

Abstract
Objective:
To determine the specificity of myositis-specific autoantibodies (MSAs) for autoimmune myopathy compared with inherited muscle diseases.
Methods:
Serum samples from 47 patients with genetically confirmed inherited muscle diseases were screened for the most common MSAs, including those recognizing TIF1γ, NXP2, Mi2, MDA5, Jo1, SRP, and HMGCR. We compared these results with the findings in a cohort of patients with dermatomyositis (DM) previously screened for anti-TIF1γ, -NXP2, -Mi2, -MDA5, and -Jo1.
Results:
Overall, the presence of anti-TIF1γ, -NXP2, -Mi2, -MDA5, or -Jo1 was 96% specific and 67% sensitive for DM compared to patients with genetic muscle diseases. No patients with inherited muscle disease had anti-SRP or anti-HMGCR autoantibodies. Only 2 patients with genetic muscle disease had a MSA. One patient with anti-Mi2 autoantibodies had both genetically confirmed facioscapulohumeral dystrophy and dermatomyositis based on a typical skin rash and partial response to immunosuppressive medications. A second patient with anti-Jo-1 autoantibodies had both genetically defined limb-girdle muscular dystrophy type 2A (i.e., calpainopathy) and a systemic autoimmune process based on biopsy-confirmed lupus nephritis, sicca symptoms, and anti-Ro52 autoantibodies.
Conclusions:
The MSAs tested for in this study are highly specific for autoimmune muscle disease and are rarely, if ever, found in patients who only have genetic muscle disease. In patients with genetic muscle disease, the presence of a MSA should suggest the possibility of a coexisting autoimmune process.
The autoimmune myopathies are a heterogeneous group of diseases characterized by proximal muscle weakness, elevated muscle enzyme levels, and muscle biopsies revealing inflammation or myofiber necrosis.1,2 The principal forms of autoimmune myopathy are dermatomyositis (DM), polymyositis (PM), and immune-mediated necrotizing myopathy (IMNM).3 Many patients with these diseases produce autoantibodies. Myositis-specific autoantibodies (MSAs) are defined by their presence in patients with autoimmune myopathy, but not in those with other rheumatic diseases.
Patients with inherited muscle disease may present with clinical features resembling myositis. The utility of MSAs to distinguish myositis from genetic muscle disease depends on them being found specifically in patients with myositis. Importantly, it was demonstrated a decade ago that 3 of the most commonly recognized MSAs at the time (anti-Jo1, -SRP, and -Mi2) were only rarely found in patients with inherited muscle disease.4 Since then, however, several new MSAs have been identified.5 These include anti-TIf1γ, -NXP2, and -MDA5; like anti-Mi2, these autoantibodies are found only in patients with DM. Similarly, anti-HMG-CoA reductase (HMGCR) autoantibodies, like anti-SRP autoantibodies, are now recognized to be associated with IMNM.6
In the current study, we have determined the prevalence of the most common MSAs in a population of patients with inherited muscle disease. A large proportion of patients with facioscapulohumeral dystrophy (FSHD) were included because muscle biopsies from these patients demonstrate inflammatory infiltrates. We also included patients with other muscular dystrophies reported to have an inflammatory component, including limb-girdle muscular dystrophies (LGMD) 1C, 2A, 2B, and 2L.
METHODS
Patients.
Sera from all 20 patients with available samples and a genetically confirmed diagnosis of inherited muscle disease evaluated between 2008 and 2013 at the Johns Hopkins Myositis Center were screened for the presence of the most common MSAs. The group consisted of 5 patients with type 2 myotonic dystrophy, 4 with FSHD, 3 with LGMD 2B, 2 with Laing distal myopathy, and one each with LGMD 1C, LGMD 2A, LGMD 2I, LGMD 2L, mitochondrial myopathy, and a valosin-containing protein gene mutation (causing inclusion body myopathy with Paget disease of bone and dementia). An additional 27 sera from patients with genetically confirmed FSHD were from the Center for Genetic Muscle Disorders at Kennedy Krieger. The results were compared with a control group of 91 patients with well-defined DM, reported previously.7
Standard protocol approvals, registrations, and patient consents.
The study protocols were approved by the Johns Hopkins Institutional Review Board and all participants signed informed consent.
Autoantibody assays.
