

Abstract
The activity of HBF4 (aqueous solution) as a catalyst in propargylation reactions is presented. Diverse types of nucleophiles were employed in order to form new C–O, C–N and C–C bonds in technical acetone and in air. Good to excellent yields and good chemoselectivities were obtained using low acid loading (typically 1 mol-%) under simple reaction conditions.
Keywords: Propargylic alcohols, Brønsted acid, Nucleophilic substitution, Friedel–Crafts reaction, Homogeneous catalysis
Introduction
The direct nucleophilic substitution of alcohols is of high interest as it provides access to a wide variety of derivatives, with the formation of water as the only by-product. Indeed, the ACS Green Chemistry Institute Pharmaceutical Roundtable identified OH activation for nucleophilic substitutions as a priority area currently used in the preparation of pharmaceutical intermediates that would greatly benefit from the development of better methods.1 Unarguably, propargylic substitutions have progressed substantially since the pioneering work of Nicholas on octacarbonyldicobalt-stabilised propargylic cations.2 The versatility of the propargylic moiety as a synthon in organic chemistry as well as its occurrence in natural products and synthetic pharmaceuticals have been the main driving forces for these advances. Furthermore, propargylic alcohols are easily prepared from the corresponding aldehydes or ketones by addition of an alkynyl anion. However, propargylic substitution reactions remain underdeveloped when compared to allylic substitutions. Diverse transition metals,3 such as ruthenium, palladium, gold or silver,4 have been successfully used in this context. However, the cost of the catalyst, together with its selectivity (metal–allenylidene vs. metal–propargylic intermediates) remain important issues to solve.
The direct displacement of “activated” alcohols – such as benzylic, allylic, and propargylic alcohols – can also be achieved using Brønsted or Lewis acids by simple SN1 reactions.5 Important advantages of Brønsted acids over Lewis acids often include lower catalytic loadings and easier handling as they are generally more stable towards oxygen and water.
Sulfonic acids are the most commonly used Brønsted acids for the nucleophilic substitution of propargylic alcohols as described in extensive work by Sanz and co-workers with p-toluenesulfonic acid.6,7 Inorganic acids, such as phosphomolybdic acid on silica, have also been studied with C-, N- and O-nucleophiles.8 Depending on the substrates, the reactions required either 10 mol-% of acid at room temperature, or 1 mol-% in refluxing toluene. An additional asset of these inorganic acids is their straightforward separation from the organic products through a simple basic workup. A common feature for all these catalytic systems is their compatibility with air and reagent-grade solvents, although they are mostly undesirable ones (toxic, costly to dispose of, such as MeNO2).
HBF4 is a common acid in academic and industrial laboratories that has found diverse applications in synthesis, either as a reagent (nucleophilic fluorination,9 synthesis of vinylidene–metal complexes10), or catalyst (amidation of olefins,11 Biginelli reaction,12 acylation of aldehydes13). In particular, the Friedel–Crafts alkylation of benzylic alcohols in the presence of an excess of HBF4·OEt2 solution at –78 °C has been reported.14 Even though high diastereoselectivities could be achieved with this methodology, the excess of acid and low reaction temperatures represent important drawbacks. Herein, we report the use of HBF4 as a highly efficient catalyst for SN1 reactions of propargylic alcohols with different nucleophiles under mild, simple reaction conditions.
Results and Discussion
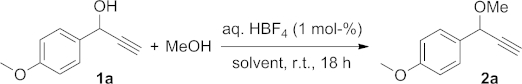
In a first step, the effect of different solvents was tested on the reaction of propargylic alcohol 1a with MeOH to give 2a with 1 mol-% of HBF4 (Table1). These reactions were run in air with 2 equiv. of MeOH and a commercially available 48 wt.-% solution of HBF4 in water as catalyst. No attempts were made to optimise the reaction times. Whereas sluggish reactions were observed in THF or in water (Table1, Entries 1 and 2), high conversions were obtained in DCM, acetonitrile, and acetone (Table1, Entries 4–6). Overall, acetone was chosen as our preferred solvent because of its greener profile.15 It is important to note that all tested solvents were technical grade, and in particular, the acetone employed in these reactions was standard laboratory washing acetone. Furthermore, the formation of fluoro derivatives or α,β-unsaturated compounds derived from a Meyer–Schuster rearrangement16 was not observed either in the model reaction or during the study of the scope of the reaction.
