

Abstract
Background:
Residual CXCR2 expression on CNS cells in Cxcr2+/−→Cxcr2−/− chimeric animals slowed remyelination after both experimental autoimmune encephalomyelitis and cuprizone-induced demyelination.
Methods:
We generated Cxcr2fl/−:PLPCre-ER(T) mice enabling an inducible, conditional deletion of Cxcr2 on oligodendrocyte lineage cells of the CNS. Cxcr2fl/−:PLPCre-ER(T) mice were evaluated in 2 demyelination/remyelination models: cuprizone-feeding and in vitro lysophosphatidylcholine (LPC) treatment of cerebellar slice cultures.
Results:
Cxcr2fl/−:PLPCre-ER(T)+ (termed Cxcr2-cKO) mice showed better myelin repair 4 days after LPC-induced demyelination of cerebellar slice cultures. Cxcr2-cKOs also displayed enhanced hippocampal remyelination after a 2-week recovery from 6-week cuprizone feeding.
Conclusion:
Using 2 independent demyelination/remyelination models, our data document enhanced myelin repair in Cxcr2-cKO mice, consistent with the data obtained from radiation chimerism studies of germline CXCR2. Further experiments are appropriate to explore how CXCR2 function in the oligodendrocyte lineage accelerates myelin repair.
Multiple sclerosis (MS) is a chronic inflammatory demyelinating disease in the CNS and remyelination often ultimately fails. Oligodendrocyte progenitor cells (OPCs) are believed to give rise to most remyelinating oligodendrocytes.1 Understanding the mechanisms that control the migration, differentiation, and proliferation of OPCs during remyelination is crucial for developing effective MS treatments.2 Chemokine receptor CXCR2 mediates a diverse array of functions, being expressed on neutrophils in the periphery and OPCs in the CNS.3,4 Inactivation of CXCR2 in the CNS in radiation chimeras promoted remyelination in both experimental autoimmune encephalomyelitis and cuprizone feeding demyelination animal models.5 Furthermore, myelin repair was improved when CXCR2 was inactivated by either CXCR2 blocking antibodies or genetic deletion in demyelinated cerebellum slice cultures.5Given the global deletion of CNS CXCR2 in radiation chimeras, our data left unclear whether CXCR2 in oligodendrocyte lineage cells was responsible for enhancing remyelination.
Our laboratory generated Cxcr2fl/fl mice in which CXCR2 could be deleted on specific cell types based on the Cre recombinase expression.6 We crossed Cxcr2fl/fl mice to PLPCreER(T), in which the inducible Cre recombinase is abundantly expressed in oligodendrocyte lineage cells to generate Cxcr2fl/fl:PLPCreER(T) mice; then we crossed Cxcr2fl/fl:PLPCreER(T) mice to Cxcr2+/− mice to generate Cxcr2fl/−:PLPCreER(T) mice. Efficient recombination of the floxed Cxcr2 gene in oligodendrocyte lineage cells was observed 4 weeks following tamoxifen injection. This genetic model was then evaluated in toxin-induced models of demyelination/remyelination, to enable analysis of precisely timed and localized regions of remyelination.
METHODS
Mice.
Cxcr2fl/fl mice (stock number 024638; strain name C57BL/6-Cxcr2tm1Rmra/J, from the Jackson Laboratory, Sacramento, CA) were crossed to PLPCreERT (from Dr. Brian Popko, The University of Chicago)7 and PLPeGFP mice (from Dr. Wendy Macklin, Lerner Research Institute, Cleveland Clinic) to generate Cxcr2fl/fl:PLPCreERT+:PLPeGFP+. For most of the experiments such as cuprizone/rapamycin experiments, we crossed germline Cxcr2+/− to Cxcr2fl/fl:PLPCreERT+:PLPeGFP+ to generate Cxcr2fl/+:PLPcreERT:PLPeGFP+ (littermate control) or Cxcr2fl/−:PLPcreERT+:PLPeGFP+ mice (Cxcr2-cKO). ROSA26mT/mG mice (stock number 007676; strain name Gt[ROSA]26Sortm4[ACTB-tdTomato,-EGFP]luo/J, the Jackson Laboratory) were crossed to PLPCreERT mice to generate ROSA26 mT/mG+:PLPCreERT+.
Standard protocol approvals.
The Institutional Animal Care and Use Committee at the Cleveland Clinic approved all mouse studies.
Genotyping.
The Cxcr2fl, Cxcr2+, Cxcr2−, PLPCreERT+, PLPeGFP+, and ROSA26 mT/mG alleles were detected as described previously.3,5–8 Primers for Cxcr2fl, Cxcr2+, Cxcr2− and detection of deletion were described previously.3,5,6 PLPCreERT (450 bp) was detected using forward primer (GATGTAGCCAGCAGCATGTC) and reverse primer (ACTATATCCGTAACCTGGAT). PLPeGFP (180 bp) was detected using forward primer (AAGTTCATCTGCACCACCG) and reverse primer (TCCTTGAAGAAGATGGTGCG). ROSAmTmG was genotyped using the following 3 primers: P4349: CTCTGCTGCCTCCTGGCTTCT; P4350: CGAGGCGGATCACAAGCAATA; and P4351: TCAATGGGCGGGGGTCGTT to identify the WT band (330 bp) and mutant band (250 bp).
