0.1 mg/mL bovine serum albumin. Reverse phase HPLC on a Nucleosil 100–5 C18 column was used to purify the reaction product, which had a shorter retention time than the starting oligonucleotide, using a linear gradient of 0–20% acetonitrile in 0.1 M triethyl-ammonium acetate (pH 7.0). The pooled fractions were concentrated and then converted to the lithium salt form using a Sep-Pak Plus C18 cartridge (Waters, Milford, MA, USA). The integrity of the AP-containing ODN was assessed by PAGE followed by Stains-All staining. To confirm the presence of the AP site in the ODN after the treatment with uracil-DNA glycosylase, samples were treated with 10% aqueous piperidine at 95 °C or annealed to the complementary oligonucleotide and treated with APE1 under the conditions described below. A PAGE analysis indicated that in both cases the material was cleaved to two shorter oligonucleotides. When needed, the modified strands were 32P-labeled using [γ-32P]ATP and bacteriophage T4 polynucleotide kinase (SibEnzyme, Novosibirsk, Russia) according to the manufacturer’s protocol and purified by 20% denaturing PAGE. ODN duplexes were prepared by annealing the modified and complementary strands in a molar ratio of 1:1. Under the conditions of stopped-flow experiments with a high Mg2+ concentration (see below), the predicted equilibrium constant of the duplex formation is 1.2 × 109 M−1, suggesting that nearly 100% of the oligonucleotides are in the duplex form (42–44).
Table 1.
Sequences of ODNs Used As Specific and Nonspecific Substrates for APE1
| shorthand | sequencea |
|---|---|
| AP-substrate | 5′C-T-C-T-C-AP-C-C-T-T-C-C3′ |
| 3′G-A-G-A-G-C-G-G-A-A-G-G5′ | |
| F-substrate | 5′C-T-C-T-C-F-C-C-T-T-C-C3′ |
| 3′G-A-G-A-G-C-G-G-A-A-G-G5′ | |
| G-ligand | 5′C-T-C-T-C-G-C-C-T-T-C-C3′ |
| 3′G-A-G-A-G-C-G-G-A-A-G-G5′ |
AP, abasic site; F, tetrahydrofuran.
APE1 Expression and Purification
Human APE1 was expressed in BL21 (DE3) pLysS E. coli cells from a pXC53 plasmid carrying a wild-type or Y171F-P173L-N174K APE1 gene. As CD spectra of the triple mutant were very close to that of the WT enzyme, the overall tertiary structure of the WT enzyme was retained (Supporting Information, Figure S1). However, the alteration in these three residues ensured near complete disruption of the active site. To purify the protein, the cells were grown in 2 L of YT × 2 broth with 50 μg/mL ampicillin at 37 °C until A600 = 0.6–0.7. After induction with isopropyl-1-thio-β-D-galacto-pyranoside for 2 h, the cells were harvested by centrifugation, resuspended in 50 mL of the buffer containing 10 mM Tris-HCl (pH 8.0), 1 mM EDTA, and 1 mM phenylmethylsulfonyl fluoride, and incubated with lysozyme (0.5 mg/mL) for 30 min at room temperature. The incubation was continued for another 20 min after the addition of NaCl to 1 M. The lysis was then completed by sonicating the suspension on ice using a Sonopuls HD 3100 ultrasound generator (Bandelin, Germany) with 10 pulses of 30 s, with a 90 s interval between pulses. The lysate was clarified by centrifugation (12,000g, 4 °C, 20 min) and diluted with 4 volumes of 25 mM potassium phosphate (pH 7.4, Buffer A). The resulting solution was applied to a 5-ml HiTrap SP HP column (GE Healthcare, Little Chalfont, UK) equilibrated in Buffer A. The column was washed with 10 mL of Buffer A supplemented with 1 M NaCl. The eluate was diluted 10-fold with Buffer A and applied to a 5-mL HiTrap Heparin HP column (GE Healthcare) equilibrated in Buffer A. The proteins were eluted with a gradient of 0–1000 mM NaCl in Buffer A; the fractions absorbing at 280 nm were collected. The purity of APE1 was 95% as judged from SDS–PAGE with Coomassie Blue staining. Expression and purification for the mutant APE1 were similar to those of the wild-type enzyme, except for the IPTG induction (30 °C, 2 h).
Stopped-Flow Fluorescence Experiments
Stopped-flow fluorescence measurements were carried out essentially as described in ref 30 using a model SX.18MV stopped-flow spectrometer (Applied Photophysics, UK) fitted with a 150 W Xe arc lamp. The fluorescence emission from the enzyme’s Trp residues (λex = 280 nm) was observed through a 320 nm long-pass filter (WG-320, Schott, Mainz, Germany). The instrument dead time was 1.38 ms. Typically, each trace shown is the average of at least five independent experiments. All experiments were carried out at 25 °C in a reaction buffer containing 50 mM HEPES-KOH (pH 7.5), 20 mM KCl,10 mM MgCl2, and 2 mM DTT. The enzyme concentration was 1.5 μM, and the substrate concentrations varied from 0.5 μM to 4 μM.
Bleaching of Enzyme Fluorescence
To correct the measured data for bleaching, the fluorescence intensities were recalculated using eq 1
| (1) |
where F is the corrected fluorescence intensity, Fobs is the observed fluorescence intensity, Fb is the background fluorescence, and kb is the coefficient determined for each substrate concentration in experiments with the noncleavable G-ligand.
Kinetic Data Analysis
Global nonlinear least-squares fitting was performed using DynaFit software (BioKin, Pullman, WA, USA) (45). The stopped-flow fluorescence traces were directly fitted by expressing the corrected fluorescence intensity (Fc) at any reaction time t as the sum of the background fluorescence (Fb) and the fluorescence intensities of each protein species:
| (2) |
where Fi(t) = fi(Ei(t)), fi is the coefficient of specific fluorescence for each discernible APE1 conformer, and (Ei(t)) is the concentration of the conformer at any given time t (i = 0 relates to the free protein and i > 0 to the protein–DNA complexes). These specific fluorescence coefficients describe only the part of fluorescence that changes due to DNA binding. Errors in all rate constant values measured by the stopped-flow assay did not exceed 10%.
Fluorescence Equilibrium Titration of APE1
To determine the dissociation constant Kd of the complex of APE1 with the product of AP site incision, the enzyme was titrated with the mixture of ODNs d(GGAAGGCGAGAG), d(CTCTC), and d(pFCCTTCC). Each point on the fluorescence titration curve was obtained by measurement of the fluorescence intensity of separate solutions (80 μL) containing APE1 (1.5 × 10−6 M) and the ODNs at the required concentration in the reaction buffer described above. The mixtures were incubated at 25 °C for 2 min, and the fluorescence spectra were measured (λex = 280 nm) using an SFM 25 spectrofluorometer (Kontron Instruments, Italy) in the shortest possible time to avoid bleaching. The value of Kd was determined from eqs 3–6:
| (3) |
| (4) |
| (5) |
| (6) |
where, F0, F1, and F2 are fluorescence intensities of APE1 with no ODNs added, at any given ODN concentration, and at the saturating ODN concentration, respectively, [Ape1]0 and [ODN]0 are the total concentrations of APE1 and the ODNs, [Ape1] and [ODN] are the concentrations of free APE1 and the ODNs, and [complex] is the concentration of the APE1–ODN complex.
Cleavage Time Course Experiments
The substrate duplexes containing a natural AP site or its F analogue were 5′-labeled with 32P and annealed to the complementary strand. Substrates (1.5 μM) were mixed with 1.5 μM enzyme in the reaction buffer (see above). The reaction was terminated at required time points by adding of a loading dye solution containing 7 M urea. Aliquots were then analyzed by 20% denaturing PAGE, the gels were imaged by autoradiography and quantified by scanning densitometry using Gel-Pro Analyzer 4.0 software (Media Cybernetics, Silver Spring, MD, USA).