Table 1.
Solvent screening for propargylation reactions.
|
||||
|---|---|---|---|---|
| Entry | Solvent | Conv. [%][a] | ||
| 1 | THF | 26 | ||
| 2 | water | 47 | ||
| 3 | toluene | 77 | ||
| 4 | MeCN | 93 | ||
| 5 | DCM | ≥ 95 | ||
| 6 | acetone | ≥ 95 | ||
1H NMR conversions.
The use of different O-nucleophiles was first explored (Scheme 1). Propargylic alcohol 1a was treated with different primary and secondary alcohols to form the expected ethers 2a–h in high yields under our standard conditions. When chiral alcohols were used, the corresponding ethers 2g and 2h were formed as a mixture of inseparable diastereoisomers. A tertiary alcohol, tBuOH, only led to low yields of the ether 2i, and the major product of that reaction (51 % conversion) formed from dimerisation of the starting propargylic alcohol (3a; vide infra for further details). On the other hand, an ortho-disubstituted aryl group was not detrimental to the reactivity of the propargylic alcohol, as exemplified with the formation of 2k. Also, unlike most transition-metal-based methodologies,3 the reaction is not limited to terminal alkynes, and alkyl (2j, 2k), aryl (2l) or silyl groups (2m, 2n) at the acetylenic position did not have any major effect on the outcome of the reaction (Scheme 1). Also, several functional groups (ketone, halogen or sulfone) were shown to be compatible with the reaction conditions. In the absence of any other nucleophile, the starting propargylic alcohol dimerised to form the symmetrical ether as a mixture of diastereoisomers (Scheme 2).
scheme 1.
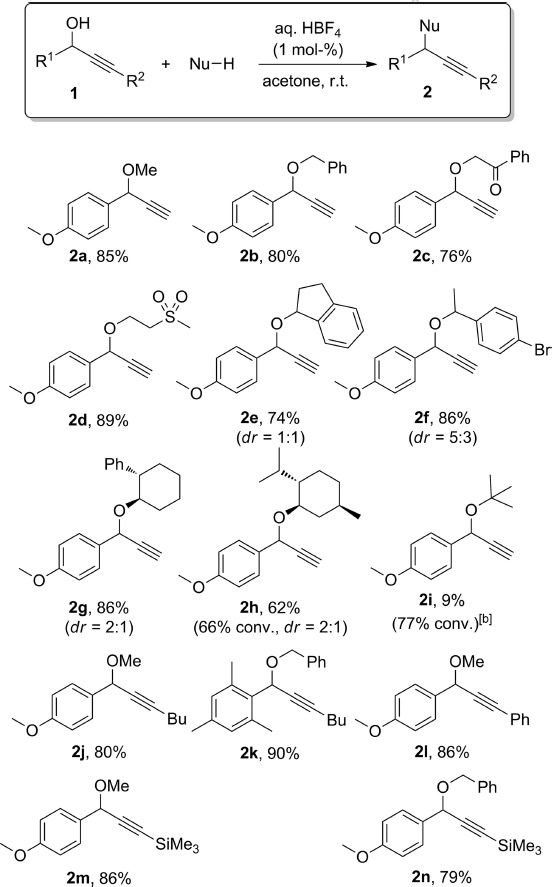
C–O bond formation with HBF4 at room temperature.[a] dr = diastereoisomeric ratio. [a] Isolated yields; 1H NMR conversions are provided in parentheses when lower than 95 %. [b] 51 % 1H NMR conversion into dimer 3a.
scheme 2.
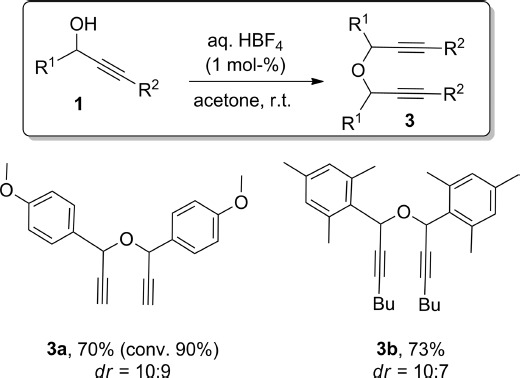
HBF4-catalysed dimerisation of propargylic alcohols.[a] dr = diastereoisomeric ratio. [a] Isolated yields; 1H NMR conversions are provided in parentheses when lower than 95 %.