Injection of tamoxifen.
Four-week-old mice were treated with 1 mg of tamoxifen (Sigma-Aldrich, St. Louis, MO; T5648) dissolved in 10% ethanol and sunflower seed oil twice a day for 5 days in a row. Eight to 10 weeks after last injection, mice were killed for flow cytometry or brain histology.
Flow cytometry.
Flow cytometry was performed as previously described3 with modification. Briefly, peripheral erythrocytes were lysed and washed in FACS buffer. After blocking, the cells were incubated with CD45APC (Biolegend, San Diego, CA; clone: 30-F11) at 4°C for 30 minutes. Cells were washed twice and resuspended for flow cytometry analysis (LSRFortessa; BD Biosciences, Franklin Lakes, NJ). FlowJo software (version 7.6 Tree Star) was used for data analysis.
Tissue processing and histologic analysis.
For histologic analysis of CNS tissues, 4 weeks after tamoxifen injection, ROSA26 mT/mG+:PLPCreERT+ mice were perfused with phosphate-buffered saline (PBS), then with 4% paraformaldehyde (PFA) solution transcardially. Brains were rapidly dissected and postfixed in 4% PFA for 24 hours at 4°C. Then brains were washed with PBS and soaked in cryoprotection buffer at least 12 hours at 4°C, and 30-μm-thick coronal sections were cut using a microtome (Leica Microsystems, Wetzlar, Germany) and kept in cryostorage buffer at freezer for future experiments. Slides were made using tissues from different areas of brain mounted with DAPI-anti-fade mounting medium (DAKO, Carpinteria, CA). Images were obtained with a Leica DM5500B microscope equipped with Q-Capture software.
Black-Gold staining.
Coronal sections of brain from control and Cxcr2-cKO 4 weeks after tamoxifen injection mice were stained with Black-Gold solution (Histo-Chem, Jefferson, AR) as previously described.3
Oligodendrocyte lineage cell sorting.
Brains from controls or Cxcr2-cKO mice were dissected from the skull after PBS perfusion. Thirty percent to seventy percent Percoll gradient was used to isolate the total brain cells after digestion with 30 U/mL papain (Roche, Basel, Switzerland; 10108014001). The total cells were stained with CD45APC (Biolegend, clone: 30-F11) after CD16/32 (Biolegend, 101310) and mouse serum blocking. Cells were sorted on a Becton Dickinson (San Jose, CA) FACSAria II SORP with blue 488 nm and red 640 nm excitation lasers running Diva 6.1., gating on CD45− and eGFP+ cells.
RNA extraction and qPCR for analysis of Cxcr2 deletion.
Deletion of CXCR2 was determined by qPCR of sorted PLPeGFP+ cells from controls and Cxcr2-cKO mice. RNA was extracted using Qiagen (Venlo, Netherlands) Qiashredder and RNAeasy kits. Fifty nanogram cDNA was analyzed by qPCR on a Roche LightCycler.3 CXCR2, PLP, and GAPDH expression was determined using the following primers: GAPDH-F: CATGGCCTTCCGTGTTCCTA; GAPDH-R: ATGCCTGCTTCACCACCTTCT; Cxcr2-F2: AGCAAACACCTCTACTACCCTCTA; Cxcr2-R2: GGGCTGCATCAATTCAAATACCA; PLP-F11: TCAGTCTATTGCCTTCCCTAGC; PLP-R11: AGCATTCCATGGGAGAACAC.
Cuprizone/rapamycin demyelination/remyelination model.
Cuprizone/rapamycin experiments were carried out by Renovo Neural (Cleveland, OH). Briefly, controls and Cxcr2-cKO mice received 0.3% (w/w) cuprizone (bis-cyclohexanone oxaldihydrazone; Sigma-Aldrich) for 6 weeks demyelination, and received intraperitoneal injections of 0.25 mg/d rapamycin (LC Laboratories, Woburn, MA)9 followed by 2 weeks of spontaneous remyelination (cuprizone feed was replaced with normal feed and rapamycin injections were halted). Thirty-micrometer coronal brain sections were obtained as explained earlier. Thirty-micrometer floating sections were immunostained with PLP antibody (gift from Dr. Bruce Trapp's laboratory, Department of Neurosciences, Lerner Research Institute) followed by biotin conjugated secondary antibody and avidin-biotin complex and development with 3,3′-diaminobenzidine. Stained tissues were imaged and analyzed to quantify percentage of area occupied by PLP staining in the hippocampus by ImageJ.
Cerebellum slice culture.
The cerebella of postnatal day 13 (P13) mice were cut into 300-μm-thick sagittal sections by a vibratome (Leica VT 1000S) and cultured at top of a Millicell insert as previously described.3 Sections were cultured with 4-hydroxytamoxifen (1 μM, 4-HT) (Sigma-Aldrich) for 2 days to delete the Cxcr2LoxP or ROSA26 mT/mGLoxP. The efficiency of PLPCreERT was determined by observing the turnover of mT to mG in ROSA26 mT/mG + PLPCreERT + mice on myelinated area, which was confirmed by myelin basic protein (MBP) antibody staining (data not shown). Sections were cultured with 0.25 mg/mL lysolecithin (Sigma-Aldrich) in complete media for 17 hours to induce the demyelination, and cultured with fresh media for an additional 4 days to allow remyelination. To evaluate the demyelination and remyelination, MBP antibody and neurofilament antibody were applied to stain the slices.