Measurement of Steady-State Kinetic Parameters
Steady-state kinetic parameters for wild-type APE1 were measured at the substrate concentrations between 10 and 1500 nM. The enzyme concentration was 0.5 nM and 1 nM for AP- and F-substrates, respectively. To determine the initial rates for the mutant APE1 reaction, 15 nM enzyme was incubated with the AP-substrate concentrations varying from 200 to 1200 nM. The KM and Vmax values were estimated using least-squares nonlinear regression analysis (46). Errors in the KM and Vmax values measured by the steady-state kinetics were within 20–50%.
RESULTS
Binding of APE1 to DNA
Several high-resolution crystal structures of APE1 in a complex with DNA have defined a minimal length of double-stranded AP-DNA required for efficient substrate binding and cleavage as eight base pairs (23, 47). For our study, we have designed 12-mer ODN duplexes containing the natural abasic site (AP-substrate) or its tetrahydrofuran analogue (F-substrate) in the middle of the modified strand (Table 1). These substrates were sufficient for essentially the full catalytic activity of APE1 and did not show appreciable end effects. The recent study has shown a noticeable difference between fluorescence emission spectra of free APE1 and the APE1 bound to the 26F-DNA duplex, indicating possible conformational changes of the nucleoprotein complex (48). In the present work, an undamaged ODN duplex containing G instead of the AP site was employed to probe the conformational dynamics of APE1 during nonspecific DNA recognition and binding steps. An effect of APE1 binding to this G-ligand on the enzyme fluorescence was evident from a decrease of the enzyme fluorescence (Supporting Information, Figure S2). The emission maximum of the free protein was 330 nm when the excitation was at 280 nm. A notable decrease in the peak emission intensity was observed in the fluorescence spectrum of APE1 after the addition of the G-ligand under the equilibrium conditions. Thus, we concluded that the APE1 conformational dynamics can be studied by the presteady-state stopped-flow approach in combination with enzyme fluorescence detection.
Stopped-Flow Single Turnover Kinetics of AP-DNA Cleavage by APE1
To begin, we registered the time courses of Trp fluorescence after mixing of wild-type APE1 with specific DNA substrates under single turnover conditions. To determine the kinetic mechanism of APE1 interactions with the natural AP site, stopped-flow experiments were carried out at concentrations of the AP-substrate between 0.5 and 4 μM in a time interval of up to 50 s. A suitable observation time was optimized from preliminary stopped-flow runs in which the registration of Trp fluorescence was continued up to 2000 s. The obtained fluorescent traces are shown in Figure 2A. The traces are suggestive of significant APE1 conformational changes corresponding to individual steps of the catalytic process within a 3 s time range. On the contrary, the fluorescence traces remained steady if APE1 was mixed with the buffer without DNA substrate (data not shown). A visual inspection of the traces reveals the existence of several transient intermediate states of the enzyme (dashed drop lines in Figure 2A, each placed approximately at a characteristic time of the respective conformational transition in the protein molecule), which are reflected in changes of the slope of tangents to the trace. The first phase is observed up to 20 ms, when the protein fluorescence decreased rapidly after the addition of the AP-substrate. This conformational transition of the APE1 molecule corresponds to the substrate binding and formation of a primary enzyme–substrate complex (ES). The second phase of the traces (20–200 ms) corresponds to a distinct minimum of the fluorescence intensity, which became flat and widened with an increase in DNA concentration, indicating the existence of a steady-state phase under the conditions of an excess of the substrate over the enzyme. This phase likely reflects the conformational rearrangement resulting in the catalytically competent conformation of the APE1 active site. In the third phase (0.2–1 s), the Trp fluorescence increased rapidly to the starting level. Subsequently, in the fourth phase, the traces remained unchangedupto50s.The third and fourth phases of the kinetic curves are most likely associated with the catalytic step followed by the release of the incised DNA product.
Figure 2.

Time-dependent fluorescence change associated with the cleavage of AP-substrate (A) and F-substrate (B) by APE1. The fluorescence traces are distributed along the signal axis for better visualization. The final concentrationofAPE1 is1.5 μM. Jagged traces represent experimental data; smooth curves are theoretically fitted. Blue dashed curves (B) represent the results of fitting of the experimental data to Scheme 5 (23). Dashed drop lines approximately correspond to different stages of kinetic Scheme 1. (C) Representative graph of the residuals for fitting of the experimental data (3.0 μM substrate).
The observed fluorescence changes were kinetically described by Scheme 1. To extract the individual rate constants, the stopped-flow fluorescence traces were directly fitted by treating the corrected fluorescence intensity as the sum of the contribution of each protein species at any reaction time (30). At first, the initial segments of the traces (0–20 ms time interval) were fitted to a one-step mechanism corresponding to bimolecular binding of the enzyme to damaged DNA. As a result, two rate constants for the forward and reverse directions (k1AP and k−1AP) had been determined. Then, the second segments of the fluorescence traces were added, and the next round of fitting was undertaken using a more complicated two-stage kinetic scheme. During each round of fitting, the parameters already determined at the previous stages were kept constant, whereas the currently analyzed parameters were variable. The quality of the fit was estimated by visual inspection of overlays of the fitted curves and the data as well as by inspection of the residuals (Figure 2C). Errors in the calculated constants were around 10%. Thus, Scheme 1 represents the minimal kinetic mechanism corresponding to the observed fluorescence changes. The values of the individual rate constants are presented in Table 2.
Scheme 1.

Kinetic Scheme for the APE1 Interaction with AP-, F-, and G-Containing DNAa
aE, APE1; S, free DNA substrate; (ES), bimolecular encounter complex; (ES)′ and (ES)″, subsequent states of the APE1-DNA complex; (EP), complex of APE1 with the DNA product; P, product of the substrate incision. Constants k1, k2, and k3 characterize the forward direction, whereas k−1, k−2, and k−3 are the rate constants for the reverse reactions; kirr corresponds to the irreversible chemical step. Kd is an equilibrium constant calculated as the k4/k−4 ratio. In the case of F-substrate cleavage, an additional stage of the mechanism is shown in red. AP, AP-substrate; F, F-substrate; G, G-ligand.
Table 2.
Rate Constants for Interactions of Wild-Type APE1 with AP-, F-, and G-Containing DNA
| rate constants | substrate (ligand) X |
||
|---|---|---|---|
| AP | F | G | |
| k1X (M −1 s −1) | (8.93 ± 0.48) × 107 | (2.0 ± (0.1) × 108 | (7.4 ± (0.5) × 10 |
| k−1X (s −1) | 6.02 ± 0.69 | 14.6 ± 0.21 | 17.4 ± (0.6 |
| k2X (s −1) | 3.24 ± 0.07 | 5.61 ± 0.15 | 0.17 ± (0.01 |
| k−2X (s −1) | 1.83 ± 0.05 | 28.6 ± 0.32 | 0.76 ± (0.02 |
| k3X (s −1) | 6.55 ± 0.18 | ||
| k−3X (s −1) | 0.15 ± 0.01 | ||
| kirrX (s −1) | 3.20 ± 0.06 | 2.05 ± 0.04 | |
| KdX (M) | (0.96 ± 0.05) × 10−6 | (5.47 ± 0.11) × 10−6 | |
| KaX (M−1)a | (4.11 ± 0.08) × 107 | (1.33 ± 0.06) × 108 | (5.2 ± (0.2) × 104a |
| KtitrWT (M) | (8.31 ± 0.10) × 10−7 | ||
Equilibrium association constant Ka was calculated from the equation for N-step binding (see Scheme 1). .