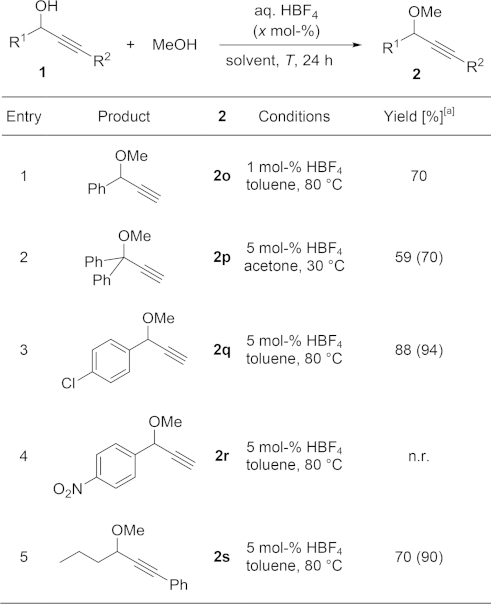
On the other hand, when R1 on the starting propargylic alcohol was not an electron-rich aryl group, no reaction was observed at room temperature. In most cases, however, the formation of the desired ethers was possible by increasing the reaction temperature and/or the acid loading (Table2). Good yields could be then obtained, except for a nitro-substituted substrate (Table2, Entry 4). It is important to note that, since no decomposition or undesired reactions were observed at room temperature, this difference in reactivity could be used to selectively functionalise a more complex molecule with two electronically dissimilar propargylic alcohols (vide infra).
Table 2.
HBF4-catalysed reaction of electron-poor/neutral propargylic alcohols.
|
Isolated yields; 1H NMR conversions are provided in parentheses when lower than 95 %; n.r. = no reaction.
We next explored the use of nitrogen nucleophiles in this substitution reaction. The inherent basicity of most amines is an obvious potential limitation of any Brønsted acid catalysed reaction as they might simply neutralise the catalyst. Our conditions, however, could be successfully applied to different carbamates and sulfonamides, as well as weakly basic anilines (Scheme 3), and the expected products 4 were prepared in good yields at room temperature, with the exception of 4e.
scheme 3.
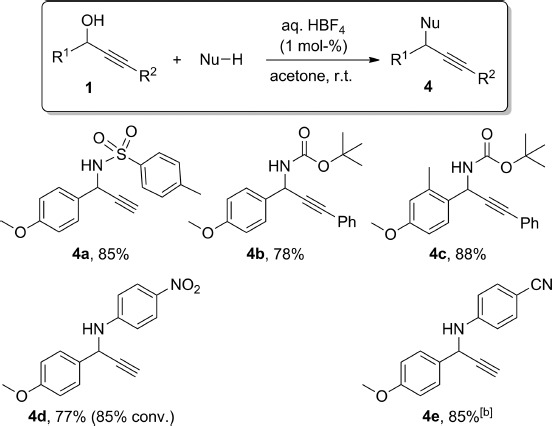
C–N bond formation with HBF4 at room temperature.[a] [a] Isolated yields, 1H NMR conversions are provided in parentheses when lower than 95 %. [b] Reaction carried out with 5 mol-% of HBF4 at 60 °C.
Carbon nucleophiles were also investigated, and diketones as well as electron-rich arenes reacted to form the expected products 5 in good to excellent yields (Scheme 4). Very similar results were obtained with pentane-2,4-dione and a variety of substituted propargylic alcohols. For the formation of 5d, with an electron-neutral aryl group at the propargylic position, a higher catalyst loading and an elevated temperature were required in order to obtain high conversions. Phenol also reacted efficiently in a Friedel–Crafts-type reaction,17 to give para-substituted derivatives 5e–g exclusively. When using 2-phenylphenol as the nucleophile, the hydroxy group had a stronger directing power than the arene, as expected (Scheme 4, compound 5h). We then tested a phenol with a second strongly activating group at the para position (4-methoxyphenol, for the formation of 5i). In this case, only the product derived from reaction at the ortho position to the phenol was isolated.
scheme 4.