Quantification of colocalization of myelin sheath with axon.
Confocal images were acquired at a magnification of 63× using a Leica SP5 confocal microscope. To quantify the myelination in the cerebellar slices, the proportion of neurofilament overlapped by myelin basic protein was analyzed using Fiji software. Images were thresholded using the autothreshold method in Fiji and a circularity filter was applied to exclude nonspecific staining and select for axons based on their cylindrical shape. The Mander overlap coefficient was calculated using the Coloc_2 plugin.10 Preprocessing and plugin implementation were combined into a macroinstruction. To confirm the validity of the analysis, the results were analyzed and determined to be independent of the area and the mean fluorescent intensity of the image. To determine the ideal method of image analysis, results obtained from the full z-stack were compared with those derived from each individual image in the plane. As expected, the values derived from the full z-stack were higher than the individual plane images, but the trends between images were consistent, suggesting that either data set can be used for analysis. During method development, colocalized images were visually assessed to confirm the accuracy of preprocessing in selecting for axonal and myelin staining. The Mander coefficient varied minimally with differences in fluorescent channel intensity during image acquisition and variation in stack thickness.
Statistical analysis.
All data are expressed as mean ± SD. One-way analyses of variance were used to evaluate the multiple comparisons by using Prism 5 (GraphPad Software, La Jolla, CA). The related expression of CXCR2 and PLP between Cxcr2-cKO and control mice was evaluated by the Student t-test. A p value <0.05 was considered significant.
RESULTS
Conditional knockout of Cxcr2 (Cxcr2-cKO) mice in the oligodendrocyte lineage.
Given previous findings that CXCR2 can be expressed in the oligodendrocyte lineage, we generated Cxcr2fl/+:PLPCreERT+ (control) and Cxcr2fl/−:PLPCreERT+ (Cxcr2-cKO) animals (figure 1). Cre recombinase was induced with intraperitoneal injections of tamoxifen.7 PCR results from genomic DNA after tamoxifen injection showed the Cxcr2 deletion (∼400 bp) was only in samples from Cxcr2fl/−:PLPCreERT+ and Cxcr2fl/+:PLPCreERT+ but not Cxcr2+/+:PLPCreERT+ or Cxcr2fl/fl mice (data not shown). Sequence analysis confirmed deletion of Cxcr2 exon 2 (data not shown).
Figure 1. Generation of a conditional Cxcr2 allele and genotype determination of conditional knockouts by PCR.
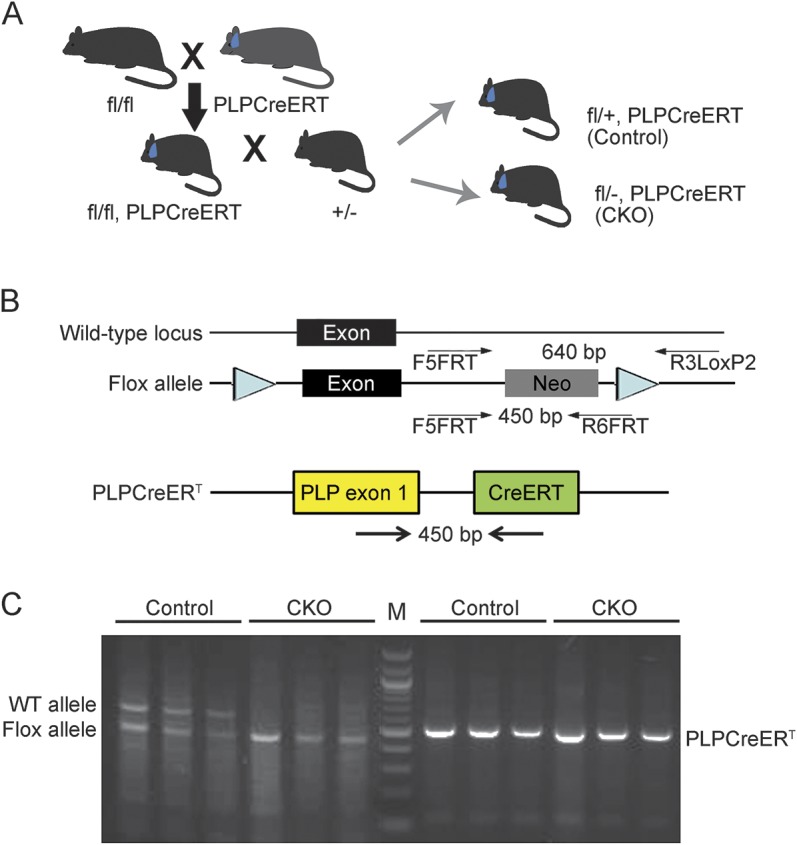
(A) Schematic diagram for Cxcr2-cKO mouse generation. (B) Schematic diagram for Cxcr2-cKO mouse genotyping. (C) Genotype determination of conditional knockouts by PCR. PCR amplification of genomic DNA using primers for the flox gene before injection of tamoxifen; the higher band (∼650 bp) indicates the wild-type allele and the lower band (∼450 bp) indicates the flox allele. PLPCreERT transgenes detection: PCR amplification of the genomic DNA using primers indicated on (B) for PLPCreERT band (∼500 bp).
