

Abstract
The Reelin signaling pathway is implicated in processes controlling synaptic plasticity and hippocampus-dependent learning and memory. A single direct in vivo application of Reelin enhances long-term potentiation, increases dendritic spine density and improves associative and spatial learning and memory. Angelman syndrome (AS) is a neurological disorder that presents with an overall defect in synaptic function, including decreased long-term potentiation, reduced dendritic spine density, and deficits in learning and memory, making it an attractive model in which to examine the ability of Reelin to recover synaptic function and cognitive deficits. In this study, we investigated the effects of Reelin administration on synaptic plasticity and cognitive function in a mouse model of AS and demonstrated that bilateral, intraventricular injections of Reelin recover synaptic function and corresponding hippocampus-dependent associative and spatial learning and memory. Additionally, we describe alteration of the Reelin profile in tissue from both the AS mouse and post-mortem human brain.
Keywords: behavior, cannulation, hippocampus, ubiquitin ligase, ventricle
Introduction
Angelman syndrome (AS) is a neuro-genetic disorder caused by disruption of the imprinted and maternally expressed UBE3A gene that occurs in approximately 1 : 12 000 live births (Steffenburg et al., 1996) and currently has no approved treatment. Children with this disorder present with developmental delay, severe speech impairment, ataxia, happy demeanor, and high seizure propensity (Williams et al., 2006). The AS mouse model was created by a null mutation of UBE3A, resulting in synaptic dysfunction and disruption in spatial and associative memory formation (Jiang et al., 1998). Specifically, there are severe hippocampal long-term potentiation (LTP) deficits and impairments in memory formation associated with spatial learning via the hidden platform water maze and associative fear conditioning (Jiang et al., 1998; van Woerden et al., 2007).
Reelin, a ligand of the lipoprotein receptor family, is involved in embryonic neuronal migration and positioning (Goffinet, 1982; Hack et al., 2002; Heinrich et al., 2006; Won et al., 2006; Jossin et al., 2007) as well as modulating adult synaptic plasticity (Niu et al., 2004; Rogers & Weeber, 2008; Rogers et al., 2011, 2013; Trotter et al., 2013). In the developing brain, Reelin is primarily secreted by the Cajal–Retzius cells in the hippocampus and cortex, and is required for proper layering of hippocampal and cortical neurons. In the adult brain, Reelin is primarily secreted by GABAergic interneurons (Alcantara et al., 1998; Pesold et al., 1998). Reelin is shown to regulate α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor insertion and to increase N-methyl-D-aspartate receptor function (Qiu et al., 2006; Rogers et al., 2011). Any disruption to the Reelin signaling pathway, whether it be to the RELN gene, one or both of its lipoprotein receptors (very-low-density lipoprotein receptor and apolipoprotein E receptor 2), or to the adaptor protein Disabled-1, results in severe hippocampal learning and memory deficits, decreased hippocampal LTP response, and decreased dendritic spine density (Howell et al., 1997; Weeber et al., 2002; Arnaud et al., 2003; D’Arcangelo, 2006; Rogers et al., 2013; Trotter et al., 2013).
Our recent research revealed the ability of Reelin to enhance LTP, increase dendritic spine density, and improve spatial and associative learning and memory in wild-type mice (Rogers et al., 2011). Furthermore, we have shown that exogenous Reelin can recover defects in the heterozygous reeler mouse, which displays significantly reduced levels of the Reelin protein compared with wild-type mice and exhibits deficits in synaptic function and learning and memory (Rogers et al., 2013). Ube3a has not been shown to be directly associated with the lipoprotein signaling system; however, there is an interesting similarity between the biochemical and behavioral changes associated with the absence of Ube3a in the AS mouse model and those associated with altered Reelin signaling. For example, both result in altered α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor surface expression, dendritic spine density, excitatory synaptic plasticity and GABAergic function and tone. Thus, if the absence of Ube3a alters common pathways involved in Reelin signaling, specifically those shown to be enhanced with Reelin supplementation, then Reelin supplementation may ameliorate the severity of the established AS mouse phenotype.
Materials and methods
Animals
The UBE3A null mutation (AS) mice, described previously (Jiang et al., 1998), and wild-type (129/SvEv) littermate control mice were originally obtained from The Jackson Laboratory (Bar Harbor, ME, USA). Mice were backcrossed to the 129/SvEv line for at least five generations. Breeding pairs consisted of female mice containing the null mutation with wild-type males to produce maternally-deficient AS offspring and wild-type littermate controls. Animals were housed with a standard 12 h light/dark cycle and supplied with food pellets and water ad libitum at the University of South Florida. Experiments were performed on 12–18-week-old mice. All animal testing procedures and care followed the NIH guidelines and were approved by the University of South Florida’s Institutional Animal Care and Use Committee.
Human tissue
Human AS brain tissue samples and age-matched controls were acquired from The National Institute of Child Health & Human Development at the University of Maryland, School of Medicine. The research protocol was approved by the Institutional Review Board of the University of South Florida, College of Medicine. Patients 1754 and 5418 were confirmed 15q11-q13 deletion cases, whereas the remaining two AS donors had no information regarding the initial diagnosis. After the collection of post-mortem tissue, individual samples were frozen in isopentane/dry ice at between −30 and −40 °C and then stored at −85 °C. Frozen post-mortem samples were collected from eight individuals ranging in age from 4 to 43 years (Table1). The storage times for the various samples ranged from approximately 6 to 11 years.
Table 1.
