

Abstract
Early‐phase clinical development in oncology has evolved dramatically with the deciphering of the human genome in 2004. Genomic analysis and the tools identifying genetically disrupted pathways within a patient's tumor have been a driving force for personalized medicine and for the development of highly targeted novel therapies. Tumors are often genetically heterogeneous, with multiple concurrent genetic abnormalities. On the other hand, tumors arising from different tissues may share identical molecular drivers.
An increased understanding of these underlying molecular drivers and the discovery of agents targeting them has resulted in a shift toward clinical trials that evaluate only those patients whose tumors contain these relevant mutations. As a result, a number of “Umbrella” trials have been conducted (i.e., BATTLE, I‐SPY2, Lung‐MAP), in which patients with a particular tumor type are profiled and preferentially assigned to receive treatment with agents specifically targeting their tumor's molecular drivers. This approach increases the chance for a patient to receive a novel targeted agent and thereby improves the odds of clinical benefit.
An alternative, but complementary, approach is the “basket” trial, wherein tumors of a variety of different histologies are treated with a specific agent on the basis of a common set of driver mutations. In this approach, the trial is centered on the molecular target for the compound, and patients are enrolled based upon the presence of relevant mutation(s). This type of trial is an efficient and rapid method for determining whether a targeted treatment might have activity across one or more tumor types. There are several other examples of this approach in development including the NCI Molecular Analysis for Therapy Choice Program (MATCH) and Pediatric MATCH programs that are anticipated to launch in 2015.1 The Signature Program, launched in March 2013 by Novartis (www.signaturetrial.com), is a unique basket trial consisting of multiple single agent protocols that enroll multiple tumor types in a tissue‐agnostic manner, with key inclusion criteria being the presence of an actionable mutation or pathway activation.
The Signature Program currently consists of eight exploratory signal‐finding studies, in which research‐qualified physicians, either in a community or academic setting, can match eligible patients with targeted therapies. Mutation profiling is done in local Clinical Laboratory Improvement Amendments (CLIA)‐certified laboratories, prior to participation in the Signature Program (Figure 1). When a patient is identified as having an actionable mutation, their oncologist contacts the Signature Trial call center to confirm the patient's potential eligibility to participate in the program. Only after this step will a protocol package, including a fixed contract, central Institutional Review Board (IRB)–approved protocol, standard budget, and standard set of informed consent forms, be sent to the site. On average, the process from contacting Novartis to new site initiation is 6 weeks, which is much shorter than the typical 34‐week site initiation timeline.2, 3
Figure 1.
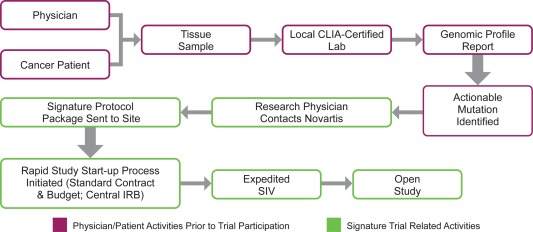
Schematic representation of the Signature Program study start‐up design. This study is intended for patients who have already had genomic profiling of their tumors in a CLIA‐certified laboratory and have been preidentified to have relevant pathway activation. Once the patient has been identified, treating physicians who are qualified investigators may contact Novartis to consider enrollment in this study. A rapid study start‐up process ensues, followed by site initiation, patient consent, and dosing. SIV, site initiation visit.
The study schema for the Signature Trial is shown in Figure 2. The primary endpoint of the Signature study is the clinical benefit rate associated with drug treatment based on local investigator assessment. The statistical design is similar for each study and utilizes a patient‐sparing approach in which a minimum of four patients is required to establish a disease cohort for a particular histology. A lack of clinical benefit seen in at least 10 patients within a particular cohort establishes futility, whereas clinical benefit observed in a cohort of 15–30 patients, depending on the strength of the response signal, will confirm a positive signal.
Figure 2.
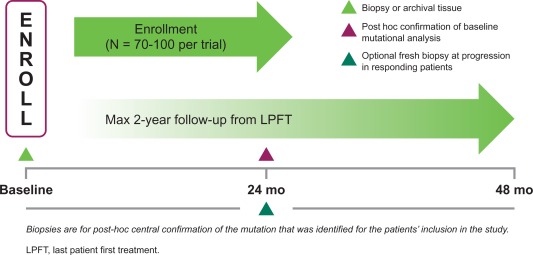
Signature Trial study schema.
A Bayesian adaptive design with a hierarchical model that allows dynamic borrowing of information across subgroups is used for the analysis of the clinical benefit rate.4 Such an analysis produces higher power or lower type I error where some commonality is observed (identical effects are not required) among the subgroups and allows evaluation of clinical benefit with very small sample sizes in each subgroup. At each interim analysis, the subgroups are evaluated for early futility or early success by comparing posterior quantities for the response rate based on the observed response data and the assumed prior distribution of response with prespecified early stopping criteria.