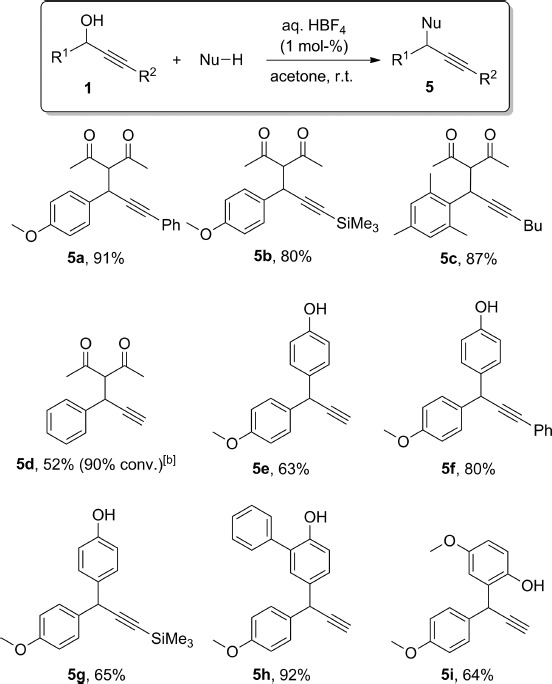
C–C bond formation with HBF4 at room temperature.[a] [a] Isolated yields; 1H NMR conversions are provided in parentheses when lower than 95 %. [b] Reaction carried out with 5 mol-% of HBF4 in MeCN at 80 °C.
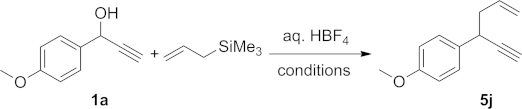
Surprisingly, allyltrimethylsilane proved to be a very poor reaction partner for the substitution of propargylic alcohols with HBF4 as the catalyst. Low conversions were obtained in either acetone or hot toluene, even when higher catalyst or nucleophile loadings were used (Table3). Overall, the best results were obtained in MeCN at 80 °C, and still product 5j could only be isolated in 50 % yield. It is important to note that alcohol 1a was stable under the studied conditions, and besides the expected product 5j, only 1a and ether 3a were evidenced in the 1H NMR spectra. Hence, no amide formation, which could potentially take place by a Ritter reaction,18,19 was observed under these conditions.
Table 3.
HBF4-mediated reactions with an allylsilane.
| ||
|---|---|---|
| Entry | Conditions | Conv [%][a] |
| 1 | 2 equiv. NuH, HBF4 (1 mol-%) acetone, r.t. | 18 |
| 2 | 2 equiv. NuH, HBF4 (5 mol-%) toluene, 80 °C | 30 |
| 3 | 3 equiv. NuH, HBF4 (5 mol-%) toluene, 80 °C | 35 |
| 4 | 2 equiv. NuH, HBF4 (5 mol-%) MeCN, 80 °C | 72 (50) |
1H NMR conversions; isolated yield is provided in parentheses.