Tissue samples from four patients with AS and four age-matched controls were used to determine the relative levels of Reelin expression
| Age | |||||||
|---|---|---|---|---|---|---|---|
| University of Maryland Brain & Tissue Bank no. | Group | Years | Days | Gender | Race | Cause of Death | Post-mortem interval (h) |
| 1824 | AS | 4 | 46 | Male | Caucasian | Reactive airway disease | 30 |
| 1754 | AS | 4 | 0 | Male | Caucasian | Drowning | 24 |
| 5418 | AS | 43 | 85 | Female | Caucasian | Subarachnoid hemorrhage | 21 |
| M3845M | AS | 27 | 157 | Female | Caucasian | Cerebral edema | 21 |
| 1499 | Control | 4 | 170 | Female | Asian | Lymphocytic myocarditis | 21 |
| 4670 | Control | 4 | 237 | Male | Caucasian | Commotio cordis | 17 |
| 4636 | Control | 43 | 52 | Female | Caucasian | Pulmonary embolism | 19 |
| 1614 | Control | 27 | 149 | Female | Caucasian | Gunshot wound to abdomen | 18 |
Reelin purification
The HEK293 cells were stably transfected with a full-length Reelin construct in pCrl vector to produce recombinant Reelin, as previously described (Weeber et al., 2002; Sinagra et al., 2005). Briefly, once confluent, cells were grown in low-glucose Dulbecco’s modified Eagle’s medium with 0.2% bovine serum albumin for 2 days, followed by media collection, sterile filtration, and concentration by Centricon Plus-80 centrifugal filter units (Millipore). Mock control media were collected from non-Reelin construct transfected HEK293 cells, and purified by the same method as above (Sinagra et al., 2005; Rogers et al., 2013). Reelin is cleaved extracellularly at two sites, resulting in the generation of three major fragments: the N-terminus to repeat 2 (roughly 180 kDa), the central fragment from repeat 3–6 (roughly 190 kDa), and the C-terminal fragment consisting of repeats 7 and 8 (roughly 80 kDa) (Nakajima et al., 1997; de Rouvroit et al., 1999; Utsunomiya-Tate et al., 2000; Jossin et al., 2004, 2007; Krstic et al., 2012; Koie et al., 2014; Trotter et al., 2014). Additionally, two intermediate fragments are produced: one consisting of the N-terminus to repeat 6 (roughly 370 kDa), and one consisting of repeats 6–8 (roughly 270 kDa) (Jossin et al., 2004). Each of these fragments and full-length Reelin are found in our Reelin preparation.
Direct bilateral ventricular injections
Mice were anesthetised with isoflurane and placed on a stereotaxic surgery apparatus (Stoelting Co.). A sagittal incision was made mid-cranium, and the skin gently pushed back to enlarge the opening. Two holes were drilled through the skull to allow passage of the Hamilton needle through the brain, into the ventricles (AP −0.35 mm, L ± 0.75 mm, and V −2.5 mm from bregma). Mice were injected bilaterally with 0.5 μL mock control or Reelin to yield a 5 nm total hemisphere concentration of Reelin at a rate of 1 μL/min, as previously established (Weeber et al., 2002; Rogers et al., 2013). The needle was then removed, holes sealed with dental cement, and incisions were sutured. Mice were observed post-operatively for 2 h in individual cages on a warm heating pad. The rectal temperature was measured daily to monitor inflammatory or infectious responses; any mouse with a temperature of 100.5 °F or higher was euthanised via CO2 inhalation. The animals were allowed to recover for 5 days. We have previously shown that this time course is optimal for behavioral examination post-intraventricular injection (Rogers et al., 2011, 2013). We have shown that Reelin expression and Disabled-1 phosphorylation are increased at 3 h post-injection, but return to baseline at 5 days post-injection (Rogers et al., 2011, 2013).
Golgi staining and in vivo dendritic spine analysis
At 5 days post-injection, brains were dissected out and hemi-bisected. Half hemispheres were processed for Golgi staining, as previously described (Chen et al., 2006), and the remaining half used for electrophysiology. Brains were sectioned to a thickness of 150 μm using a Vibratome (VT1000S; Leica). Images (at 63× magnification) of hippocampal CA1 pyramidal neurons were taken via bright-field microscopy (Axioplan 2, Zeiss). Images were analysed (AxioVision, Zeiss) and spines counted along a 20 μm span by an experimenter blinded to the experimental parameters.
Western blotting
Brain tissue samples of the hippocampus and cortex were homogenised in radioimmunoprecipitation assay buffer and protease and phosphatase inhibitors (Millipore: 50 nm Tris/HCl, pH 7.4, 1 mm EDTA, 150 nm NaCl, 1% NP-40, 0.25% deoxycholic acid) (Sigma: 1× phosphatase inhibitors I and II, 1× complete protease inhibitors, 1× phenylmethylsulfonyl fluoride). The protein concentration was measured via bicinchoninic acid assay (Pierce, Rockford, IL, USA) and standardised to 2 μg/μL. Equal amounts of protein and Laemmli buffer (Bio-Rad) were loaded into an 18-well, pre-cast polyacrylamide 4–15% gradient gel (Bio-Rad). After transfer, blots were blocked in 5% non-fat dry milk in 1× Tris-buffered saline for 1 h at room temperature (23 ± 2 °C), and then primary G-10 (Millipore) at 1 : 2000 (identifies 450, 370, and 180 kDa Reelin fragments), antibody 142 (Millipore) at 1 : 1000 (identifies 450 and 250 kDa fragments), or ab12 + 14 (generously provided by Andre Goffinet, Université catholique de Louvain, Brussels, Belgium; identifies 450, 370, and 180 kDa Reelin fragments) at 1 : 1000 in block was applied overnight at 4 °C with agitation. After three, 10 min rinses in Tris-buffered saline containing 0.1% Tween 20 (Sigma-Aldrich), the blot was incubated with horseradish peroxidase-conjugated goat anti-mouse (SouthernBiotech) at 1 : 2000, or rabbit anti-goat (Life Technologies) at 1 : 2000, secondary. The same protocols were used to probe the membranes with β-actin (Cell Signaling Technology). Membranes were then washed as before, and protein expression visualised via enhanced chemiluminescence treatment and exposed on film. Protein band concentrations were analysed using ImageJ by evaluating the pixel density. Densitometry was performed using β-actin levels as standard controls.