The quantitative analysis of fluorescence changes suggests that the mechanism of AP-DNA cleavage by APE1 includes at least four steps. The bimolecular encounter between the enzyme and AP-DNA is described by the forward and reverse rate constants of (8.93 ± 0.48) × 107 M−1 s−1 and 6.02 ± 0.69 s−1, respectively. The equilibrium association constant KaAP is (1.48 ± 0.08) × 107 M−1, showing a high affinity of APE1 for the damaged DNA. The second segment of the fluorescence traces is described in the reaction mechanism by a reversible step with the rate constants k2AP = 3.24 ± 0.07 s−1 and k−2AP = 1.83 ± 0.05 s−1. The third step where the fluorescence increases is irreversible and may correspond to a catalytic step accompanied by a return of the enzyme in the catalytically inactive form. The observed rate constant of this step kirrAP is3.20 ± 0.06 s−1. Thus, our presteady-state kinetic data are in good agreement with the notion of very fast DNA incision by human APE1. However, the use of a short DNA duplex results in a rather slow rate of catalysis compared with the recently reported data for longer substrates (36). The fourth reaction step includes reversible dissociation of APE1 bound to the cleaved duplex to the free enzyme and three individual ODNs. This equilibrium characterizes the protein’s affinity for reaction products with the calculated constant KdAP = (0.96 ± 0.05) × 10−6 M, indicating the formation of a stable enzyme-product complex. The APE1 interaction with the incised duplex was also estimated from fluorescent equilibrium titration experiments (see below). The kinetic mechanism described above includes a minimal number of transient states of the protein molecule sufficient to describe the traces that we have registered using the stopped-flow approach.
Stopped-Flow Single Turnover Kinetics of F-DNA Cleavage by APE1
(3-Hydroxytetrahydrofuran-2-yl)-methyl phosphate (F) is a synthetic analogue of the natural AP site lacking the C1′ hydroxyl group. F is more stable than the natural AP site and has been used in a wide variety of applications, including investigations of DNA repair enzymes (31, 47, 49–51). In the present study, a duplex carrying the F moiety instead of a natural AP site has been employed as another specific substrate for APE1. To determine the kinetic mechanism of specific substrate cleavage by the wild-type protein, stopped-flow experiments were carried out at increasing concentrations of the F-substrate (Figure 2B). The observed fluorescence traces were consistent with a multistage catalytic process taking up to 10 s. In general, the changes in fluorescence during the binding and processing of the F-substrate were similar to those for the AP-substrate. However, a careful examination of the middle segment of the traces allowed us to identify a more complex fluorescence dynamics of APE1 cleaving the F-substrate. An additional stage was observed on the fluorescence traces at 0.03–1 s, becoming the most apparent at DNA concentrations higher than 1.5 μM. By global fitting, we have found that the minimal catalytic mechanism includes five steps and four transient states of the APE1 molecule along the reaction pathway (Scheme 1). The values for the individual kinetic constants are presented in Table 2. The first segment of the traces is characterized by a decrease in the fluorescence intensity until 30 ms, consistent with primary binding and (ES) complex formation. The rate constants of the forward and reverse reactions are typical for bimolecular encounters, (2.0 ± 0.1) × 107M−1 s−1and 14.6 ± 0.2 s−1, respectively. Two following equilibrium stages, observed from 30 ms to 1 s, reflect the steps of mutual adjustment of the structures of APE1 and the F-substrate, leading sequentially to (ES)′ and (ES)″ transient complexes. Accordingly, the bimolecular encounter makes possible an intramolecular rearrangement, resulting in a catalytically competent conformation of the APE1 active site. The subsequent rise in the Trp fluorescence, characterized by the lowest rate constant kirrF = 2.05 ± 0.04 s−1, most likely represents the final catalytic step, in which the product is formed. The steady fluorescence intensity level detected after 10 s of registration means that the reaction has attained equilibrium between the enzyme–product complex and free APE1 protein.
The stopped-flow data for natural AP site processing reveal significant differences in the reaction rates compared to those of the F-substrate. In the case of the F-substrate, the steady Trp fluorescence intensity is achieved in 10 s, whereas the fluorescent traces for the AP-substrate cleavage demonstrate a steady level after 2 s of registration. In terms of reaction rates, the cleavage of the modified AP site lacking its C1′ hydroxyl group is ~1.6 times slower than the natural AP site incision process.
Kinetics of APE1 Interactions with Undamaged DNA
We have used an undamaged DNA duplex containing a G residue instead of the AP site as the nonspecific ligand for APE1. Stopped-flow analysis of APE1 interactions with this uncleavable G-ligand was expected to enable the detection of APE1 conformational changes associated with DNA binding and search for the lesion, independently of the chemical step. In these experiments, the concentration of APE1 was 1.5 μM, and the concentrations of the G-ligand were varied in the range 1.5–4.0 μM. Representative fluorescence traces obtained at the increasing G-ligand concentrations are shown in Figure 3. The process of APE1 binding to the undamaged duplex was essentially complete by 1 s. Despite the relatively low amplitude of the fluorescence changes, the data could be fitted by a two-step kinetic mechanism, and the appropriate rate constants were estimated (Table 2). The nonspecific binding of APE 1 to the undamaged DNA duplex was described by forward and reverse constants of (ES) and (ES)′ complex formation (Scheme 1). The general association constant KaG value was estimated as (5.2 ± 0.2) × 104 M−1 s−1. Thus, the relative affinity of APE1 for specific and nonspecific ODNs in terms of Ka values differs by more than 2 orders of magnitude. Fluorescence changes occurring during the binding of the G-ligand most likely correspond to the precatalytic steps in the processes of AP and F incision.
Figure 3.

Stopped-flow fluorescent traces of APE1 binding the nonspecific G-ligand. The final concentration of APE1 is 1.5 μM. Jagged traces represent experimental data; smooth curves are theoretically fitted. Inset: the same fluorescence traces presented on a log scale.
Analysis of the Reaction Product Formation
The pre-steady-state stopped-flow analysis permits the detection of conformational changes associated with different transient states of the enzyme molecule. In order to associate the conformational transitions with the particular chemical steps of the APE1 catalytic cycle, we have analyzed the accumulation of the nicked product by denaturing PAGE (Figure 4, top panel). As expected, autoradiograms representing the time courses of processing of AP- and F-substrates by APE1 demonstrate rapid accumulation of the product. Specifically, the conversion of 95% of the AP-substrate into the product took only 5 s and that of the F-substrate, 10 s (Figure 4, bottom panel), close to the first time point (3 s) that could be reliably sampled with manual mixing. The fast formation of the DNA product observed by PAGE analysis is consistent with our stopped-flow results.
Figure 4.