All sera were tested for autoantibodies recognizing the 7 most common targets of MSAs: Jo1, TIF1γ, NXP2, MDA5, Mi2, SRP, and HMGCR. Anti-Jo1 and anti-HMGCR antibodies were determined using commercially available ELISA kits (Inova Diagnostics, San Diego, CA). MDA5, NXP2, SRP54, and Mi2 antibodies were assayed by immunoprecipitation using 35S-methionine-labeled proteins generated by in vitro transcription and translation (IVTT) from the appropriate cDNAs as described.8 All IVTT immunoprecipitates were electrophoresed on sodium dodecyl sulfate–polyacrylamide gels and detected by fluorography. Initial screening for TIF1γ antibodies was performed by immunoprecipitation from HeLa cells labeled with 35S-methionine as previously described.9 In 8 cases, a protein of 140–160 kD was immunoprecipitated from HeLa cells. Since TIF1γ has a molecular weight of ∼155 kD, we subsequently retested these 8 sera using an assay that specifically reads out anti-TIF1γ antibodies. This was done by immunoprecipitation using lysates made from cells transiently transfected with TIF1γ cDNA, followed by detection by immunoblotting as described previously.8 Of note, autoantibody testing of the participants with genetic muscle disease was performed in the same laboratory using the same methods and reagents as previously reported for the control DM cohort.7
RESULTS
The specificities, sensitivities, positive predictive values, and negative predictive values for anti-TIF1γ, -NXP2, -Mi2, -MDA5, and -Jo1 in DM compared to inherited muscle disease are shown in the table. The specificity and sensitivity of individual autoantibodies ranged from 98% to 100% and from 5% to 27%, respectively. Taken together, these antibodies were 96% specific and 67% sensitive for DM compared to genetic muscle disease.
Table.
MSAs in autoimmune compared to genetic muscle disease
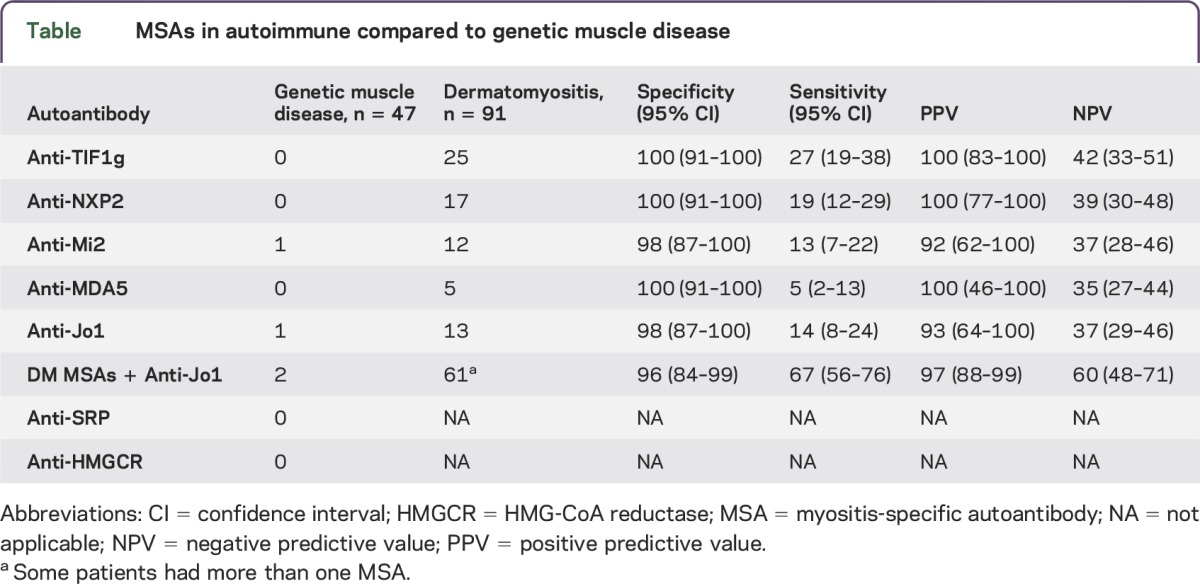
None of the 47 participants with inherited muscle diseases tested positive for antibodies recognizing TIF1γ, NXP2, MDA5, SRP, or HMGCR (table). A single patient with FSHD tested positive for Mi2. Interestingly, this female patient had genetically confirmed FSHD when referred to the Johns Hopkins Myositis Center for suspected dermatomyositis based on the presence of characteristic rashes with resolution of the rash and partial improvement in proximal muscle weakness with immunosuppressive therapy. A female patient with LGMD 2A tested positive for Jo1 autoantibodies. This patient had a muscle biopsy revealing a mild necrotizing myopathy without prominent inflammation; this is a common finding in LGMD and is also the predominant histopathologic feature in ∼15% of Jo-1-positive patients.7 Of note, this patient had manifestations of widespread autoimmunity including biopsy-proven lupus nephritis, sicca symptoms (i.e., dry mouth and positive Schirmer test), positive antinuclear antibodies (1:2,560), and positive anti-Ro. However, she did not have other manifestations of the antisynthetase syndrome, such as arthritis or interstitial lung disease.