Extracellular recordings
At 5 days post-injection a cohort of mice were killed and the hippocampi were dissected out to be used in hippocampal LTP experimentation, as previously described (Weeber et al., 2002). Briefly, field excitatory post-synaptic potentials were recorded from hippocampal area CA1 in the stratum radiatum using theta-burst stimulation (five trains of four pulses at 200 Hz separated by 200 ms, repeated a total of six times with a 10 s intertrain interval) to elicit LTP.
Behavioral experimentation
All behavior testing was performed serially in the following order.
Rotarod was used to assess motor coordination, motor learning and stamina. For four trials over two consecutive days, mice were placed on the 3 cm diameter rotarod (Ugo Basile, Italy) at an initial rotation speed of 4 rpm. The rod was accelerated to 40 rpm over 5 min. Mice were tested for latency to fall off the rod.
Open field behavior was assessed to determine general locomotor activity and anxiety. Mice were placed in an open field chamber (40 × 40 × 27 cm) for 15 min. The ANY-Maze animal activity system (Stoelting Co.) was used to monitor immobility, movement, and distance traveled. Anxiety levels were assessed by comparing time spent in the perimeter vs. time spent in the center.
The elevated plus maze was used to assess anxiety levels in the mice. The elevated plus maze was elevated 40 cm off the floor, and consisted of four arms: two (30 × 5 cm) open, well-lit arms opposite each other and two (30 × 5 × 15 cm) enclosed arms opposite each other, with each arm attached to a common open square center platform (4.5 cm). Mice were placed in the center platform and allowed 5 min to explore. A digital camera (XV-BP330, Panasonic) was used to monitor activity, and the ANY-Maze animal activity system (Stoelting Co.) was used to record and analyse mice behavior. The total time spent in open arms vs. closed arms was measured, as well as freezing (immobility for at least 2 consecutive seconds). Anxiety levels were assessed by time spent in the open arms vs. closed arms.
The hidden platform water maze was used to assess spatial learning and memory. A 10 cm diameter platform was submerged just below the surface in a 1.2 m diameter pool filled with opaque water, deep enough that mice could not touch the bottom. Large visual cues were positioned around the room. Mice were placed in the pool and allowed to swim for a maximum of 60 s to find the platform. Training consisted of five training days with four trials per day, separated by a 15 min intertrial interval. On days six and eight, the platform was removed and the ANY-Maze video-tracking software (Stoelting Co.) was used to track each mouse’s swim pattern for 60 s (probe trials).
Contextual fear conditioning was conducted to evaluate fear-based learning and memory. Fear conditioning training was conducted in a square sound attenuation chamber (25 × 25 cm) with a wire grid flooring. In training, mice were placed in the chamber with background white noise and allowed to explore for 3 min before the conditioned stimulus was presented (90 dB tone) for 30 s. At 28 s, the unconditioned stimulus [a mild (0.5 mA) foot shock] was administered for a total of 2 s. After a period of 90 s, a second conditioned stimulus/unconditioned stimulus pairing was conducted, followed by another 90 s period, for a total of 7 min. Contextual fear conditioning was conducted 24 h after the training in the same chamber, with no tone, for 3 min, and freezing was assessed. Cued fear conditioning was conducted following the contextual trial, in which the chamber was altered by scent, lighting, and floor texture. The mice were placed into the altered chamber and given a 3 min habituation phase (no tone) followed by the 90 dB conditioned stimulus (tone) for the last 3 min of the test. Behavior was monitored via ANY-Maze software (Stoelting Co.). Freezing was assessed as immobility for at least 2 s.
Statistical analysis
The Student’s two-tailed t-test was used to analyse open field, elevated plus maze, rotarod, and Reelin protein levels, with significance assigned at P < 0.05 (Prism Software). The dendritic spine density, hippocampal LTP, fear conditioning, and the hidden platform water maze data were analysed using a one-way anova followed by Tukey’s Multiple Comparison post-hoc test, set at a significance of P < 0.05. N refers to the number of animals used in all tests including behavioral testing, western blotting, and LTP analysis.
Results
Dendritic spine density
It has been established that AS mice exhibit reduced apical dendritic spine density in both the hippocampus and cortex of adult mice (Dindot et al., 2008). We have recently shown that Reelin supplementation results in increased hippocampal CA1 dendritic spine density in treated wild-type and Reelin heterozygous mice (Rogers et al., 2011, 2013). Therefore, we sought to determine if Reelin supplementation could recover the apical dendritic spine reduction in the AS mouse. Consistent with previous reports, mock treated AS mice had a significantly reduced apical dendritic spine density compared with mock treated wild-type littermates (P < 0.05). Intraventricular Reelin infusion in AS mice significantly improved the apical dendritic spine density, restoring the spine density to wild-type numbers (Fig.1) (F2,171 = 5.423, P < 0.01).
Figure 1.
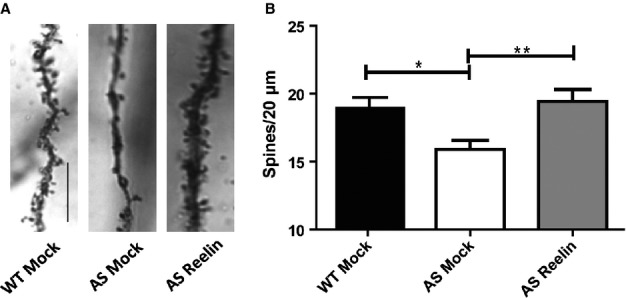
Dendritic spine density is increased in the AS mouse model following a single injection of Reelin. (A) Light micrograph images from Golgi-impregnated hippocampal pyramidal neurons from mock treated wild-type mice (WT Mock), mock treated AS mice (AS Mock) and AS mice at 5 days after Reelin injection (AS Reelin). Scale bar: 10 μm. (B) Quantification of hippocampus area CA1 apical dendrites. Spine density of AS Mock mice is significantly reduced compared with WT Mock littermates. Reelin rescues spine density in the AS mice, restoring it to wild-type levels. WT Mock and AS Reelin, n = 10 slices; AS mock control, n = 11 slices; P < 0.05. *P < 0.05 and **P < 0.01.