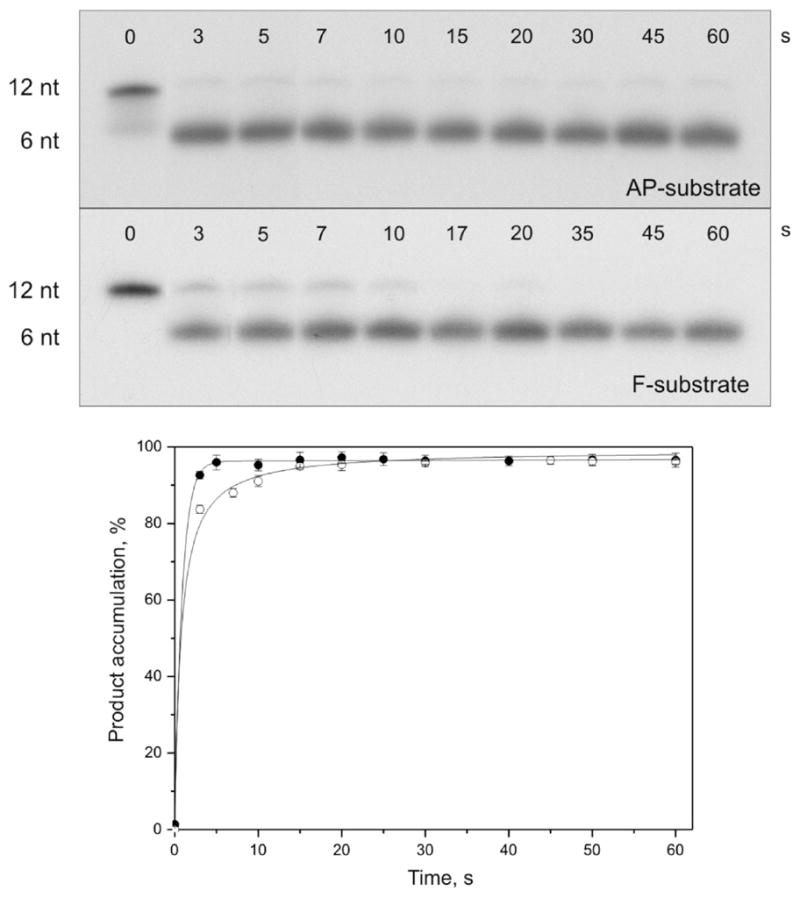
Cleavage of 32P-labeled AP- and F-substrates by APE1. Top panel, analysis by 20% PAGE; autoradiograms are shown. Reaction mixtures (10 μL) contained 1.5 μM substrate and 1.5 μM enzyme. Bottom panel, time course of the cleaved product accumulation; ●, AP-substrate incision; ○, F-substrate incision. Error bars reflect the standard error of the mean from three independent experiments.
Stopped-Flow Kinetics of AP-DNA Cleavage by the Mutant APE1
The Tyr171 residue of APE1 was reported to bind the a basic nucleotide of damaged DNA in a proper position for catalysis (39, 40) and to coordinate the Mg2+ ion within the active site (35). It was shown that the Y171F substitution dramatically reduces the rate of the catalytic reaction. In the present study, we have attempted to determine more precisely which steps of the APE1 kinetic mechanism are influenced by disruption of the active site including Tyr171. Two series of stopped-flow traces were registered at increasing concentrations of AP- and F-containing DNA duplexes (1.0–4.0 μM) under nearly single turnover conditions (Figure 5). The fluorescence traces were nearly identical for both AP- and F-substrates, and represented a two-phase decrease of Trp fluorescence in the 1.5 ms–1500 s time range. Of particular interest here is the observation that the fluorescence intensity of mutant APE1 was not restored to its initial value at these times, indicating either the existence of a stable complex between the protein and the DNA products or the absence of catalytic activity or very low reaction rate. The fluorescent traces describing the cleavage of both substrates by mutant APE1 showed a rather low signal-to-noise ratio, making their quantitative treatment difficult. Immediately after the fast mixing of reagents, the interaction started with an exponential decay up to 100 s, which was followed by the slow step until 12 min for the AP-substrate and 20 min for the F-substrate. These changes of Trp fluorescence may correspond to the processes of substrate recognition and binding. The registration process was terminated after 2000 s because of the strong bleaching of Trp fluorescence, displaying no conformational changes that could be attributed to the catalytic step. Fitting of these data to a two-step mechanism (Scheme 2) yielded the individual rate constants presented in Table 3. The equilibrium association constants KaAP and KaF were calculated from the ratio of the forward and reverse rate constants of the (EMutS) complex formation. The calculated Ka values for both specific substrates indicate that the affinity of the modified enzyme for damaged DNA is 1 order of magnitude lower than that of wild-type APE1 (Table 3).
Figure 5.

Stopped-flow kinetics of mutant APE1 interactions with specific substrates. Traces of Trp fluorescence observed during the interaction of 3 μM mutant APE1 with the AP-substrate (A) and F-substrate (B). Dashed drop lines approximately correspond to different stages of the kinetics in Scheme 2.
Scheme 2.
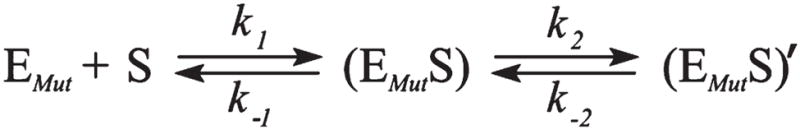
Kinetic Scheme for Interactions of the Triple Mutant with AP- and F-Substratesa
aEMut, mutant APE1; S, free DNA substrate; (EMutS), bimolecular encounter complex; (EMutS)′, subsequent state of the mutant APE1-DNA complex.
Table 3.
Rate Constants for Interactions of Mutant APE1 with AP- and F-Containing DNA
| rate constants | mutant APE1/AP-substrate | mutant APE1/F-substrate |
|---|---|---|
| k1 (M −1 s −1) | (1.78 ± 0.07) × 103 | (5.4 ± 0.2) × 103 |
| k−1 (s −1) | (1.51 ± 0.05) × 10−3 | (2.1 ± 0.1) × 10−3 |
| k2 (s −1) | (2.2 ± 0.1) × 10−4 | (1.5 ± 0.01) × 10−4 |
| k−2 (s −1) | (5.4 ± 0.2) × 10−3 | (1.12 ± 0.01) × 10−3 |
| Ka (M−1) | (4.81 ± 0.02) × 104 | (3.44 ± 0.01) × 105 |
| Ktitr (M) | (2.9 ± 0.4) × 10−6 |
PAGE Analysis of DNA Substrate Incision by the Mutant APE1
The endonuclease activity of the mutant APE1 toward 32P-labeled AP- and F-containing DNA duplexes was also assayed by 20% denaturing PAGE. When the concentrations of the enzyme and the substrate were similar to those used in stopped-flow experiments, the accumulation of cleavage products was evident after 10–15 min of incubation, and the reaction was nearly complete after 90–150 min (Figure 6, top panel). It is clear from these data that the rate of AP site hydrolysis catalyzed by the modified APE1 is notably lower compared to that of the wild-type enzyme. Incision of the AP-DNA by an equimolar amount of the mutant protein requires at least 2 h instead of 5 s for WT APE1 (Figure 6). Despite the unusually low DNA cleavage rate, the amount of the incision product reached 90% within 2.5 h. Therefore, the loss of the Tyr171 hydroxyl group and two additional alterations within the APE1 active site disturbs correct substrate binding and greatly reduces the efficiency of DNA cleavage but preserves the ability of the mutant enzyme to incise damaged DNA. The observed low rate of the mutant APE1 turnover probably would not let us to reliably register transient intermediates corresponding to the steps of substrate incision and product release under the conditions of stopped-flow experiments. Similar to the wild-type APE1, processing of the F-substrate by the mutant enzyme took more time as compared to that for AP-DNA.
Figure 6.

Cleavage of 32P-labeled AP- and F-substrates by the mutant APE1. Top panel, analysis by 20% PAGE; autoradiograms are shown. Reaction mixtures (10 μL) contained 3 μM substrate and 3 μM enzyme. Bottom panel, time course of the cleaved product accumulation; ●, AP-substrate incision; ○, F-substrate incision. Error bars reflect the standard error of the mean from three independent experiments.