DISCUSSION
Since antibodies recognizing Jo1, SRP, and Mi2 were shown to be specific for autoimmune myopathy compared with genetic muscle diseases,4 several other MSAs have been discovered. Some of these new antibodies are even more common in patients with myositis than those previously studied. For example, in the Johns Hopkins Myositis Cohort, TIf1γ and NXP2 were found in 27% and 19% of patients with DM, respectively; Mi2 was present in just 13% of patients with DM.7 Similarly, in patients with necrotizing myopathies in the Johns Hopkins cohort, antibodies recognizing HMGCR were found in 42%, whereas anti-SRP was found in just 16%.9
In this study, we demonstrate that the myositis autoantibodies we tested for were only found in 2 of 47 (4.2%) participants with inherited muscle diseases. These included one patient with anti-Mi2 and one with anti-Jo-1, both of which are found in patients with DM. In contrast, among 91 patients with DM tested, 61 (67%) had antibodies recognizing TIF1g, NXP2, Mi2, Jo-1, or MDA5.7 Thus, the sensitivity and specificity for these antibodies in DM compared to genetic muscle disease is 67% and 96%, respectively.
Anti-SRP and anti-HMGCR antibodies are found in patients with IMNM and were not found in any patients with inherited muscle disease. Thus, these 2 antibodies appear to be 100% specific for IMNM compared to those with inherited muscle diseases. Of note, the presence of one of these antibodies is what usually confirms the diagnosis of IMNM. In patients with a necrotizing muscle biopsy who do not have an antibody, the diagnosis is often in doubt; a significant number of these patients may have an undiagnosed genetic disease. Given the uncertainty in diagnosing antibody-negative IMNM, we did not attempt to define the sensitivity of anti-SRP and -HMGCR antibodies for IMNM.
There are vanishingly few patients among more than 1,000 in the Johns Hopkins Myositis Center cohort that we would currently consider to have PM. Most patients referred to our clinic with an established or suspected diagnosis of PM are ultimately diagnosed with inclusion body myositis, IMNM, a genetic muscle disease, or the antisynthetase syndrome (which is defined by the presence of Jo-1 or another antisynthetase antibody). Other groups have also confirmed the exceptional rarity of PM.10,11 Consequently, we have not attempted to determine the sensitivity and specificity of MSA testing in PM.
Importantly, the participant with FSHD and the DM autoantibody Mi-2 was diagnosed with DM prior to autoantibody testing. This is not the first patient to be described with both a genetic and autoimmune muscle disease. Indeed, a patient with LGMD 2L caused by anoctamin-5-gene mutations was shown to also have anti-HMGCR antibodies.12 Like our FSHD/DM participant, this patient improved with immunosuppressive treatment, supporting the conclusion that he had both genetic and autoimmune muscle diseases. It is also notable that the LGMD 2A patient who tested positive for Jo-1 antibodies in the current study had evidence of systemic autoimmunity, if not other typical manifestations of the antisynthetase syndrome. Taken together, these examples suggest that a positive MSA test in a patient with genetic muscle disease may not be a false-positive. Rather, such a result may suggest that there is a coexisting autoimmune process. We suspect that in these cases, the presence of both an autoimmune muscle disease and a genetic muscle disease is not serendipitous. Rather, we hypothesize that high levels of myositis autoantigens known to be expressed by regenerating muscle cells13–15 might fuel an autoimmune process in susceptible patients (e.g., those with specific immunogenetic risk factors).
This study has several limitations. First, not all forms of inherited myopathy were represented in our cohort, and it may be that myositis-specific autoantibodies could be found in one or more of these diseases. Second, we tested only for the most common myositis-specific autoantibodies; it is possible that some of the rarer myositis autoantibodies could be found in patients with muscular dystrophies. Third, the positive and negative predictive values shown in the table may not be accurate for other patient populations with different relative proportions of the different types of muscle disease.
These limitations notwithstanding, this study shows that MSAs are only infrequently found in patients with genetic muscle diseases and have a high specificity for patients with autoimmune myopathy. These findings support the use of MSA testing to help distinguish autoimmune from genetic muscle diseases. However, clinicians should be aware that in rare cases, patients testing positive for an MSA could also have a genetic muscle disease that will not respond to immunosuppressive therapy.
ACKNOWLEDGMENT
Dr. Kathryn Wagner supplied vital reagents for the study.
GLOSSARY
- DM
dermatomyositis
- FSHD
facioscapulohumeral dystrophy
- HMGCR
HMG-CoA reductase
- IMNM
immune-mediated necrotizing myopathy
- IVTT
in vitro transcription and translation
- LGMD
limb-girdle muscular dystrophies
- MSA
myositis-specific autoantibody
- PM
polymyositis
AUTHOR CONTRIBUTIONS
Andrew Mammen: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, acquisition of data, statistical analysis, study supervision. Livia Casciola-Rosen: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, acquisition of data, study supervision, obtaining funding. Thomas E. Lloyd: drafting/revising the manuscript, contribution of vital reagents/tools/patients. Lisa Christopher-Stine: study concept or design, contribution of vital reagents/tools/patients, acquisition of data. Kathryn R. Wagner: study concept or design.