Behavioral analysis
The general locomotion and anxiety levels were examined in AS mock treated and Reelin-treated mice beginning at 5 days after single injection. General locomotor behavior, as measured by the open field (Fig.2A and B), and anxiety behavior, as measured by the open field and elevated plus maze, were unaltered by the Reelin treatment (Fig.2C–E, respectively). Several reports demonstrate motor coordination deficits in the AS mouse model using the rotarod test (Jiang et al., 1998; van Woerden et al., 2007). We observed no difference in motor coordination assessed by the rotarod test among mock treated and Reelin-treated AS mice (Fig.2F). These results suggest that a single injection of Reelin did not affect general locomotion, anxiety, or motor coordination in the AS mouse model.
Figure 2.

Reelin injection in AS mice does not affect general locomotion, anxiety or motor coordination. (A) Open field: distance traveled. (B) Open field: time spent immobile. (C) Open field: time spent in center vs. perimeter. (D) Open field: distance traveled in center vs. perimeter. Single injection of Reelin did not affect the general locomotion or anxiety of AS mice. (E) Elevated plus maze: total time spent in open arms, closed arms and center. Anxiety levels in AS mice are unaffected by an injection of Reelin. (F) Rotarod: mice were tested for latency to fall off the rod. Motor coordination, motor learning and stamina were not affected by an injection of Reelin. n = 9/group, P > 0.05 all test comparisons. AS Mock, mock treated AS mice; AS Reelin, AS mice at 5 days after Reelin injection.
Long-term potentiation
Hippocampal LTP is the predominant method used as a measure of synaptic plasticity. The AS mouse model displays severe deficits in the induction of high-frequency stimulation-induced LTP in hippocampal CA1 pyramidal neurons when elicited with multiple established protocols compared with wild-type controls (Jiang et al., 1998; Weeber et al., 2003; van Woerden et al., 2007; Gustin et al., 2010; Daily et al., 2011; Kaphzan et al., 2012; Filonova et al., 2014). Consistent with previous reports, we found that mock treated AS mice demonstrated significant deficits in theta-burst-induced hippocampal LTP compared with mock treated wild-type littermates (Fig.3). Remarkably, the same protocol to elicit LTP in AS mice injected with Reelin 5 days earlier resulted in a very significantly enhanced expression of LTP, enhancement that significantly exceeded the level of LTP achieved by mock treated wild-type littermates (F2,30 = 79.93, P < 0.0001). Given the significant increase in LTP observed after Reelin injection in AS mice, we investigated whether there was a concomitant improvement in hippocampus-dependent learning and memory tasks.
Figure 3.
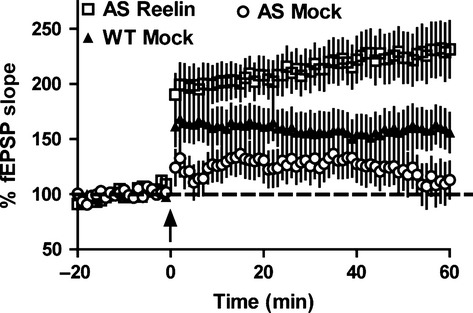
Single Reelin injection enhances LTP in AS mice. Hippocampal LTP was elicited via theta-burst stimulation (five bursts of 200 Hz separated by 200 ms, repeated six times with 10 s between the six trains; arrow) after 20 min baseline recording. Mock treated AS mice (AS Mock) display deficits in LTP compared with mock treated wild-type littermates (WT Mock). A single injection of Reelin in AS mice significantly enhanced hippocampal LTP to even greater than wild-type levels. n = 5 mice per genotype; P < 0.0001; anova. fEPSP, field excitatory post-synaptic potential; AS Reelin, AS mice at 5 days after Reelin injection.
Hippocampus-dependent learning and memory
Contextual fear conditioning was used to assess the mice for their ability to associate an aversive stimulus with their environment. All treatment groups performed similarly during training (data not shown). However, AS mice consistently demonstrate a deficit in contextual fear conditioning (Jiang et al., 1998; van Woerden et al., 2007; Daily et al., 2011). In accordance with these previous reports, we found significant differences among the three treatment groups during the context probe test (F2,34 = 10.96, P < 0.001). Post-hoc analysis revealed that mock treated AS mice exhibited a significant decrease in freezing behavior during the probe trial at 24 h after training (P < 0.01; Fig.4A), whereas Reelin-treated AS mice were indistinguishable from mock treated wild-type littermates. The degree of freezing displayed by the Reelin-treated AS mice compared with mock treated AS mice (F2,30 = 79.93, P < 0.0001) suggested that Reelin treatment restored the ability to associate a context with an unconditioned stimulus. Consistent with previous studies reporting cued fear conditioning to be unaltered in AS mice (Jiang et al., 1998; van Woerden et al., 2007), we observed no change in the amount of freezing in response to the cue in either the mock treated or Reelin-treated AS mice groups compared with mock treated wild-type littermates (Fig.4B).
Figure 4.
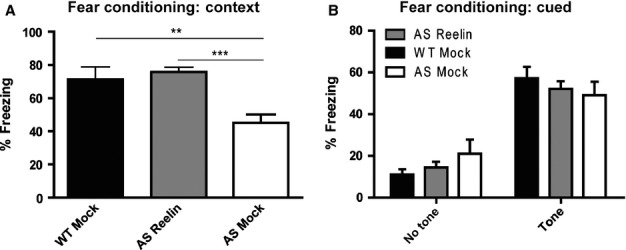
AS mice given a single injection of Reelin display recovered associative learning and memory. (A) Mock treated AS mice (AS Mock) display reduced freezing during context testing performed at 24 h after training compared with mock treated wild-type littermates (WT Mock). Reelin injection rescued freezing behavior of AS mice during context testing. WT Mock and AS Mock, n = 10; AS mice at 5 days after Reelin injection (AS Reelin), n = 17; **P < 0.01 and ***P < 0.001. (B) No differences in freezing behavior were observed among the groups during cued fear conditioning testing performed at 24 h after training.