Tamoxifen treatment induces deletion of CXCR2 on oligodendrocyte lineage cells but not on peripheral blood cells in Cxcr2-cKO mice.
CXCR2 antibodies have been nonspecific for immunohistochemistry.3 ROSA26mT/mG mice express Tomato red (mT) before recombination and green fluorescent protein (mG) after recombination.8 To examine the efficiency and specificity of the PLPCreERT Cre driver, we crossed ROSA26AmT/mG reporter mice8 to Cxcr2fl/−:PLPCreERT+ mice. Four weeks following tamoxifen injection, green fluorescent parenchymal cells were present exclusively in CNS white matter, such as the striatum of ROSA26 mT/mG+:PLPCreERT+ mice compared to noninjected mice (ROSA26 mT/mG+:PLPCreERT+ mice) (figure 2A). We verified that no recombination was observed in microglia (CD45dim cells isolated from brain single cell suspensions over Percoll gradients) (figure 2B). As well, there were no eGFP+ cells in peripheral blood of ROSA26 mT/mG+:PLPCreERT+ mice after tamoxifen injections (figure e-1 at Neurology.org/nn). We isolated oligodendrocyte lineage cells11 and immediately ex vivo examined CXCR2 mRNA by qPCR. Cxcr2-cKO mice showed 87% reduction of CXCR2 mRNA (normalized to PLP) compared to littermate controls, with about 15% of residual CXCR2 expression as compared to germline Cxcr2−/− mice (figure 2C). Taken together, these results showed that recombination in PLPCreERT mice occurred in CNS oligodendrocyte lineage cells.
Figure 2. Induced deletion of CXCR2 on oligodendrocyte lineage cells after tamoxifen injection in Cxcr2-cKO mice.

(A) Striatum from ROSA26 mT/mG+:PLPCreERT+ mice with or without tamoxifen injection (4 weeks after injection) reveals the expression of tomato red and GFP by fluorescence microscopy. These data represent at least 3 independent experiments. Scale bar: 25 μM, n = 3 mice each experiment. (B) Brain cells from B6, ROSA26 mT/mG+:PLPCreERT+ mice with or without tamoxifen injection were isolated by Percoll gradient, stained with CD45 antibody, and analyzed by flow cytometry. These data represent at least 3 independent experiments. (C) Brain cells from control and Cxcr2-cKO mice 4 weeks after tamoxifen injection were isolated by Percoll gradient. The PLPeGFP-positive cells were sorted from 6 pooled mice per group. Total RNA was extracted from sorted cells and qPCR was performed for CXCR2 and PLP genes. These data represent 3 technical replicates.
Similar myelin content in tamoxifen-treated Cxcr2fl/+:PLPCreERT+ and Cxcr2fl/−:PLPCreERT+ mice.
Cxcr2fl/−:PLPCreERT+ mice were born in the expected Mendelian ratios. Normal size, behavior, and lifespan were shown for these mice. Both Cxcr2fl/−:PLPCreERT+ males and females were able to mate and rear pups until weaning, and were indistinguishable from Cxcr2fl/+:PLPCreERT+ littermates. We previously reported that adult germline Cxcr2−/− mice showed some alterations in the oligodendrocyte lineage, myelin, and white matter,12 which were associated with runting, poor breeding, and reduced lifespan. To exclude confounds related to baseline myelination, we examined CNS tissues of Cxcr2-cKO mice for myelin integrity. Myelin histochemical staining revealed no difference between tamoxifen-treated Cxcr2fl/+:PLPCreERT+ and Cxcr2fl/−:PLPCreERT+ mice (figure 3A). Furthermore, we found similar PLP expression by isolated oligodendrocyte-lineage cells in both tamoxifen-treated Cxcr2fl/+:PLPCreERT+ and Cxcr2fl/−:PLPCreERT+ mice (figure 3B). These results indicated that induced deletion of CXCR2 on oligodendrocyte lineage cells did not alter the myelin content in tamoxifen-treated Cxcr2fl/−:PLPCreERT+ mice.
Figure 3. Similar myelin content in tamoxifen-treated control and Cxcr2-cKO mice.

(A) Brain tissues from control and Cxcr2-cKO mice 4 weeks after tamoxifen injection were stained with Black-Gold solution to analyze extent of myelination. The data shown include corpus callosum (on left) and hippocampi (on right). Scale bar: 100 μM, n = 6 mice each group. (B) Brain cells from control and Cxcr2-cKO mice (6 mice each group) 4 weeks after tamoxifen injection were isolated by Percoll gradient, then PLPeGFP positive cells were sorted. Total RNA was extracted from sorted cells, and qPCR was performed for GAPDH and PLP genes. These data represent 3 technical replicates.
CXCR2 deficiency on oligodendrocyte lineage cells accelerates remyelination in lysophosphatidylcholine-treated cerebellar slice cultures.