STUDY FUNDING
Supported in part by the Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the NIH. These studies were funded by NIH grant R56A062615 (L.C.-R.). The Johns Hopkins Rheumatic Disease Research Core Center, where the assays were performed, is supported by NIH grant P30-AR-053503.
DISCLOSURE
A. Mammen is on the advisory board for aTYR Pharmaceuticals and Biogen, is on the editorial board for Experimental Neurology and Arthritis and Rheumatism, and has patented the anti-HMGCR antibody test and receives royalties from it. L. Casciola has patented the anti-HMGCR antibody test and receives royalties from it, received research support from the NIH and Jerome L. Greene Foundation. T.E. Loyd received research support from Novartis, NIH, Muscular Dystrophy Association, Robert Packard Center for ALS Research, and ALSA. L. Christopher-Stine is on the scientific advisory board for Novartis, Medimmune, Walgreen's/Option Care, Mallinckrodt, and Idera Pharmaceuticals; received travel funding and/or speaker honoraria from Inova Diagnostics; has patented the anti-HMGCR antibody test and received royalties from it; has consulted for Mallinckrodt Pharmaceuticals, Astellas, Ono Pharma UK, and Marathon Pharmaceuticals; received research support from NuFactor, Walgreens, IgG America, MedProRx, NHLBI, and Donald B. and Dorothy L. Stabler Foundation; and has been an expert witness for McAloon and Friedman, Schaub Ahmuty, Citrin and Spratt, and The Chartwell Law Offices. K.R. Wagner is on the scientific advisory board for SOLID GT, FSH Society, and PPMD DSMB of CINRG; and received research support from NICHD, NCATS, and Team Saij. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Mammen AL. Dermatomyositis and polymyositis: clinical presentation, autoantibodies, and pathogenesis. Ann NY Acad Sci 2010;1184:134–153. [DOI] [PubMed] [Google Scholar]
- 2.Rider LG, Miller FW. Deciphering the clinical presentations, pathogenesis, and treatment of the idiopathic inflammatory myopathies. JAMA 2011;305:183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoogendijk JE, Amato AA, Lecky BR, et al. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10–12 October 2003, Naarden, the Netherlands. Neuromuscul Disord 2004;14:337–345. [DOI] [PubMed] [Google Scholar]
- 4.Hengstman GJ, van Brenk L, Vree Egberts WT, et al. High specificity of myositis specific autoantibodies for myositis compared with other neuromuscular disorders. J Neurol 2005;252:534–537. [DOI] [PubMed] [Google Scholar]
- 5.Casciola-Rosen L, Mammen AL. Myositis autoantibodies. Curr Opin Rheumatol 2012;24:602–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum 2011;63:713–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pinal-Fernandez I, Casciola-Rosen L, Christopher-Stine L, Corse A, Mammen A. The prevalence of individual histopathologic features varies according to autoantibody status in muscle biopsies from dermatomyositis patients. J Rheumatol 2015;42:1448–1454. [PMC free article] [PubMed] [Google Scholar]
- 8.Fiorentino DF, Chung LS, Christopher-Stine L, et al. Most patients with cancer-associated dermatomyositis have antibodies to nuclear matrix protein NXP-2 or transcription intermediary factor 1gamma. Arthritis Rheum 2013;65:2954–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum 2010;62:2757–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amato AA, Griggs RC. Unicorns, dragons, polymyositis, and other mythological beasts. Neurology 2003;61:288–289. [DOI] [PubMed] [Google Scholar]
- 11.van der Meulen MF, Bronner IM, Hoogendijk JE, et al. Polymyositis: an overdiagnosed entity. Neurology 2003;61:316–321. [DOI] [PubMed] [Google Scholar]
- 12.Claeys KG, Gorodinskaya O, Handt S, et al. Diagnostic challenge and therapeutic dilemma in necrotizing myopathy. Neurology 2013;81:932–935. [DOI] [PubMed] [Google Scholar]
- 13.Casciola-Rosen L, Nagaraju K, Plotz P, et al. Enhanced autoantigen expression in regenerating muscle cells in idiopathic inflammatory myopathy. J Exp Med 2005;201:591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mammen AL, Casciola-Rosen LA, Hall JC, Christopher-Stine L, Corse AM, Rosen A. Expression of the dermatomyositis autoantigen mi-2 in regenerating muscle. Arthritis Rheum 2009;60:3784–3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mohassel P, Rosen P, Casciola-Rosen L, Pak K, Mammen AL. Expression of the dermatomyositis autoantigen transcription intermediary factor 1 gamma in regenerating muscle. Arthritis Rheumatol 2015;67:266–272. [DOI] [PMC free article] [PubMed] [Google Scholar]