The hidden platform water maze was used to assess spatial learning and memory. During water maze training, there were no statistically significant differences among the ability of mice to learn to locate the platform (Fig.5A). At 24 h post-training, we also found that there were no differences among groups in their ability to remember the platform location (data not shown), consistent with previous results (Daily et al., 2011). However, at 72 h post-training, there was a significant difference among the groups when looking at the preference for the target quadrant and actual platform crossings. Mock treated AS mice displayed significant reductions in both their number of target platform crossings (P < 0.01; Fig.5B) and time spent searching in the target quadrant (P < 0.001; Fig.5C). A single injection of Reelin rescued these phenotypes in spatial learning and memory in the AS mice when compared with mock treated AS mice (P < 0.001).
Figure 5.

A single injection of Reelin recovers spatial learning and memory in the AS mouse model. (A) All groups performed similarly during hidden platform water maze training. (B) The mean number of platform location crossings during the 72 h probe trial shown for the training quadrant and the corresponding locations in other quadrants. (C) Time spent in each quadrant at 72 h post-training. All mice displayed a preference for the pool quadrant in which the platform was located during training. However, mock treated AS mice (AS Mock) demonstrate reduced preference for the target quadrant compared with mock treated wild-type littermates (WT Mock) that can be enhanced by a single injection of Reelin. WT Mock and AS mice at 5 days after Reelin injection (AS Reelin), n = 10; AS mock control, n = 8; **P < 0.01; ***P < 0.001.
The Reelin expression profile
Our electrophysiology and behavior results, combined with the phenotypic similarities between the AS mouse model and the haplo-insufficiency Reelin mouse model, suggested to us that we examine the endogenous Reelin protein profile in the cortex and hippocampus of the AS mouse. Interestingly, we found that the Reelin protein profile was altered in both the hippocampus and cortex of the AS mouse brain. Specifically, we observed a decrease in the 180 kDa fragment of Reelin [Fig.6A and B; t9 = 2.963, P < 0.05; Fig.6C and D; t6 = 4.240, P < 0.05]. We examined post-mortem human brain tissue obtained from individuals with AS and also found the Reelin protein profile to be altered in the cortex when compared with age-matched control tissue. Remarkably, we observed a reduction in several of the Reelin fragments [Fig.6E and F; 180 kDa: t6 = 2.852, P < 0.05; 250 kDa: t6 = 2.485, P < 0.05; 370 kDa: t6 = 2.498, P < 0.05], as well as full-length Reelin [450 kDa: t6 = 3.025, P < 0.05] protein in human AS cortical tissue. These data are the first to suggest that UBE3A maternal deficiency is coincident with alteration in the Reelin protein profile.
Figure 6.
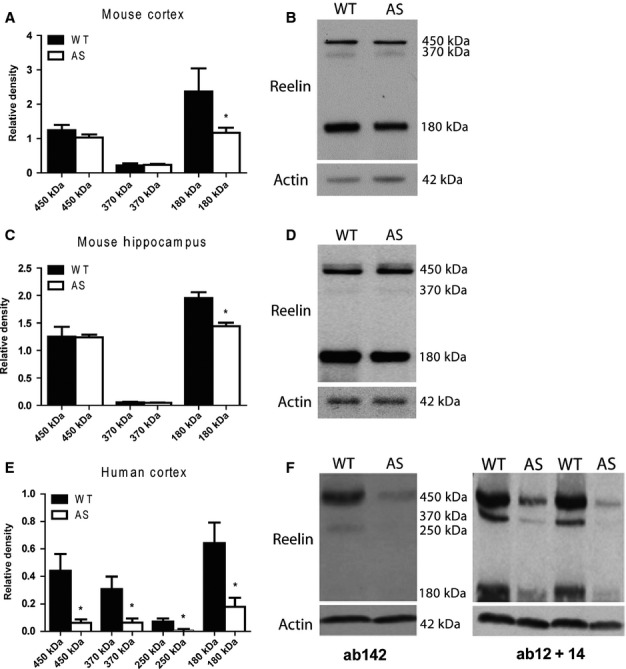
Reelin protein levels are altered in the AS mouse model and in human post-mortem brain. Quantification and representative western blot of the Reelin protein profile in (A and B) AS mouse cortex and (C and D) AS mouse hippocampus. The 180 kDa fragment of Reelin is significantly decreased in the cortex and hippocampus of AS mice. Wild-type (WT), n = 4; AS, n = 8; P < 0.05. (E and F) Quantitative and representative western blot of the Reelin protein profile in AS human post-mortem cortical tissue. All measurable Reelin protein fragments were significantly reduced in the AS tissue samples compared with age-matched control tissue. n = 4/group; *P < 0.05.
Discussion
The molecular mechanisms underlying the cognitive disruption in AS are unclear. To date, there is no indication that in-utero alterations in brain development underlie the AS phenotype. Use of the AS mouse model has suggested that biochemical alterations such as decreased activity of Ca2+/calmodulin-dependent protein kinase II, activity-regulated cytoskeleton-associated protein and extracellular signal-regulated kinases; physical deficits in dendritic spine density; and changes in α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid and N-methyl-D-aspartate receptor function controlling synaptic plasticity may contribute to the clinical features of the syndrome. The Reelin signaling system appears to modulate nearly all of the molecular components listed above, can recover the phenotype of the heterozygous Reelin haplo-insufficiency model, and shows a profound enhancement of learning and memory following injection of purified Reelin protein in wild-type mice. Thus, we sought to determine if Reelin treatment could recover the synaptic plasticity and learning and memory defects observed in the AS mouse model.