During the past 18 months, over 300 patients have been enrolled in the Signature Program at 224 study sites.2 The targeted agents that have been evaluated so far include BJG398 (FGFR), binimetinib (RAS/RAF/MEK/ERK and NF1), buparlisib (PI3K/PTEN), ceritinib (ALK/ROS1), dovitinib (multiple receptor tyrosine kinases), encorafenib (BRAF), sonidegib (PTCH/SMO), and ribociclib (CDK4/CDK6/p16/CCND1/ CCND3). The majority of these compounds are in the early stages of development. Standardization of the design approach has allowed new protocols to be added to the program easily as novel compounds reach a point early in development where broad testing across multiple tumors is appropriate.
From the perspective of rapid and efficient development of novel targeted therapies and the ability to quickly define the broadest clinical value for these molecules, the Signature Program offers several clear advantages over traditional trial approaches. For example, in the absence of a fixed number of study sites, any physician with adequate research capability can identify and nominate patients for an appropriate protocol. Speed of accrual is, therefore, not limited by study site number, but rather by physician awareness of the studies and the level of ongoing genetic screening of their cancer patients. Existing data indicate that study start‐up and site maintenance costs can range as high as $50,000 per site and, across the pharmaceutical industry, ∼30% of opened trial sites do not enroll patients.5 By eliminating unproductive sites, and focusing only on patients whose tumors harbor relevant mutations, the Signature Program minimizes costs while maximizing the accumulation of clinical data. Moreover, the program optimizes the opportunity for clinical benefit, which encourages participation by oncologists and their patients.
The results from trials in the Signature Program are limited to the identification of early clinical signals in a heterogeneous population. Tissue‐specific studies with appropriate comparator arms would be initiated in the event that a signal is identified. In the Signature Program, it is also possible that the initial patient enrolled at a site may not be dosed within 6 weeks because their disease progresses prior to enrollment. However, once the site is open, this limitation is removed and patients can be enrolled into the program without delay. The program is also limited by the possibility that too few patients will accrue in a particular indication to allow for a definitive determination of futility or efficacy. Furthermore, as is the case in many studies, the determination of patients' overall survival is influenced by subsequent therapies.
Experience with the program thus far suggests that an institutional program champion is critical to effective implementation of this novel approach. Tumor‐specific trials are limited in scope and number, so when faced with a patient who has a relevant actionable tumor mutation and has failed on standard therapies, physicians are motivated to open their site and begin treatment quickly. The Signature Program study design can provide rapid signal finding that benefits both patients and drug development.
The Signature Program has been a unique approach, focusing specifically on patients' molecular data and offering early pipeline agents to patients with targetable mutations, regardless of the tumor histology, within a single study. Moreover, its flexible design retains the power to analyze accrued indication‐specific cohorts. As these cohorts are not predetermined, their accrual rate is a barometer for future confirmatory trial feasibility. Since sites are not preselected, the program facilitates the enrollment of patients with rare tumor types at local sites, thereby more fully engaging both academic and community practices and reducing travel for patients. This type of program may also accelerate the drug development process by efficiently matching the correct therapy to the most appropriate patient. Already, promising results have been observed for some patients, and these will be the topic of future publications. Furthermore, a Signature‐based approach can be incorporated more broadly into confirmatory trials in the future.
ACKNOWLEDGMENT
Financial support for medical editorial assistance was provided by Novartis Pharmaceuticals. The authors thank Daniel Hutta, PhD (Articulate Science) for medical editorial assistance with this manuscript.
CONFLICT OF INTEREST
B.P.K., E.S., S.S., C.L., S.S., and A.S. are employees of Novartis Pharmaceuticals. D.B. is co‐owner of Berry Consultants, LLC, a company that designs adaptive clinical trials for pharmaceutical and medical device companies and international consortia.
AUTHOR CONTRIBUTIONS
B.P.K. and E.S. contributed equally to this work.
References
- 1. Redig, A.J. & Janne, P.A. Basket trials and the evolution of clinical trial design in an era of genomic medicine. J. Clin. Oncol. 33, 975–977 (2015). [DOI] [PubMed] [Google Scholar]
- 2. Giles, F. et al The signature program, a distinctive tissue agnostic trial model for molecularly pre‐selected hematological and solid tumor patients [abstract 4818]. Blood 124, 4818 (2014). [Google Scholar]
- 3. Lamberti, M.J. , Brothers, C. , Manak, D. & Getz, K. Benchmarking the study initiation process. Ther. Innovat. Regul. Sci. 47, 101–109 (2013). [DOI] [PubMed] [Google Scholar]
- 4. Berry, S.M. , Broglio, K.R. , Groshen, S. & Berry, D.A. Bayesian hierarchical modeling of patient subpopulations: efficient designs of phase II oncology clinical trials. Clin. Trials 10, 720–734 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Steensma, D.P. & Kantarjian, H.M. Impact of cancer research bureaucracy on innovation, costs, and patient care. J. Clin. Oncol. 32, 376–378 (2014). [DOI] [PubMed] [Google Scholar]