Interaction of Wild-Type and Mutant APE1 with the Reaction Product
The affinity of APE1 for the cleavage product was analyzed by fluorescent titration of the enzyme with an equimolecular mixture of short ODNs representing the products of the F-substrate incision and forming a duplex carrying a single nick in one strand (Figure 7, insert). We have used this approach in order to estimate the stability of the APE1 –product complex by an independent method. Figure 7 demonstrates the changes in the fluorescence intensity of APE 1 and its triple mutant during the titration. The data were fitted by a one-site binding model (Scheme 3). The calculated value of the equilibrium constant Ktitr, (8.31 ± 0.10)×10−7 M for APE1, indicates the formation of a stable complex between the protein and nicked DNA (see Table 1). These observations are in general agreement with the obtained stopped-flow value of Kd constants for the AP- and F-substrate cleavage. Comparison of this dissociation constant with the recently reported Kd value of 4.8 × 10−7 M for the product of a 30-mer duplex cleavage (36) evidences only a 2-fold decrease in the APE1 affinity upon shortening the substrate DNA from 30 to 12 base pairs. For the mutant APE1, the calculated value of Ktitr was (2.9 ± 0.4) × 10−6 M, suggesting that the mutant protein maintains the ability to bind the incised product, albeit less efficiently.
Figure 7.

Change in Trp fluorescence during titration with oligonucleotides corresponding to the product of F-substrate cleavage. Experimental data for wild-type and mutant APE1’s are shown by filled and open circles, respectively. [P], concentration of the products.
Scheme 3.
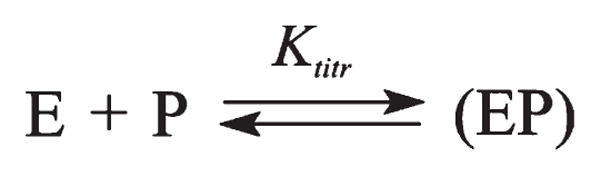
Equilibrium Representing the formation a Complex(EP)between the Protein (E) and the Cleavage Products(P)
Steady-State Kinetic Parameters for Wild-Type and Mutant APE1
In order to compare the kinetic constants obtained from our presteady-state stopped-flow experiments, with the steady-state Michaelis–Menten parameters, we have measured the KM and kcat values for the AP- and F-substrates. The KM values for the processing of AP- and F-substrates by the wild-type enzyme were 93 and 98 nM, respectively (Table 4). The observed kcat value for the processing of the F-substrate (0.6 s−1) indicates a 2-fold decrease compared to that of the AP-substrate (1.25 s−1). In the case of mutant APE1, we were able to measure the steady-state kinetic parameters only for the AP-substrate incision. Attempts to extract Michaelis–Menten parameters for the F-substrate incision were unsuccessful because of very slow reaction rates under the conditions we used. As shown in Table 4, the KM value for the mutant protein was 220 nM with a kcat of 1.53×10− s−1. The observed 2.5-fold increase in the KM value seems to arise from a somewhat reduced general affinity of mutant APE1 for DNA (compare with the 3.5-fold decrease in the affinity of the mutant enzyme for the reaction product, as described in the previous section). Therefore, our findings suggest that the lower catalytic rate contributes more to a significant loss of the enzyme efficiency conferred by the triple mutation including Y171F than does weak substrate binding. The values of KMSF and kcatSF were calculated from the stopped-flow data using the Volkenstein graph method (52) (Table 4). The similarity of Michaelis–Menten parameters determined by these two methods confirms the reliability of transient kinetic constants evaluated by the stopped-flow approach.
Table 4.
Steady-State Kinetic Parameters for Cleavage of AP- and F-Substrates by Wild-Type and Mutant APE1
| enzyme/substrate | KM (nM) | kcat (s−1) | kcat/KM (M−1 s−1) | KMSF (nM)a | kcatSF (s−1) | kcat SF/KM SF (M−1 s −1) |
|---|---|---|---|---|---|---|
| WT APE1/AP | 93 | 2.8 | 3 × 107 | 55 | 1.25 | 2.27 × 107 |
| WT APE1/F | 98 | 1.0 | 1×107 | 46 | 0.6 | 1.3 × 107 |
| mutant APE1/AP | 220 | 1.53 × 10−4 | 0.63 × 103 |
Values of KMSF, kcatSF, and kcatSF/KMSF were calculated from the presteady-state kinetic parameters using graph theory approach (see Results).
DISCUSSION
Several studies of the APE1 mechanism were reported in the literature (25, 40, 53), including our recent work (36). In the first single-turnover study of APE1 (24), explicit binding and dissociation kinetics allowed the authors to suggest a Briggs-Haldane mechanism (Scheme 4) with kon =5.3 × 107 M−1 s−1, koff = 0.04 s−1 (Kd = 0.8 nM), k2 = 10 s−1, and very rapid product release (k3 ≫ 10 s−1). It was also shown that both His309 and Tyr171 are intimately involved in the catalysis by this enzyme (39, 54). Recently, Maher and Bloom published the first presteady-state kinetic analysis of the cleavage of an F-containing substrate (25). They showed that the cleavage rate of APE1 is so fast that it could not be measured using quench-flow kinetics (minimally, 850 s−1). The most important conclusion from the latter study (which investigated the wild-type as well as H309N and D210A mutants) is that a slow step, which occurs after chemistry but before dissociation, limits the steady state incision activity to 2–10 s−1. The authors postulated the existence of a conformational change, which occurs after the cleavage of DNA, resulting in kinetic Scheme 5. Nevertheless, the role of conformational transitions of this enzyme in its action was not clear, particularly due to the high rate of the catalytic reaction.
Scheme 4.

Minimal Kinetic Scheme Consistent with a Briggs–Haldane Mechanism (from Ref 24)
Scheme 5.

Enzymatic Scheme for DNA Cleavage by APE1 Protein, Derived from the Results Reported and Discussed in Ref 25
In the present study, we have applied the stopped-flow presteady-state technique with fluorescence detection to reveal conformational transitions in the molecule of human APE1 corresponding to intermediate states of the catalytic cycle. We decided to employ the shortest DNA substrates that still can be normally incised by APE1 because of concerns that longer DNA sequences, such as the 30-mer used in our previous study (36), might lead to significant increases in the reaction rate under BER conditions, making detection of the transient intermediates by the stopped-flow method difficult. Truncation of the DNA substrates to a minimal length enabled us to detect multiple changes in Trp fluorescence of APE1 during its interaction with damaged DNA. The 12-mer ODN substrates were designed to avoid the effect of enzyme sliding along DNA. The natural abasic site and its tetrahydrofuran analogue were used as lesions specifically recognized by the APE1 protein. The APE1 molecule contains seven Trp residues, which can be used as fluorescent reporter groups changing their fluorescence intensity with conformational rearrangements in the protein molecule. In the APE1 structure, the Trp267 and Trp280 residues are located in the vicinity of a hydrophobic pocket of the active site, with the Trp280 residue interacting with the abasic sugar in the course of its recognition and binding (48, 55). Thus, these two residues are the best candidates that can account for changes in the fluorescent signal of APE1 along the catalytic pathway. The results obtained under single turnover conditions show that APE1 undergoes drastic conformational transitions upon binding and cleavage of all types of DNA substrates used (Figures 2–3).
It was found that the minimal kinetic model for the natural AP site incision consists of four stages corresponding to three different transient states of APE1 (Scheme 1). When the enzyme is complexed with the AP-substrate, the catalytic cycle is completed within 3 s, permitting the detection of three conformational transitions before the products are released. Because of a slower cleavage rate in the case of the F-substrate, an additional conformational transition of the precatalytic complex has been resolved, and the process has been described by a five-step mechanism. In contrast, conformational dynamics of the APE1 protein during nonspecific binding of an undamaged DNA duplex includes only two steps. Scheme 1 summarizes several kinetic models for APE1 interactions with AP-, F-, or G-containing DNA. The quantitative analysis of the data has provided the values of rate constants for each stage of these mechanisms (Table 1). The observed values of kirr constants for the processing of specific substrates (kirrAP = 3.2 s−1; kirrF = 2 s−1) suggest very fast catalysis. The determined parameters show that the AP-substrate is processed faster than F-substrate. Thus, our results are indicative of conformational flexibility of the APE1 active site.