The increase in synaptic function achieved by Reelin supplementation is attributed to Reelin’s signaling effect on the post-synaptic neuron. The Reelin signaling pathway is involved in the modulation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor insertion into the membrane, as well as the phosphorylation of N-methyl-D-aspartate receptors on the post-synaptic membrane that is most easily observed following short-term Reelin application (Ghosh & Greenberg, 1995; Chen et al., 2005; Qiu et al., 2006). The long-term effects of Reelin injection are less clear. Wild-type mice treated with Reelin show increased spine density in apical dendritic spines. Interestingly, we see a significant change in apical dendritic spine density in the AS treated mice. It is possible that this modest increase in apical dendrites is sufficient to allow LTP recovery with a modest high-frequency-induced LTP protocol. This increase in spine density may be coupled with alterations in receptor function. For example, we find that chronic treatment with Reelin reduces the number of CA1 silent synapses in vitro (Qiu et al., 2006). Thus, the modulation of specific synapses coupled with increased potential apical synaptic connections may result in greater functional synapses.
The Ube3a protein is an E3-type ubiquitin ligase that targets specific proteins for degradation. Techniques detecting protein ubiquitination and protein expression profiling have identified only a handful of potential Ube3a targets. The localisation to the nucleus of two of the three Ube3a isotypes and its ability to bind to nuclear receptors suggest a role for Ube3a outside the lysosomal pathway (Nawaz et al., 1999; Dindot et al., 2008; Miao et al., 2013). Moreover, we find that significant biochemical changes can be seen only after induced neuronal activity (Greer et al., 2010; Filonova et al., 2014). Thus, identifying biomolecules underlying the AS synaptic plasticity defect can often rely on examination of a single specific protein. In the present study we sought to measure Reelin protein levels in the cortex and hippocampus prior to Reelin injection in order to ensure similar levels compared with our previous wild-type injections. Surprisingly, we detected a significant alteration in the Reelin protein profile in both the cortex and hippocampus, specifically of the 180 kDa fragment. Even more remarkable was that we also observed an alteration in the Reelin protein profile in human AS post-mortem cortical tissue compared with typical age-matched tissue. In the human tissue it appears that there is a reduction across all measurable Reelin protein fragments. Unfortunately, there are only two post-mortem brains with hippocampal tissue available and we were unable to verify a Reelin reduction in this region in those samples. It is unclear if the reduced fragments of Reelin are due to a decrease in Reelin expression or a change in Reelin metabolism. Reelin metabolism appears to be a complex process but it is poorly understood; however, we recently determined that tissue plasminogen activator is partially responsible for Reelin cleavage (Trotter et al., 2014). It is unlikely that Ube3a directly targets Reelin or tissue plasminogen activator and more likely that lower Reelin levels may result from reduced neuronal activity (Levenson et al., 2006).
In the larger context of biochemically-mediated cognitive enhancement, these studies exemplify the potential of Reelin to have a positive in vivo effect in a mouse model for a human mental retardation syndrome. The actions of Reelin to modify the molecular players involved in synaptic function are well known. However, it is unclear which metabolic fragments of Reelin have the in vivo effect shown in these studies and the mechanisms that allow a single Reelin treatment to influence learning and memory days after treatment are also unclear. Future studies to better understand Reelin’s long-lasting actions may provide a therapeutic with broad applications.
Our previous studies demonstrate that adeno-assocated virus transfection of the adult AS mouse model with a UBE3A construct could significantly ameliorate the synaptic plasticity and learning and memory defects (Daily et al., 2011). In the present study we similarly recover the AS phenotype in adult AS mice with the protein Reelin. Similar Reelin-treated enhancement is observed via similar biochemical and behavioral changes seen in the wild-type treated compared with AS treated animals (Table2). Taken together, these results support the hypothesis that the brain dysfunction and defects in learning and memory associated with AS are not due to developmental synaptic abrogation and are reversible. Furthermore, it raises the possibility that targeting the Reelin signaling system or associated signaling pathways may be an effective therapeutic target in the treatment of the major phenotypes of human AS.
Table 2.
Percent change from mock treated control animals
| Biochemical/behavioral changes | Wild-type Reelin-treated, % | Ube3a-deficient Reelin-treated, % |
|---|---|---|
| CA1 apical spine density | 42.22 ↑* | 28.85 ↑ |
| LTP (last 10 min) | 76.71 ↑* | 99.48 ↑ |
| Contextual fear conditioning (% freezing) | 35.29 ↑* | 66.98 ↑ |
| Hidden platform water maze (target quadrant entries) | 58.41 ↑* | 64.50 ↑ |
Data from Rogers et al. (2011).
Acknowledgments
This work was supported by the National Institutes of Health (AG035379-05) and the Foundation for Angelman Syndrome Therapeutics.