To evaluate the effect of conditional deletion of CXCR2 during demyelination/remyelination after lysophosphatidylcholine (LPC) treatment, we employed a modified in vitro cerebellar slice culture model.5 Because cerebellar slices lack the ability to metabolize tamoxifen, an active metabolite of tamoxifen, 4-hydroxytamoxifen (4-HT), was used to induce CXCR2 deletion in cultured Cxcr2fl/−:PLPCreERT+ tissue. These experiments are depicted schematically in figure 4A: Cxcr2fl/+:PLPCreERT+ and Cxcr2fl/−:PLPCreERT+ P13 cerebellar slices were incubated with 4-HT for 2 days before exposure to LPC for 17 hours to induce demyelination. LPC was then removed and the slices were cultured for an additional 4 days before analysis of remyelination. Slices were collected at 17 hours after LPC and at 4 days after LPC removal, to evaluate demyelination and remyelination using double labeling with MBP and neurofilament antibodies (figure 4B). As expected, cerebellar slices showed dramatic demyelination with a lot of myelin debris and demyelinated fibers after 17 hours of LPC treatment (figure 4B, left column). Slices from control mice and from Cxcr2-cKO mice showed equivalent robust demyelination (figure 4B, left column, figure 4C; p = 0.21). At day 4 of recovery, significant remyelination was observed in both control mice and Cxcr2-cKO mice slice cultures (figure 4B, right column, figure 4C; p < 0.01). Remyelination in slices from Cxcr2-cKO mice (figure 4B right column) was significantly more efficient, as compared to control mice at 4 days of recovery (figure 4B, right column, figure 4C; p = 0.01). Control mice and Cxcr2-cKO mice slices incubated without LPC for 9 days in culture (the cumulative time span of the experimental protocol) showed equivalent myelin integrity (data not shown). These data imply that conditional inactivation of CXCR2 on oligodendrocyte lineage cells enhances the efficiency of remyelination, but does not affect demyelination in vitro.
Figure 4. CXCR2 deficiency on oligodendrocyte lineage cells causes rapid and more complete remyelination in an in vitro model of demyelination/remyelination using LPC-treated cerebellar slice cultures.
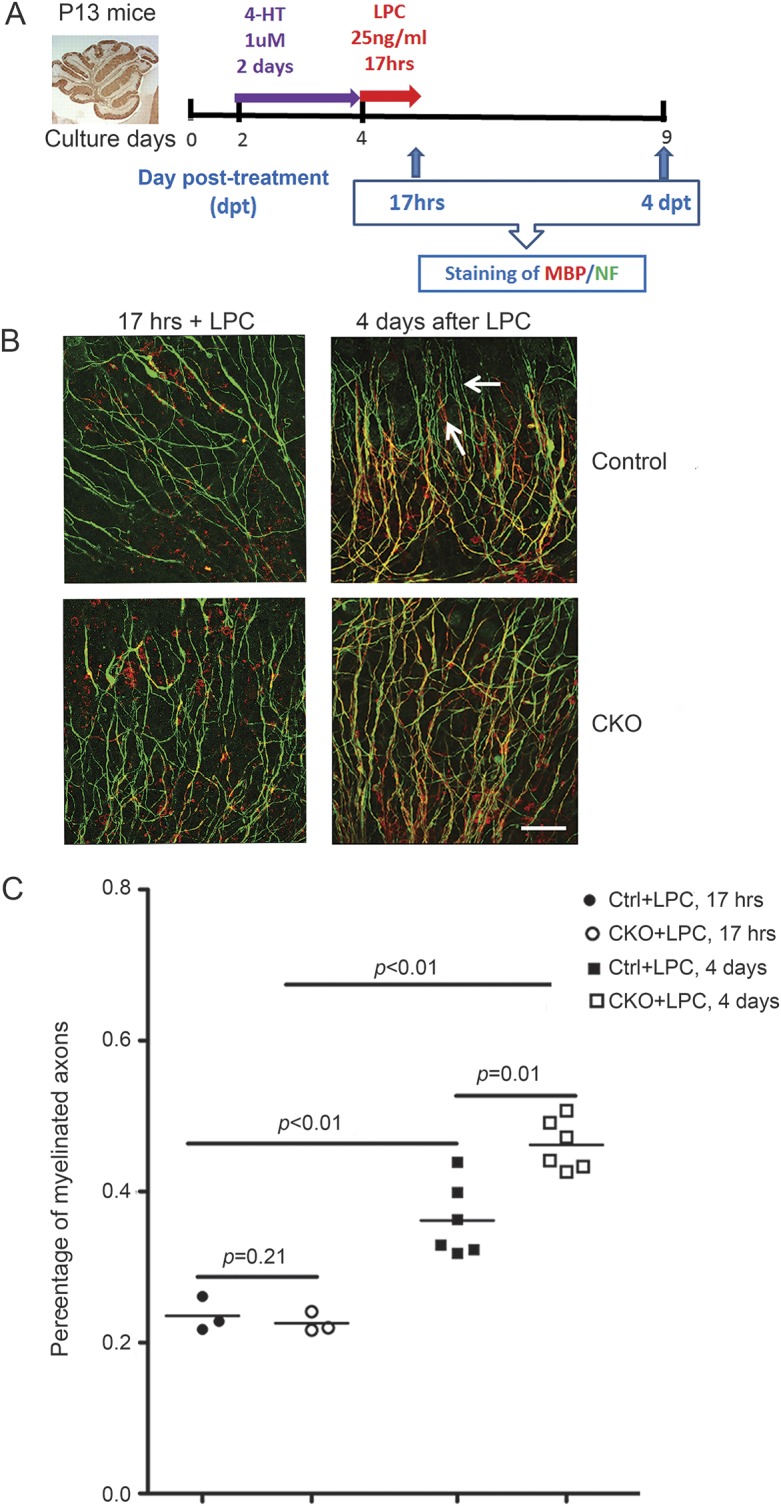
(A) Schematic diagram for slice culture on control and cKO mice. (B) Cerebellar slice from lysophosphatidylcholine (LPC) treated (17 hours) and 4 days after LPC treated from control and cKO mice were collected and dual labeled with myelin basic protein (MBP) (red) and neurofilament (green), with myelinated fibers appearing yellow in merged images. At 17 hours, all slices are demyelinated, showing abundant green unmyelinated nerve fibers and scant yellow myelinated fibers. Four days after removing LPC, better remyelination is shown in slices from cKO mice compared to control mice. (C) The customized Fiji software used to quantify the colocalization of myelin sheath on axons as percentage of myelinated axons is described in detail in the Methods. Scale bar, 25 μm. Arrow: Unmyelinated axons. These data represent 5 independent experiments. Each experiment included 3–4 mice each group.