According to the X-ray structural data, APE1-abasic DNA interactions are characterized by a number of events, including insertion of protein’s loops into both the major and minor grooves of the DNA, flipping of the abasic nucleotide into the hydrophobic pocket of APE1, and kinking of the DNA helix. Stopped-flow detection of changes in the Trp fluorescence of APE1 in the course of the natural AP site incision enabled us to accurately discern the conformational transitions of the APE1 molecule reflecting specific substrate binding and formation of the precatalytic complex and the enzyme–product complex. However, due to a high reaction rate it was impossible to discriminate transient states that precede the formation of the catalytically competent complex. This obstacle has been bypassed using the tetrahydrofuran AP site analogue instead of the natural AP site. As mentioned above, stopped-flow kinetics of the F-substrate incision process has shown a reaction rate 1.6-fold slower compared to that of the AP-substrate. The fluorescence traces describing the F-substrate cleavage by APE1 reveal an additional intermediate state of the protein molecule in the 25–100 ms time range. Additionally, monitoring of DNA substrate movement by 2-aminopurine fluorescence reveals distortion of the DNA helix at approximately the same time (manuscript in preparation). On the basis of these findings, we suggest that the observed enhancement of Trp fluorescence at the second stage of APE 1-catalyzed processing of the F-substrate represents the process of abasic sugar eversion resulting in DNA distortion, characterized with the rate constants k2F = 5.61 s−1 and k3F = 6.55 s−1, respectively. Of particular interest here is the fact that the decrease in the incision rate for the F-substrate is essentially due to a reduction of the enzyme efficiency at steps 2–4 of Scheme 1, while the efficiency of the bimolecular encounter is the same for the AP- and F-substrates. Therefore, although APE1 is known to recognize and incise a wide variety of abasic units (47), our data indicate that noncovalent contacts between the amino acid residues in the APE1 active site and C1-hydroxyl group of the natural AP site contribute to the recognition of the optimal AP-DNA substrates. These contacts may involve Ala230, the main chain carbonyl of which is poised to accept a hydrogen bond from the α-epimer of the natural AP site (8). Notably, the ring of Trp280 is located only 4 Å away from C1′ in the structure of APE 1 bound to F-containing DNA (8); the interactions of Trp with substituents at C1′ can explain the differences in the fluorescence signals between our AP-substrate and F-substrate. Thus, the use of the synthetic abasic site analogue F in addition to the natural AP site has provided new important insights into the APE1 mechanism.
Recent reports indicate that Tyr171 in the APE1 active site is important for discrimination between DNA with an AP site and without one (35, 38, 39). This residue, together with other amino acid residues, also participates in the Mg2+ coordination, through a hydrogen-bonding network (37). Mutations of Tyr171 resulted in reduced incision activity (56). The side chain of Asn174 forms hydrogen bonds with O5′ of the target phosphate on the 5′ side of the APE1 active site (8). Mutation at this position is likely to lead to disruption of the active site. On the basis of these data, we have constructed the APE1 active site mutant Y171F-P173L-N174K. Comparison of 3D structures of the WT APE1 and its triple mutant is presented in Figure 8. Our presteady-state kinetic analysis has revealed that the removal of the hydroxyl group of Tyr171 and alteration of two additional residues at the active site result in a 1.8 × 104-fold drop in the rate of substrate cleavage (Table 4). For this reason, we could not follow the whole reaction pathway in the stopped-flow experiments. Nevertheless, the products of abasic DNA incision by the APE1 mutant were shown to eventually accumulate as revealed by 20% denaturing PAGE analysis. On the basis of a rather modest decrease in the affinity for the substrate according to the stopped-flow data, we do not agree with the earlier hypothesis (35, 38, 39) that these residues, in particular, the hydroxyl group of Tyr171, are absolutely required for the proper AP site recognition and binding. Rather, the observed efficiency of AP-substrate conversion to the product by mutant APE1 indicates that the catalytic step of the mechanism is the main one affected by this substitution.
Figure 8.
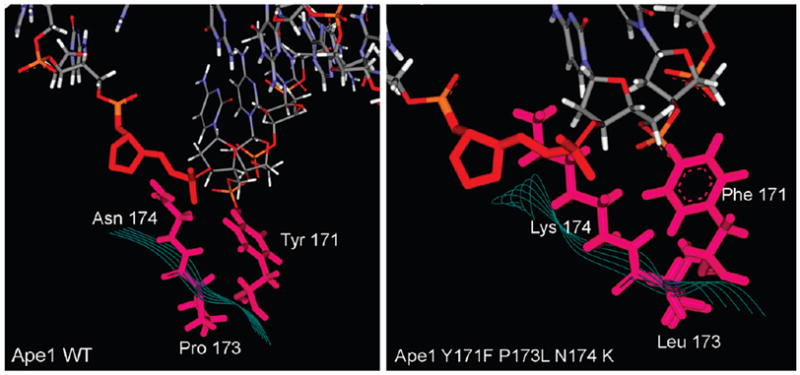
Structure of the wild-type formofAPE1 in comparison with the structure of the active site mutant Y171F-P173L-N174K. Models are based on PDB file 1DEW (8).
Trp fluorescence kinetic studies cannot be used to directly determine the rate of formation of the DNA incision product. Therefore, kinetic Schemes 1 and 2, proposed inthis work, reflect mainly the behavior of the APE1 molecule. It can be seen from Table 2 that the rate constants of the step reflected in the increase in fluorescence and the release of the free enzyme, kirrX, are 3.20 s−1 and 2.05 s−1 for the AP- and F-substrate, respectively. The rate constant of this step coincides with the rate constant of an irreversible step limiting the overall reaction rate as measured in ref 25. Considering that the stopped-flow assay does not directly register product accumulation, we propose that this irreversible step may correspond to a phosphodiester bond hydrolysis reaction and/or some conformational transition in the APE1 molecule, resulting in the release of the product ODNs. Scheme 1, which we use to describe the conversion of specific AP- and F-substrates, is identical to Scheme 5 proposed by Maher and Bloom for the AP-substrate (25) and introduces one extra step for the F-substrate (cf. solid and dashed fit curves in Figure 2). Thus, our data do not contradict the hypothesis that the rate-limiting step of the reaction is an irreversible conformational change in the complex of APE1 with the cleaved DNA (EP) (25). Calculation of Michaelis–Menten kcat produced the values 2.8 and 1.0 s−1 for the AP- and F-substrate, respectively, in good agreement with the irreversible rate constants for both DNA substrates and literature reports (24). Thus, the obtained values of kcat and kirr confirm that the irreversible step likely reflects the chemical incision of the substrate.
The stopped-flow fluorescence study of human APE1 protein presented here has revealed conformational mobility of the enzyme in the course of recognition and catalytic incision of AP- or F-containing DNA. The APE1 molecule was shown to undergo at least four conformational transitions, including nonspecific encounter complex formation, mutual adjustment of the enzyme and DNA substrate structures for catalysis, catalytic incision of the substrate, and release of the enzyme from its complex with the product. Our findings also suggest that the C1′-hydroxyl moiety of the abasic site is required for the most effective recognition and catalysis. Detailed characterization of amino acid side chains which are responsible for the coordination of the abasic sugar moiety is the object of future studies.
Supplementary Material
Acknowledgments
We thank Dr. Dmitri Pyshnyi and Mrs. Tatiana Bushueva for the synthesis of oligonucleotide substrates.
Footnotes
This work was supported in part by grants from the Siberian Branch of the Russian Academy of Sciences (Nos.: 28, 38, 48, 90); the Russian Foundation for Basic Research (10-04-00070, 08-04-00334), the Russian Ministry of Education and Science (NS-3185.2010.4, State Contracts 02.740.11.0079 and 02.740.11.5012); and the Presidium of the Russian Academy of Sciences (22.21).