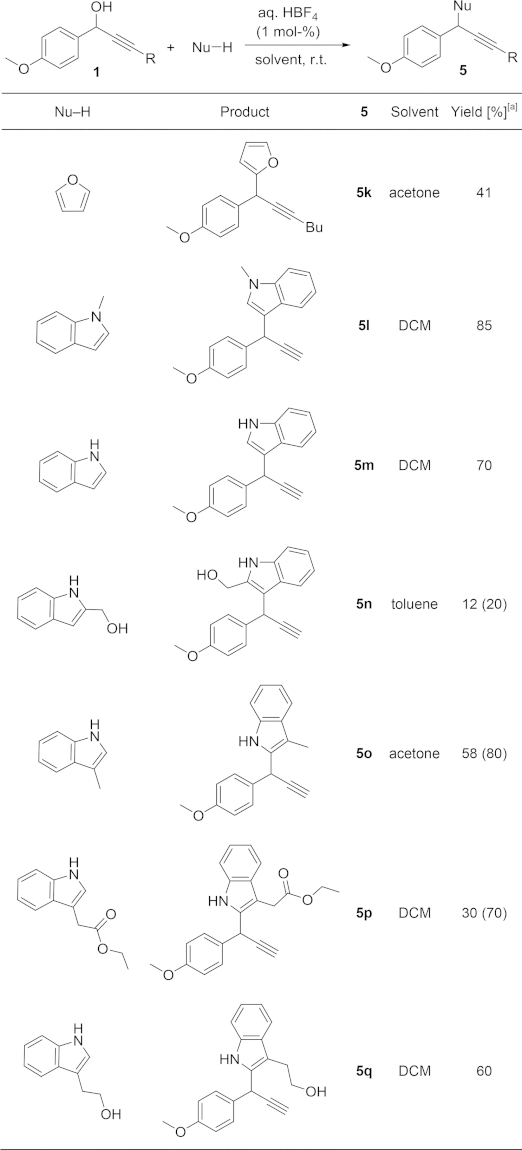
We next moved to electron-rich heterocycles (Table4). When furan was used as nucleophile, the 1H NMR spectrum of the crude reaction mixture showed the formation of a complex mixture of products. Prompt purification allowed the isolation of 5k in moderate yield, but it is important to note that 5k still decomposed rapidly after purification. These observations were perhaps not surprising as many furan derivatives are well-known to be acid-sensitive. This is also the case for some indoles, but as it can be seen in Table4, better yields were obtained with this important family of heterocyclic nucleophiles.20
Table 4.
HBF4-catalysed propargylation reactions of heterocycles.
|
Isolated yields; 1H NMR conversions are provided in parentheses when lower than ≥ 95 %.
Reaction carried out with 5 mol-% of HBF4 in toluene at 80 °C.
In order to investigate the influence of substitution on the nucleophile, different indole derivatives were treated with the same propargylic alcohol 1a. 1-Methyl- and 1H-indoles reacted regioselectively at C-3, as expected, leading to the formation of 5l and 5m, respectively, in good yields. Related indol-2-ylmethanol, in contrast, only gave very low conversions. This might be due to a higher instability under acidic conditions or its low solubility either in acetone or DCM. Even under more forcing conditions (5 mol-% of acid at 80 °C), no evidence for reaction of the hydroxy group could be detected. We next screened different indoles with a substituent at C-3. Gratifyingly, products 5o–q, derived from a Friedel–Crafts reaction at C-2 could be prepared under very simple reaction conditions. To the best of our knowledge, this is the first example of the synthesis of 2,3-disubstituted indoles by Brønsted acid catalysed propargylation reactions.21
Some of the reactions in Table4 were carried out in DCM, instead of acetone, to avoid the formation of undesired by-products. It has previously been reported that indoles can react with ketones or aldehydes as electrophiles under acidic conditions.22 Indeed, when 1-methylindole was treated with propargylic alcohol 1a under our standard conditions in acetone, the expected product 5l was formed preferentially, but it was contaminated with bis(indole) 6 (Scheme 5A). The high yield obtained of 5l indicates that the indole reacts preferentially with the propargylic cation formed from 1a and that the reaction between the excess of indole and acetone is quite sluggish.
scheme 5.
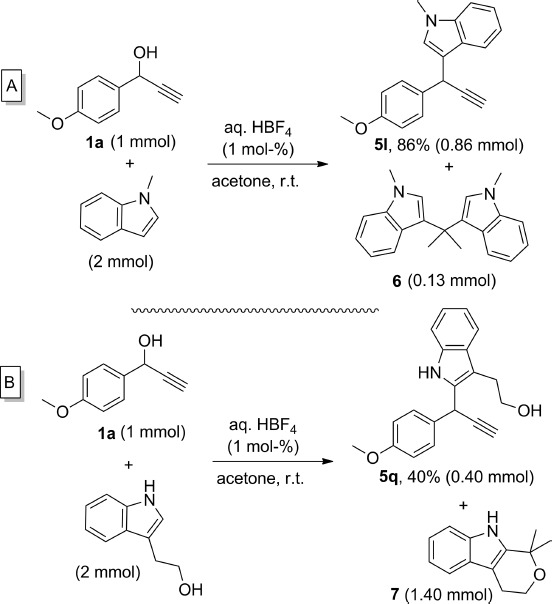
Undesired reactions of indoles in acetone. Isolated yields are provided.