Deficient CXCR2 in oligodendrocyte lineage cells promotes remyelination in an in vivo cuprizone/rapamycin demyelination model.
We3 and others found that demyelination and recovery overlap markedly during the 6-week cuprizone feeding protocol, complicating evaluation of remyelination. For the present study, we contracted with Renovo Neural (http://www.renovoneural.com/cuprizone-model-of-remyelination), whose modified protocol using rapamycin injection during cuprizone feeding eliminates this confound. This protocol provides for blinded and standardized demyelination/remyelination assays and examination of gray as well as white matter (figure 5A). Both tamoxifen-treated Cxcr2fl/+:PLPCreERT+ and Cxcr2fl/−:PLPCreERT+ mice showed equivalent severe hippocampal demyelination (14.8% PLP–immunoreactive [IR] area fraction in both groups; n = 8) after cuprizone feeding/rapamycin injection for 6 weeks as monitored by quantitative PLP immunohistochemistry (figure 5, B and C). Two weeks after cuprizone feeding, Cxcr2fl/−:PLPCreERT+ mice showed modestly but significantly enhanced remyelination (26.2% PLP-IR area fraction, n = 15) compared to tamoxifen-treated Cxcr2fl/+:PLPCreERT+ mice (24.1% PLP-IR area fraction, n = 15) (figure 5, B and C; p < 0.05). The hippocampi of tamoxifen-treated Cxcr2fl/+:PLPCreERT+ and Cxcr2fl/−:PLPCreERT+ mice were equal in area (data not shown). These results demonstrate that CXCR2 expression on oligodendrocyte lineage cells impaired myelin repair in both in vivo and in vitro models.
Figure 5. CXCR2 deficiency on oligodendrocyte lineage cells accelerates remyelination in a cuprizone/rapamycin-induced demyelination model.

(A) Schematic diagram for cuprizone feeding on control and Cxcr2-cKO mice. (B) Control and Cxcr2-cKO mice 4 weeks after tamoxifen injection were fed cuprizone diet and received daily IP rapamycin injections for 6 weeks. Tissues were collected after cuprizone feeding for 6 weeks or after 2 weeks recovery from 6 weeks cuprizone. PLP antibody staining for myelin shows equivalent demyelination severity of the hippocampi in tamoxifen-treated control and Cxcr2-cKO mice at 6 weeks of cuprizone/rapamycin treatment. Tamoxifen-treated Cxcr2-cKO mice show significantly accelerated remyelination at 2 weeks compared to tamoxifen-treated control mice. Scale bars: 200 μm. (C) Percent of PLP occupied area in the hippocampal area after cuprizone treatment shows no significant differences between these 2 groups at 6 weeks feeding. Percent of PLP occupied area in the hippocampal area shows significant differences between tamoxifen-treated control and Cxcr2-cKO mice remyelination, 2 weeks post recovery from 6 weeks demyelination. *p < 0.05. n = 8 per group for 6 weeks demyelination; n = 15 per group for 2 weeks remyelination.
DISCUSSION
CXCR2 expression on varied cell types is proposed to play crucial roles in diverse diseases from cancer to chronic inflammatory disease to metabolic syndrome. In inflammatory CNS disease, CXCR2 exerts a particularly wide spectrum of functions given its expression both on OPCs and neutrophils. Reports have documented varied effects in models of demyelination, remyelination, host defense against pathogens, and in a viral model of CNS demyelination.3,5,13,14 However, for reasons noted above, it has been challenging to examine the functions of CXCR2 on oligodendroglia by using germline CXCR2 knockout mice.