Glossary
- AS
Angelman syndrome
- LTP
long-term potentiation
- UBE3A
ubiquitin protein ligase E3A
References
- Alcantara S, Ruiz M, D’Arcangelo G, Ezan F, de Lecea L, Curran T, Sotelo C. Soriano E. Regional and cellular patterns of reelin mRNA expression in the forebrain of the developing and adult mouse. J. Neurosci. 1998;18:7779–7799. doi: 10.1523/JNEUROSCI.18-19-07779.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud L, Ballif BA, Förster E. Cooper JA. Fyn tyrosine kinase is a critical regulator of disabled-1 during brain development. Curr. Biol. 2003;13:9–17. doi: 10.1016/s0960-9822(02)01397-0. [DOI] [PubMed] [Google Scholar]
- Chen Y, Beffert U, Ertunc M, Tang T-SS, Kavalali ET, Bezprozvanny I. Herz J. Reelin modulates NMDA receptor activity in cortical neurons. J. Neurosci. 2005;25:8209–8216. doi: 10.1523/JNEUROSCI.1951-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, Jing D, Bath KG, Ieraci A, Khan T, Siao CJ, Herrera DG, Toth M, Yang C, McEwen BS, Hempstead BL. Lee FS. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006;314:140–143. doi: 10.1126/science.1129663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daily JL, Nash K, Jinwal U, Golde T, Rogers J, Peters MM, Burdine RD, Dickey C, Banko JL. Weeber EJ. Adeno-associated virus-mediated rescue of the cognitive defects in a mouse model for Angelman syndrome. PLoS One. 2011;6:e27221. doi: 10.1371/journal.pone.0027221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Arcangelo G. Reelin mouse mutants as models of cortical development disorders. Epilepsy Behav. 2006;8:81–90. doi: 10.1016/j.yebeh.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Dindot SV, Antalffy BA, Bhattacharjee MB. Beaudet AL. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet. 2008;17:111–118. doi: 10.1093/hmg/ddm288. [DOI] [PubMed] [Google Scholar]
- Filonova I, Trotter JH, Banko JL. Weeber EJ. Activity-dependent changes in MAPK activation in the Angelman Syndrome mouse model. Learn. Memory. 2014;21:98–104. doi: 10.1101/lm.032375.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. Greenberg ME. Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science. 1995;268:239–247. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- Goffinet AM. The embryonic development of the cerebellum in normal and reeler mutant mice. Anat. Embryol. 1982;168:73–86. doi: 10.1007/BF00305400. [DOI] [PubMed] [Google Scholar]
- Greer PL, Hanayama R, Bloodgood BL, Mardinly AR, Lipton DM, Flavell SW, Kim TK, Griffith EC, Waldon Z, Maehr R, Ploegh HL, Chowdhury S, Worley PF, Steen J. Greenberg ME. The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell. 2010;140:704–716. doi: 10.1016/j.cell.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustin RM, Bichell TJ, Bubser M, Daily J, Filonova I, Mrelashvili D, Deutch AY, Colbran RJ, Weeber EJ. Haas KF. Tissue-specific variation of Ube3a protein expression in rodents and in a mouse model of Angelman syndrome. Neurobiol. Dis. 2010;39:283–291. doi: 10.1016/j.nbd.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack I, Bancila M, Loulier K, Carroll P. Cremer H. Reelin is a detachment signal in tangential chain-migration during postnatal neurogenesis. Nat. Neurosci. 2002;5:939–945. doi: 10.1038/nn923. [DOI] [PubMed] [Google Scholar]
- Heinrich C, Nitta N, Flubacher A, Muller M, Fahrner A, Kirsch M, Freiman T, Suzuki F, Depaulis A, Frotscher M. Haas CA. Reelin deficiency and displacement of mature neurons, but not neurogenesis, underlie the formation of granule cell dispersion in the epileptic hippocampus. J. Neurosci. 2006;26:4701–4713. doi: 10.1523/JNEUROSCI.5516-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell BW, Gertler FB. Cooper JA. Mouse disabled (mDab1): a Src binding protein implicated in neuronal development. EMBO J. 1997;16:121–132. doi: 10.1093/emboj/16.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, Sweatt JD. Beaudet AL. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 1998;21:799–811. doi: 10.1016/s0896-6273(00)80596-6. [DOI] [PubMed] [Google Scholar]
- Jossin Y, Ignatova N, Hiesberger T, Herz J, de Rouvroit CL. Goffinet AM. The central fragment of Reelin, generated by proteolytic processing in vivo, is critical to its function during cortical plate development. J. Neurosci. 2004;24:514–521. doi: 10.1523/JNEUROSCI.3408-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jossin Y, Gui L. Goffinet AM. Processing of Reelin by embryonic neurons is important for function in tissue but not in dissociated cultured neurons. J. Neurosci. 2007;27:4243–4252. doi: 10.1523/JNEUROSCI.0023-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaphzan H, Hernandez P, Jung JI, Cowansage KK, Deinhardt K, Chao MV, Abel T. Klann E. Reversal of impaired hippocampal long-term potentiation and contextual fear memory deficits in Angelman syndrome model mice by ErbB inhibitors. Biol. Psychiat. 2012;72:182–190. doi: 10.1016/j.biopsych.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koie M, Okumura K, Hisanaga A, Kamei T, Sasaki K, Deng M, Baba A, Kohno T. Hattori M. Cleavage within Reelin repeat 3 regulates the duration and range of the signaling activity of Reelin protein. J. Biol. Chem. 2014;289:12922–12930. doi: 10.1074/jbc.M113.536326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krstic D, Rodriguez M. Knuesel I. Regulated proteolytic processing of Reelin through interplay of tissue plasminogen activator (tPA), ADAMTS-4, ADAMTS-5, and their modulators. PLoS One. 2012;7:e47793. doi: 10.1371/journal.pone.0047793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson JM, Roth TL, Lubin FD, Miller CA, Huang I-C, Desai P, Malone LM. Sweatt JD. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J. Biol. Chem. 2006;281:15763–15773. doi: 10.1074/jbc.M511767200. [DOI] [PubMed] [Google Scholar]