Even if both compounds can be separated by column chromatography the formation of 6 remains undesirable, and hence acetone was avoided as a solvent for these reactions. More problematic was the reaction of 1a with tryptophol as nucleophile (Scheme 5B). In this case the expected product could only be isolated in 40 % yield because of a competitive oxa-Pictet–Spengler acid-catalysed cyclocondensation of tryptophol with the solvent,23 which consumed 70 % of the available nucleophile.
Next, two competition experiments were performed to exploit the particular activity of HBF4 in propargylation reactions (Scheme 6). Firstly, relatively electron-rich alcohol 1a reacted selectively in the presence of 1g, bearing a chloro substituent at the para position of the phenyl ring (Scheme 6A). Also, the unexpected diminished reactivity of allylsilanes as nucleophiles was exploited when 1a was treated with 2 equiv. of benzyl alcohol and 2 equiv. of allyltrimethylsilane. Only 2b, derived from the reaction with benzyl alcohol, was formed under these conditions. Importantly, these chemoselectivities are not possible when using other Brønsted acids reported for this transformation, such as p-toluenesulfonic acid,6a or phosphomolybdic acid.8a
scheme 6.
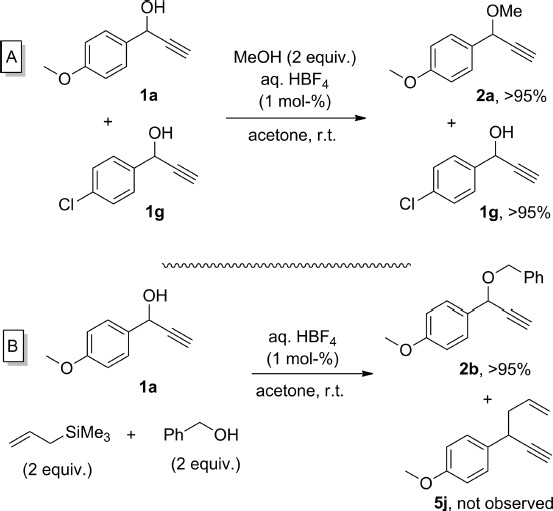
Competition experiments with HBF4. 1H NMR conversions are provided.
Finally, a gram-scale reaction was performed to further demonstrate the applicability of this reaction, and compound 5r was isolated in high yield when using our optimised conditions (Scheme 7).
scheme 7.
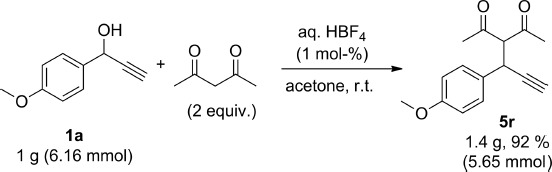
Gram-scale reaction.
Conclusions
The scope and limitations of HBF4 as a practical catalyst for propargylation reactions have been explored. In general, good to excellent yields for the formation of C–O, C–N and C–C bonds were obtained under exceptionally simple reaction conditions. Challenging substrates such as electron-poor propargylic alcohols, or acid-sensitive indoles could be used with this methodology, even if slightly more forcing conditions were sometimes required. All reactions were carried out in air and in technical solvents, and the acid used was a commercially available aqueous solution. All the reactions were also completely regioselective, and no allene products were observed in any case. Furthermore, many of the reactions were extremely clean, and the desired products could be isolated analytically pure without the need for further purification after a simple aqueous workup (i.e., 2a, 2j, 2l–o, 5b–c). Overall, this is a convenient and powerful methodology that does not employ a costly metal catalyst. The full potential of HBF4 in this context remains to be uncovered. For instance, we were pleased to see that an allylic alcohol also reacted with MeOH at room temperature in very high yields (Scheme 8). Further applications of this synthetic protocol are currently being investigated in our laboratory.
scheme 8.