Abbreviations: AP, abasic; APE1, apurinic/apyrimidinic endonuclease; BER, base excision repair; F, (3-hydroxytetrahydrofuran-2-yl)methyl phosphate; ODN, oligodeoxyribonucleotide.
SUPPORTING INFORMATION AVAILABLE
Circular dichroism spectra were performed on the wild-type APE1 protein at both 20 °C and at 27 °C and on the mutant protein (Y171F-P173L-N174K) at 20 °C. The intrinsic fluorescence spectrum of free APE1 and APE1–G-ligand complex were recorded. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lindahl T, Nyberg B. Rate of depurination of native deoxyribonucleic acid. Biochemistry. 1972;11:3610–3618. doi: 10.1021/bi00769a018. [DOI] [PubMed] [Google Scholar]
- 2.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 3.Nakamura J, Swenberg JA. Endogenous apurinic/apyrimidinic sites in genomic DNA of mammalian tissues. Cancer Res. 1999;59:2522–2526. [PubMed] [Google Scholar]
- 4.Dianov GL, Sleeth KM, Dianova II, Allinson SL. Repair of abasic sites in DNA. Mutat Res. 2003;531:157–163. doi: 10.1016/j.mrfmmm.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 5.Lindahl T. Suppression of spontaneous mutagenesis in human cells by DNA base excision-repair. Mutat Res. 2000;462:129–135. doi: 10.1016/s1383-5742(00)00024-7. [DOI] [PubMed] [Google Scholar]
- 6.Lindahl T, Barnes DE. Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol. 2000;65:127–133. doi: 10.1101/sqb.2000.65.127. [DOI] [PubMed] [Google Scholar]
- 7.Demple B, Sung JS. Molecular and biological roles of Ape1 protein in mammalian base excision repair. DNA Repair. 2005;4:1442–1449. doi: 10.1016/j.dnarep.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 8.Mol CD, Izumi T, Mitra S, Tainer JA. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature. 2000;403:451–456. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- 9.Wilson DM, III, Barsky D. The major human abasic endonuclease: formation, consequences and repair of abasic lesions in DNA. Mutat Res. 2001;485:283–307. doi: 10.1016/s0921-8777(01)00063-5. [DOI] [PubMed] [Google Scholar]
- 10.Fan J, Wilson DM., III Protein-protein interactions and posttranslational modifications in mammalian base excision repair. Free Radical Biol Med. 2005;38:1121–1138. doi: 10.1016/j.freeradbiomed.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 11.Ischenko AA, Saparbaev MK. Alternative nucleotide incision repair pathway for oxidative DNA damage. Nature. 2002;415:183–187. doi: 10.1038/415183a. [DOI] [PubMed] [Google Scholar]
- 12.Abate C, Patel L, Rauscher FJ, III, Curran T. Redox regulation of fos and jun DNA-binding activity in vitro. Science. 1990;249:1157–1161. doi: 10.1126/science.2118682. [DOI] [PubMed] [Google Scholar]
- 13.Gaiddon C, Moorthy NC, Prives C. Ref-1 regulates the transactivation and pro-apoptotic functions of p53 in vivo. EMBO J. 1999;18:5609–5621. doi: 10.1093/emboj/18.20.5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xanthoudakis S, Curran T. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J. 1992;11:653–665. doi: 10.1002/j.1460-2075.1992.tb05097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xanthoudakis S, Miao G, Wang F, Pan YC, Curran T. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 1992;11:3323–3335. doi: 10.1002/j.1460-2075.1992.tb05411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xanthoudakis S, Smeyne RJ, Wallace JD, Curran T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc Natl Acad Sci USA. 1996;93:8919–8923. doi: 10.1073/pnas.93.17.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y, Shupenko CC, Melo LF, Strauss PR. DNA repair protein involved in heart and blood development. Mol Cell Biol. 2006;26:9083–9093. doi: 10.1128/MCB.01216-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Silber JR, Bobola MS, Blank A, Schoeler KD, Haroldson PD, Huynh MB, Kolstoe DD. The apurinic/apyrimidinic endonuclease activity of Ape1/Ref-1 contributes to human glioma cell resistance to alkylating agents and is elevated by oxidative stress. Clin Cancer Res. 2002;8:3008–3018. [PubMed] [Google Scholar]
- 19.Liu L, Yan L, Donze JR, Gerson SL. Blockage of abasic site repair enhances antitumor efficacy of 1,3-bis-(2-chloroethyl)-1-nitrosourea in colon tumor xenografts. Mol Cancer Ther. 2003;2:1061–1066. [PubMed] [Google Scholar]
- 20.McNeill DR, Wilson DM., III A dominant-negative form of the major human abasic endonuclease enhances cellular sensitivity to laboratory and clinical DNA-damaging agents. Mol Cancer Res. 2007;5:61–70. doi: 10.1158/1541-7786.MCR-06-0329. [DOI] [PubMed] [Google Scholar]
- 21.Walker LJ, Craig RB, Harris AL, Hickson ID. A role for the human DNA repair enzyme HAP1 in cellular protection against DNA damaging agents and hypoxic stress. Nucleic Acids Res. 1994;22:4884–4889. doi: 10.1093/nar/22.23.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang D, Luo M, Kelley MR. Human apurinic endonuclease 1 (APE1) expression and prognostic significance in osteosarcoma: enhanced sensitivity of osteosarcoma to DNA damaging agents using silencing RNA APE1 expression inhibition. Mol Cancer Ther. 2004;3:679–686. [PubMed] [Google Scholar]
- 23.Mol CD, Hosfield DJ, Tainer JA. Abasic site recognition by two apurinic/apyrimidinic endonuclease families in DNA base excision repair: the 3′ ends justify the means. Mutat Res. 2000;460:211–229. doi: 10.1016/s0921-8777(00)00028-8. [DOI] [PubMed] [Google Scholar]
- 24.Strauss PR, Beard WA, Patterson TA, Wilson SH. Substrate binding by human apurinic/apyrimidinic endonuclease indicates a Briggs-Haldane mechanism. J Biol Chem. 1997;272:1302–1307. doi: 10.1074/jbc.272.2.1302. [DOI] [PubMed] [Google Scholar]
- 25.Maher RL, Bloom LB. Pre-steady-state kinetic characterization of the AP endonuclease activity of human AP endonuclease 1. J Biol Chem. 2007;282:30577–30585. doi: 10.1074/jbc.M704341200. [DOI] [PubMed] [Google Scholar]
- 26.Chance B, Gibson QH, Eisenhardt RH, Lonberg-Holm KK. Rapid Mixing and Sampling Techniques. Science. 1964;146:1697–1699. doi: 10.1126/science.146.3652.1697. [DOI] [PubMed] [Google Scholar]
- 27.Johnson KA. Advances in transient-state kinetics. Curr Opin Biotechnol. 1998;9:87–89. doi: 10.1016/s0958-1669(98)80089-x. [DOI] [PubMed] [Google Scholar]
- 28.Northrop DB, Simpson FB. Beyond enzyme kinetics: direct determination of mechanisms by stopped-flow mass spectrometry. Bioorg Med Chem. 1997;5:641–644. doi: 10.1016/s0968-0896(97)00020-5. [DOI] [PubMed] [Google Scholar]
- 29.Galletto R, Jezewska MJ, Bujalowski W. Kinetic mechanism of rat polymerase beta-dsDNA interactions. Fluorescence stopped-flow analysis of the cooperative ligand binding to a two-site one-dimensional lattice. Biochemistry. 2005;44:1251–1267. doi: 10.1021/bi0487037. [DOI] [PubMed] [Google Scholar]