Here we used spatiotemporal ablation of CXCR2 in CNS oligodendrocyte lineage cells by crossing CXCR2fl/fl mice to PLPCreERT mice. No CXCR2 antibodies demonstrate adequate specificity for immunohistochemical detection of CXCR2 in CNS tissues.3 As an alternative, we crossed the ROSA26 mT/mG mice to Cxcr2fl/−:PLPCreERT+ mice, to monitor the cellular localization and efficiency of Cre-mediated recombination after tamoxifen injection. Four weeks after tamoxifen injection, green fluorescent parenchymal cells were expressed exclusively in the CNS white matter of ROSA26 mT/mG+:PLPCreERT+ mice compared to noninjected mice (figure 2A). Furthermore, qPCR from sorted PLP+ cells from Cxcr2fl/−:PLPCreER(T) mice showed reduction of CXCR2 message by 87% (figure 2C). Flow cytometry verified that there was no recombination in other CNS cell types such as microglia (CD45dim) and astrocyte or neuron cells (CD45neg) or in peripheral blood cells of ROSA26 mT/mG+:PLPCreERT+ mice. Together, these data verified that inducible deletion of CXCR2 in Cxcr2-cKO mice occurs selectively in CNS oligodendrocyte lineage cells.
Next, Cxcr2-cKO mice in in vivo and in vitro models of demyelination/remyelination were evaluated to examine the effect of selectively deleting CXCR2 on oligodendrocyte lineage cells on remyelination. Because it has been reported that varied cell types present in the CNS either in vitro or in vivo express CXCR2, including OPCs, microglia, astrocytes, neural stem cells, and neurons, it remained unclear on which cell types CXCR2 expression hindered myelin repair. By examining Cxcr2-cKO mice in a modified demyelination/remyelination in vitro cerebellar slice culture model, we showed that conditional inactivation of CXCR2 on oligodendrocyte lineage cells enhanced the efficiency of remyelination. Furthermore, by applying Cxcr2-cKO mice in a modified cuprizone-induced demyelination model, we documented significant myelin repair in the hippocampus after recovery from extensive demyelination caused by 6 weeks' cuprizone feeding. These data demonstrated that CXCR2 expression specifically on oligodendrocyte lineage cells impairs myelin repair both in vivo and in vitro. Future studies will be required to address how the absence of CXCR2 on oligodendrocyte lineage cells promotes remyelination, so that pathways to enhance repair and protective functions of oligodendrocyte lineage cells can be translated for patient benefit.
Cxcr2fl/fl mice broaden the research into CXCR2 function through specific cell-type Cre recombination to induce the deletion of the pleiotropic gene product. After we crossed PLPCreER(T), the inducible Cre recombinase that is abundantly expressed in oligodendrocyte lineage cells, to Cxcr2fl/fl mice to generate Cxcr2-cKO, efficient recombination of the floxed Cxcr2 gene in oligodendrocyte lineage cells was observed 4 weeks following tamoxifen injection. Cxcr2-cKO mice showed better myelin repair 4 days after LPC-induced demyelination of cerebellar slice cultures. Cxcr2-cKOs also displayed enhanced hippocampal remyelination after a 2-week recovery from 6-week cuprizone feeding. Using 2 independent demyelination/remyelination models, our data document enhanced myelin repair in Cxcr2-cKO mice, consistent with the data obtained from radiation chimerism studies of germline CXCR2. Further experiments are appropriate to explore how CXCR2 function in the oligodendrocyte lineage accelerates myelin repair.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Dr. Hitoshi Komuro and Dr. Dolores Hambardzumyan (Department of Neurosciences, Lerner Research Institute) for assistance with slice culture; Dr. Satish Medicetty and the late Dr. YiZhe Sun (Renovo Neural, Inc) for helping with the cuprizone model; Dr. Maria Smith and Raymond Gaines (Department of Neurosciences, Lerner Research Institute) for providing Gt(ROSA)26Sortm4(ACTB-tdTomato,−EGFP)luo/J (ROSA26 mT/mG) mice; and the LRI-Flow Core and Dr. Judith Drazba (LRI-Imaging Core) for providing service.
GLOSSARY
- IR
immunoreactive
- LPC
lysophosphatidylcholine
- MBP
myelin basic protein
- MS
multiple sclerosis
- OPC
oligodendrocyte progenitor cell
- PBS
phosphate-buffered saline
- PFA
paraformaldehyde
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
L.L. was responsible for executing the research project, performing the experiment and the statistical analyses, and writing the manuscript. L.S. performed day-to-day experiments. B.P. developed and deployed methods to quantify colocalized immunoreactivity of MBP and NF. B.B., B.H., S.S., A.C., A.L., C.Z., and C.R. worked jointly on extensive modification of published protocols for cutting tissues and staining, as well as image acquisition and quantification. R.M.R. directed the overall project from its conception, designed (with L.L.) all experiments, and revised the manuscript. The final manuscript was read and approved by all authors.
STUDY FUNDING
Supported by National Institutes of Health grants R01NS032151 (R.M.R) and NS051400 (R.M.R).