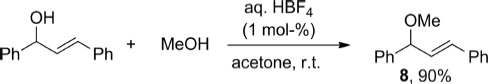
Reaction of an allylic alcohol with HBF4.
Experimental Section
General Procedure for the Nucleophilic Substitution of Propargylic Alcohols: In a vial fitted with a screw cap and a stirring bar, the propargylic alcohol 1 (1 mmol), nucleophile (2 mmol) and technical acetone (2 mL) were introduced. An aqueous solution of HBF4 (48 wt.-%, 1.2 μL,1 mol-%) was then added, and the reaction mixture was stirred at room temperature for 18 h. The reaction was quenched with a saturated aqueous solution of NaHCO3 and the mixture extracted with ethyl acetate. The combined organic extracts were washed with brine, dried with anhydrous MgSO4, filtered, and concentrated under reduced pressure to give the title compound. If needed, the crude product was then purified by column chromatography.
Acknowledgments
This research was financially supported by the Imperial College London and University College London. The Royal Society and the Engineering and Physical Sciences Research Council (EPSRC) (EP/K030760/1) are acknoledged for further funding (RG140579). The Xunta de Galicia (Spain) is acknowledged for a post-doctoral fellowship to E. B. (I2C plan). A. S. V. thanks the Universidad de Valencia (Spain) for an Erasmus scholarship. E. T. thanks the Ministère de l’Education Nationale et de l’Enseignement Supérieur (France) and the École Nationale Supérieure de Chimie de Rennes (ENSCR) for the allocation of a mobility and an Eramus scholarship. LondonCat at ICL and UCL is acknowledged for a summer studentship to S.-H. L. We thank GlaxoSmithKline (GSK) for supplying the nucleophiles used for the preparation of products 2c–g, 5n and 5q.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1.Constable DJC, Dunn PJ, Hayler JD, Humphrey GR. Linderman RJ, Lorenz K, Manley J, Pearlman BA, Wells A, Zaks A, Zhang TY. Green Chem. 2007;9:411–420. J. L. Leazer Jr., [Google Scholar]
- 2.pp. 4163–4165.; Lochwood RF, Nicholas KM. Tetrahedron Lett. 1977;18 [Google Scholar]; Nicholas KH. Acc. Chem. Res. 1987;20 [Google Scholar]
- 3.For reviews, see: pp. 1911–1925.; Zhu Y, Sun L, Lu P, Wang Y. ACS Catal. 2009;4 [Google Scholar]; Bauer EB. Synthesis. 2014;44 [Google Scholar]; Miyake Y, Uemura S, Nishibayashi Y. ChemCatChem. 2012;1 [Google Scholar]; Detz RJ, Hiemstra H, van Maarseveen JH. Eur. J. Org. Chem. 2009 [Google Scholar]
- 4.Pennell MN, Turner PG, Sheppard TD. Chem. Eur. J. 2012;18:4748–4758. doi: 10.1002/chem.201102830. [DOI] [PubMed] [Google Scholar]
- 5.For a review, see: Emer E, Sinisi R, Capdevila MG, Petruzziello D, De Vincentis F, Cozzi PG. Eur. J. Org. Chem. 2011:647–666. [Google Scholar]
- 6.pp. 1383–1386.; Sanz R, Martínez A, Álvarez-Gutiérrez JM, Rodríguez F. Eur. J. Org. Chem [Google Scholar]; Sanz R, Miguel D, Martínez A, Álvarez-Gutiérrez JM, Rodríguez F. Org. Lett. 2006;9 doi: 10.1021/ol0631298. [DOI] [PubMed] [Google Scholar]; Sanz R, Miguel D, Álvarez-Gutiérrez JM, Rodríguez F. Synlett. 2007 [Google Scholar]; Sanz R, Miguel D, Martínez A, Gohain M, García-García P, Fernández-Rodríguez MA, Álvarez E, Rodríguez F. Eur. J. Org. Chem. 2010 [Google Scholar]