- Miao S, Chen R, Ye J, Tan G-H, Li S, Zhang J, Jiang Y-H. Xiong Z-Q. The Angelman syndrome protein Ube3a is required for polarized dendrite morphogenesis in pyramidal neurons. J. Neurosci. 2013;33:327–333. doi: 10.1523/JNEUROSCI.2509-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K, Mikoshiba K, Miyata T, Kudo C. Ogawa M. Disruption of hippocampal development in vivo by CR-50 mAb against reelin. Proc. Natl. Acad. Sci. USA. 1997;94:8196–8201. doi: 10.1073/pnas.94.15.8196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawaz Z, Lonard DM, Smith CL, Lev-Lehman E, Tsai SY, Tsai M-J. O’Malley BW. The Angelman syndrome-associated protein, E6-AP, is a coactivator for the nuclear hormone receptor superfamily. Mol. Cell. Biol. 1999;19:1182–1189. doi: 10.1128/mcb.19.2.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu S, Renfro A, Quattrocchi CC, Sheldon M. D’Arcangelo G. Reelin promotes hippocampal dendrite development through the VLDLR/ApoER2-Dab1 pathway. Neuron. 2004;41:71–84. doi: 10.1016/s0896-6273(03)00819-5. [DOI] [PubMed] [Google Scholar]
- Pesold C, Impagnatiello F, Pisu MG, Uzunov DP, Costa E, Guidotti A. Caruncho HJ. Reelin is preferentially expressed in neurons synthesizing gamma-aminobutyric acid in cortex and hippocampus of adult rats. Proc. Natl. Acad. Sci. USA. 1998;95:3221–3226. doi: 10.1073/pnas.95.6.3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu S, Zhao LF, Korwek KM. Weeber EJ. Differential reelin-induced enhancement of NMDA and AMPA receptor activity in the adult hippocampus. J. Neurosci. 2006;26:12943–12955. doi: 10.1523/JNEUROSCI.2561-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JT. Weeber EJ. Reelin and apoE actions on signal transduction, synaptic function and memory formation. Neuron Glia Biol. 2008;4:259–270. doi: 10.1017/S1740925X09990184. [DOI] [PubMed] [Google Scholar]
- Rogers JT, Rusiana I, Trotter J, Zhao L, Donaldson E, Pak DT, Babus LW, Peters M, Banko JL, Chavis P, Rebeck GW, Hoe H-SS. Weeber EJ. Reelin supplementation enhances cognitive ability, synaptic plasticity, and dendritic spine density. Learn. Memory. 2011;18:558–564. doi: 10.1101/lm.2153511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JT, Zhao L, Trotter JH, Rusiana I, Peters MM, Li Q, Donaldson E, Banko JL, Keenoy KE, Rebeck GW, Hoe HS, D’Arcangelo G. Weeber EJ. Reelin supplementation recovers sensorimotor gating, synaptic plasticity and associative learning deficits in the heterozygous reeler mouse. J. Psychopharmacol. 2013;27:389–395. doi: 10.1177/0269881112463468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rouvroit CL, de Bergeyck V, Cortvrindt C, Bar I, Eeckhout Y. Goffinet AM. Reelin, the extracellular matrix protein deficient in reeler mutant mice, is processed by a metalloproteinase. Exp. Neurol. 1999;156:214–217. doi: 10.1006/exnr.1998.7007. [DOI] [PubMed] [Google Scholar]
- Sinagra M, Verrier D, Frankova D, Korwek KM, Blahos J, Weeber EJ, Manzoni OJ. Chavis P. Reelin, very-low-density lipoprotein receptor, and apolipoprotein E receptor 2 control somatic NMDA receptor composition during hippocampal maturation in vitro. J. Neurosci. 2005;25:6127–6136. doi: 10.1523/JNEUROSCI.1757-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffenburg S, Gillberg CL, Steffenburg U. Kyllerman M. Autism in Angelman syndrome: a population-based study. Pediatr. Neurol. 1996;14:131–136. doi: 10.1016/0887-8994(96)00011-2. [DOI] [PubMed] [Google Scholar]
- Trotter J, Lee GH, Kazdoba TM, Crowell B, Domogauer J, Mahoney HM, Franco SJ, Müller U, Weeber EJ. D’Arcangelo G. Dab1 is required for synaptic plasticity and associative learning. J. Neurosci. 2013;33:15652–15668. doi: 10.1523/JNEUROSCI.2010-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotter JH, Lussier AL, Psilos KE, Mahoney HL, Sponaugle AE, Hoe HS, Rebeck GW. Weeber EJ. Extracellular proteolysis of reelin by tissue plasminogen activator following synaptic potentiation. Neuroscience. 2014;274:299–307. doi: 10.1016/j.neuroscience.2014.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utsunomiya-Tate N, Kubo K-I, Tate S-I, Kainosho M, Katayama E, Nakajima K. Mikoshiba K. Reelin molecules assemble together to form a large protein complex, which is inhibited by the function-blocking CR-50 antibody. Proc. Natl. Acad. Sci. USA. 2000;97:9729–9734. doi: 10.1073/pnas.160272497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeber EJ, Beffert U, Jones C, Christian JM, Forster E, Sweatt JD. Herz J. Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J. Biol. Chem. 2002;277:39944–39952. doi: 10.1074/jbc.M205147200. [DOI] [PubMed] [Google Scholar]
- Weeber EJ, Jiang YH, Elgersma Y, Varga AW, Carrasquillo Y, Brown SE, Christian JM, Mirnikjoo B, Silva A, Beaudet AL. Sweatt JD. Derangements of hippocampal calcium/calmodulin-dependent protein kinase II in a mouse model for Angelman mental retardation syndrome. J. Neurosci. 2003;23:2634–2644. doi: 10.1523/JNEUROSCI.23-07-02634.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CA, Beaudet AL, Clayton-Smith J, Knoll JH, Kyllerman M, Laan LA, Magenis RE, Moncla A, Schinzel AA, Summers JA. Wagstaff J. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am. J. Med. Genet. A. 2006;140:413–418. doi: 10.1002/ajmg.a.31074. [DOI] [PubMed] [Google Scholar]
- van Woerden GM, Harris KD, Hojjati MR, Gustin RM, Qiu S, de Avila Freire R, Jiang YH, Elgersma Y. Weeber EJ. Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat. Neurosci. 2007;10:280–282. doi: 10.1038/nn1845. [DOI] [PubMed] [Google Scholar]
- Won SJ, Kim SH, Xie L, Wang Y, Mao XO, Jin K. Greenberg DA. Reelin-deficient mice show impaired neurogenesis and increased stroke size. Exp. Neurol. 2006;198:250–259. doi: 10.1016/j.expneurol.2005.12.008. [DOI] [PubMed] [Google Scholar]