DISCLOSURE
L. Liu and L.C. Spangler report no disclosures. B. Prager received research support from Howard Hughes Medical Institute. B. Benson received research support from Biogen Idec and Case Western Reserve University. B. Hu and S. Shi report no disclosures. A. Love received research support from Messiah College. C. Zhang and M. Yu report no disclosures. A.C. Cotleur is employed by Biogen. R.M. Ransohoff received speaker honoraria from Biogen Idec and Novartis; received speaker honoraria and/or travel funding from Special Guest & Speaker Max-Planck Institute Retreat Ringberg Germany; Invited Plenary Speaker 1st Keystone Symposium on MS, Santa Fe, NM; Grand Rounds in Neurology, Johns Hopkins University, Baltimore; Keynote Lecture, Opening of Neuropathology Institute, Freiburg, Germany; Plenary Lecture, Kick-off meeting for Neurodegeneration Research Center, Bonn; Invited Seminar, Institute for Research in Biomedicine, Bellinzona, Switzerland; Invited Speaker, FDA/NYAS Conference: “Cytokine Therapies,” New York; Invited External Speaker “Annual Japanese MS Workshop” Kyushu, Japan; Visiting Professor, Dept. of Neurology, Kyushu University, Fukuoka, Japan; Consultant, Nature “Landscape” editorial meeting on Neuroimmunology; Invited Seminar, GSK Hammersmith Hospital, London; Invited Speaker 12th International Symposium “Signaling at the BBB,” London, England; Co-organizer/Speaker “Chemokines and Chemokine Receptors in the CNS,” Rome; Grand Rounds in Neurology, Ottawa General Hospital, Ontario, Canada; Invited Speaker, Keystone Symposium “Alzheimer's Disease,” Colorado; 0 NIH EUREKA Review IRGs; Invited Speaker, ASN annual meeting, Santa Fe, NM; Invited Speaker, Nikolas Histiocytosis-X Symposium, Athens, Greece; Invited speaker, FOCIS Symposium “Interventional Immunology,” Boston; Keynote Speaker, Neuroimmunology Symposium, Kyoto, Japan; Workshop Chair, International Congress of Immunology, Kobe, Japan; Plenary Lecture, PACTRIMS, Bali, Indonesia; Symposium Organizer and Chair ANA annual meeting, San Francisco, CA; Workshop Chair, Special Interest Groups, ANA, San Francisco, CA; Invited Speaker, Theodor Kocher Institute, Bern, Switzerland; Invited Speaker/Session Chair, Cell Press Symposium “Inflammation in Disease,” Lisbon; Invited Speaker, Brazilian Society of Pharmacology 42nd Annual Meeting, Brazil; External Reviewer, DFG “Centers of Excellence” Applications, Frankfurt, Germany; and Invited Speaker, AAAS/Abelson Symposium “Defeating AD,” Washington DC; is on the editorial board and was a former editor for Neurology: Neuroimmunology & Neuroinflammation; has consulted for Pfizer and Teva; is on the medical advisory board for Guthy-Jackson Charitable Foundation; received research support from Chemocentryz, Biogen Idec, National MS Society, NIH, Nancy Davice Center Without Walls, and Williams Family Foundation for MS Research; and owns stock or stock options in SAB and Chemocentryx. This research was conducted at the Cleveland Clinic's Lerner Research Institute before A.C. and R.M.R. moved to Biogen. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med 1998;338:278–285. [DOI] [PubMed] [Google Scholar]
- 2.Franklin RJ. Why does remyelination fail in multiple sclerosis? Nat Rev Neurosci 2002;3:705–714. [DOI] [PubMed] [Google Scholar]
- 3.Liu L, Belkadi A, Darnall L, et al. CXCR2-positive neutrophils are essential for cuprizone-induced demyelination: relevance to multiple sclerosis. Nat Neurosci 2010;13:319–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsai HH, Frost E, To V, et al. The chemokine receptor CXCR2 controls positioning of oligodendrocyte precursors in developing spinal cord by arresting their migration. Cell 2002;110:373–383. [DOI] [PubMed] [Google Scholar]
- 5.Liu L, Darnall L, Hu T, Choi K, Lane TE, Ransohoff RM. Myelin repair is accelerated by inactivating CXCR2 on nonhematopoietic cells. J Neurosci 2010;30:9074–9083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu L, Li M, Spangler LC, et al. Functional defect of peripheral neutrophils in mice with induced deletion of CXCR2. Genesis 2013;51:587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doerflinger NH, Macklin WB, Popko B. Inducible site-specific recombination in myelinating cells. Genesis 2003;35:63–72. [DOI] [PubMed] [Google Scholar]
- 8.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis 2007;45:593–605. [DOI] [PubMed] [Google Scholar]
- 9.Dutta R, Chomyk AM, Chang A, et al. Hippocampal demyelination and memory dysfunction are associated with increased levels of the neuronal microRNA miR-124 and reduced AMPA receptors. Ann Neurol 2013;73:637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.White DJ, Kazimiers T, Schindelin J. Coloc_2-Standardized, modular, colocalization analysis. Available at: http://fiji.sc/Coloc_2. Accessed July 16, 2013. [Google Scholar]
- 11.Butovsky O, Jedrychowski MP, Moore CS, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neuroscience 2014;17:131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Padovani-Claudio DA, Liu L, Ransohoff RM, Miller RH. Alterations in the oligodendrocyte lineage, myelin, and white matter in adult mice lacking the chemokine receptor CXCR2. Glia 2006;54:471–483. [DOI] [PubMed] [Google Scholar]
- 13.Carlson T, Kroenke M, Rao P, Lane TE, Segal B. The Th17-ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J Exp Med 2008;205:811–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hosking MP, Lane TE. ELR(+) chemokine signaling in host defense and disease in a viral model of central nervous system disease. Front Cell Neurosci 2014;8:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.