- 7.2008. pp. 3148–3151.; Pan Y, Zheng F, Lin H, Zhan Z. J. Org. Chem. 74 doi: 10.1021/jo8027533. [DOI] [PubMed] [Google Scholar]; Liu Y-L, Liu L, Wang Y-L, Han Y-C, Wang D, Chen YJ. Green Chem. 2009;10 [Google Scholar]; Yue H-L, Wei W, Li M-M, Yang Y-R, Ji J-X. Adv. Synth. Catal. 2011;353 [Google Scholar]
- 8.2008. pp. 1448–1455.; Srihari P, Sunder JS, Bhunia DC, Mandal SS, Yadav JS. Synth. Commun. 2008;38 [Google Scholar]; Yadav JS, Reddy BVS, Pandurangam T, Rao KVR, Praneeth K. Madavi C, Kunwar AC. Tetrahedron Lett. 2008;49 G. G. K. S. N. Kumar, [Google Scholar]
- 9.Pasceri R, Bartrum HF, Hayes CJ, Moody CJ. Chem. Commun. 2012;48:12077–12079. doi: 10.1039/c2cc37284c. [DOI] [PubMed] [Google Scholar]
- 10.Esteruelas MA, Lahoz FJ, López AM, Oñate E, Oro LA. Organometallics. 1994;13:1669–1678. [Google Scholar]
- 11.Reddy BVS, Reddy NS, Madan Ch, Yadav JS. Tetrahedron Lett. 2010;51:4827–4829. [Google Scholar]
- 12.Chen WY, Qin SD, Jin JR. Catal. Commun. 2007;8:123–126. [Google Scholar]
- 13.Kamble VT, Bandgar BP, Joshi NS, Jamode VS. Synlett. 2006:2719–2722. [Google Scholar]
- 14.Stadler D, Bach T. Chem. Asian J. 2008;3:272–284. doi: 10.1002/asia.200700241. [DOI] [PubMed] [Google Scholar]
- 15.Henderson RK, Jiménez-González C, Constable DJC, Alston SR, Inglis GGA, Fisher G, Sherwood J, Binksa SP, Curzons AD. Green Chem. 2011;13:854–862. [Google Scholar]
- 16.2009. pp. 4149–4158.; Engel DA, Dudley GB. Org. Biomol. Chem. 7 doi: 10.1039/b912099h. [DOI] [PubMed] [Google Scholar]; Swaminathan S, Narayan KV. Chem. Rev. 1971;71 [Google Scholar]
- 17.1973. pp. 1501–1507.; Bandini M, Tragni M. Org. Biomol. Chem. 7 doi: 10.1039/b823217b. [DOI] [PubMed] [Google Scholar]; Olah GA. Friedel–Crafts Chemistry. New York: Wiley Interscience; 2009. [Google Scholar]
- 18.Sanz R, Martínez A, Guilarte V, Álvarez JM, Rodríguez F. Eur. J. Org. Chem. 2007:4642–4545. [Google Scholar]
- 19.pp. 4045–4048.; Ritter JJ, Minieri PP. J. Am. Chem. Soc. 70 doi: 10.1021/ja01192a022. [DOI] [PubMed] [Google Scholar]; Ritter JJ, Kalish J. J. Am. Chem. Soc. 1948;70 doi: 10.1021/ja01192a023. [DOI] [PubMed] [Google Scholar]
- 20.1948. pp. 803–852.; Ishikura M, Yamada K. Nat. Prod. Rep. 1996;26 doi: 10.1039/b820693g. [DOI] [PubMed] [Google Scholar]; Sundberg RJ. Indoles. San Diego: Academic Press; 2009. [Google Scholar]
- 21.For a silver-mediated example, see: Hao L, Pan Y, Wang T, Lin M, Chen L, Zhan Z-p. Adv. Synth. Catal. 2010;352:3215–3222. -toluenesulfonic acid was tested as catalyst. [Google Scholar]
- 22.pp. 3347–3351.; Bergman J, Högberg S, Lindström J. Tetrahedron. 1970;26 [Google Scholar]; Jackson AH, Prasitpan N, Shannon PV, Tinker AC. J. Chem. Soc. Perkin Trans. 1. 1987 [Google Scholar]
- 23.pp. 2043–2046.; Zhang X, Li X, Lanter JC, Sui Z. Org. Lett. 2005;7 doi: 10.1021/ol050623n. For a review, see: [DOI] [PubMed] [Google Scholar]; Larghi EL, Kaufman TS. Synthesis. 2006.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information