- 30.Fedorova OS, Nevinsky GA, Koval VV, Ishchenko AA, Vasilenko NL, Douglas KT. Stopped-flow kinetic studies of the interaction between Escherichia coli Fpg protein and DNA substrates. Biochemistry. 2002;41:1520–1528. doi: 10.1021/bi011524u. [DOI] [PubMed] [Google Scholar]
- 31.Koval VV, Kuznetsov NA, Zharkov DO, Ishchenko AA, Douglas KT, Nevinsky GA, Fedorova OS. Pre-steady-state kinetics shows differences in processing of various DNA lesions by Escherichia coli formamidopyrimidine-DNA glycosylase. Nucleic Acids Res. 2004;32:926–935. doi: 10.1093/nar/gkh237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuznetsov NA, Koval VV, Zharkov DO, Vorobjev YN, Nevinsky GA, Douglas KT, Fedorova OS. Pre-steady-state kinetic study of substrate specificity of Escherichia coli formamidopyrimidine--DNA glycosylase. Biochemistry. 2007;46:424–435. doi: 10.1021/bi060787r. [DOI] [PubMed] [Google Scholar]
- 33.Kuznetsov NA, Koval VV, Zharkov DO, Nevinsky GA, Douglas KT, Fedorova OS. Kinetics of substrate recognition and cleavage by human 8-oxoguanine-DNA glycosylase. Nucleic Acids Res. 2005;33:3919–3931. doi: 10.1093/nar/gki694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuznetsov NA, Koval VV, Nevinsky GA, Douglas KT, Zharkov DO, Fedorova OS. Kinetic conformational analysis of human 8-oxoguanine-DNA glycosylase. J Biol Chem. 2007;282:1029–1038. doi: 10.1074/jbc.M605788200. [DOI] [PubMed] [Google Scholar]
- 35.Beernink PT, Segelke BW, Hadi MZ, Erzberger JP, Wilson DM, III, Rupp B. Two divalent metal ions in the active site of a new crystal form of human apurinic/apyrimidinic endonuclease, Ape1: implications for the catalytic mechanism. J Mol Biol. 2001;307:1023–1034. doi: 10.1006/jmbi.2001.4529. [DOI] [PubMed] [Google Scholar]
- 36.Timofeyeva NA, Koval VV, Knorre DG, Zharkov DO, Saparbaev MK, Ishchenko AA, Fedorova OS. Conformational dynamics of human AP endonuclease in base excision and nucleotide incision repair pathways. J Biomol Struct Dyn. 2009;26:637–652. doi: 10.1080/07391102.2009.10507278. [DOI] [PubMed] [Google Scholar]
- 37.Nguyen LH, Barsky D, Erzberger JP, Wilson DM., III Mapping the protein-DNA interface and the metal-binding site of the major human apurinic/apyrimidinic endonuclease. J Mol Biol. 2000;298:447–459. doi: 10.1006/jmbi.2000.3653. [DOI] [PubMed] [Google Scholar]
- 38.Melo LF, Mundle ST, Fattal MH, O’Regan NE, Strauss PR. Role of active site tyrosines in dynamic aspects of DNA binding by AP endonuclease. DNA Repair. 2007;6:374–382. doi: 10.1016/j.dnarep.2006.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mundle ST, Fattal MH, Melo LF, Coriolan JD, O’Regan NE, Strauss PR. Novel role of tyrosine in catalysis by human AP endonuclease 1. DNA Repair. 2004;3:1447–1455. doi: 10.1016/j.dnarep.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 40.Mundle ST, Delaney JC, Essigmann JM, Strauss PR. Enzymatic mechanism of human apurinic/apyrimidinic endonuclease against a THF AP site model substrate. Biochemistry. 2009;48:19–26. doi: 10.1021/bi8016137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoehn ST, Turner CJ, Stubbe J. Solution structure of an oligonucleotide containing an abasic site: evidence for an unusual deoxyribose conformation. Nucleic Acids Res. 2001;29:3413–3423. doi: 10.1093/nar/29.16.3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Breslauer KJ, Frank R, Blocker H, Marky LA. Predicting DNA duplex stability from the base sequence. Proc Natl Acad Sci USA. 1986;83:3746–3750. doi: 10.1073/pnas.83.11.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sugimoto N, Nakano S, Yoneyama M, Honda K. Improved thermodynamic parameters and helix initiation factor to predict stability of DNA duplexes. Nucleic Acids Res. 1996;24:4501–4505. doi: 10.1093/nar/24.22.4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gelfand CA, Plum GE, Grollman AP, Johnson F, Breslauer KJ. Thermodynamic consequences of an abasic lesion in duplex DNA are strongly dependent on base sequence. Biochemistry. 1998;37:7321–7327. doi: 10.1021/bi9803372. [DOI] [PubMed] [Google Scholar]
- 45.Kuzmic P. Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 46.House JE. Principles of Chemical Kinetics. 2. Academic Press; New York: 2007. [Google Scholar]
- 47.Wilson DM, III, Takeshita M, Grollman AP, Demple B. Incision activity of human apurinic endonuclease (Ape) at abasic site analogs in DNA. J Biol Chem. 1995;270:16002–16007. doi: 10.1074/jbc.270.27.16002. [DOI] [PubMed] [Google Scholar]
- 48.Yu E, Gaucher SP, Hadi MZ. Probing conformational changes in Ape1 during the progression of base excision repair. Biochemistry. 2010;49:3786–3796. doi: 10.1021/bi901828t. [DOI] [PubMed] [Google Scholar]
- 49.Takeshita M, Chang CN, Johnson F, Will S, Grollman AP. Oligodeoxynucleotides containing synthetic abasic sites. Model substrates for DNA polymerases and apurinic/apyrimidinic endonucleases. J Biol Chem. 1987;262:10171–10179. [PubMed] [Google Scholar]
- 50.Rachofsky EL, Seibert E, Stivers JT, Osman R, Ross JB. Conformation and dynamics of abasic sites in DNA investigated by time-resolved fluorescence of 2-aminopurine. Biochemistry. 2001;40:957–967. doi: 10.1021/bi001665g. [DOI] [PubMed] [Google Scholar]
- 51.Berquist BR, McNeill DR, Wilson DM., III Characterization of abasic endonuclease activity of human Ape1 on alternative substrates, as well as effects of ATP and sequence context on AP site incision. J Mol Biol. 2008;379:17–27. doi: 10.1016/j.jmb.2008.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Volkenstein MV, Goldstein BN. Allosteric enzyme models and their analysis by the theory of graphs. Biochim Biophys Acta. 1966;115:478–485. doi: 10.1016/0304-4165(66)90446-6. [DOI] [PubMed] [Google Scholar]
- 53.Oezguen N, Schein CH, Peddi SR, Power TD, Izumi T, Braun W. A “moving metal mechanism” for substrate cleavage by the DNA repair endonuclease APE-1. Proteins. 2007;68:313–323. doi: 10.1002/prot.21397. [DOI] [PubMed] [Google Scholar]
- 54.Lucas JA, Masuda Y, Bennett RA, Strauss NS, Strauss PR. Single-turnover analysis of mutant human apurinic/apyrimidinic endonuclease. Biochemistry. 1999;38:4958–4964. doi: 10.1021/bi982052v. [DOI] [PubMed] [Google Scholar]
- 55.Kaneda K, Sekiguchi J, Shida T. Role of the tryptophan residue in the vicinity of the catalytic center of exonuclease III family AP endonucleases: AP site recognition mechanism. Nucleic Acids Res. 2006;34:1552–1563. doi: 10.1093/nar/gkl059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Erzberger JP, Wilson DM., III The role of Mg2+ and specific amino acid residues in the catalytic reaction of the major human abasic endonuclease: new insights from EDTA-resistant incision of acyclic abasic site analogs and site-directed mutagenesis. J Mol Biol. 1999;290:447–457. doi: 10.1006/jmbi.1999.2888. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.