

Abstract
Herein we report that unsaturated N-sulfonamides undergo a Rh(III)-catalyzed allylic C(sp3)-H activation followed by insertion with an exogenous internal alkyne. The reaction generates [3.3.0], [4.3.0] and [5.3.0] azabicyclic structures with excellent diastereoselectivity. Deuterium-labeling experiments implicate a 1,3-Rh shift as a key step in the mechanism.
Keywords: Rhodium(III), azabicycles, C(sp3)-H insertion, Rh 1, 3-migration, 4π-electrocyclization
Graphical Abstract
Allylic C-H bond activation by Rh(III) catalyst precedes alkyne insertion leading to an unexpected azabicycle structure in good yield and excellent diastereoselectivity. The mechanism of the reaction was interrogated with deuterium labelling experiments and is proposed to involve a 1,3-Rh shift followed by an electrocyclization event.
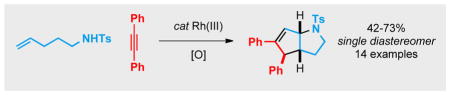
Transition metal-catalyzed C-H bond activation has emerged as a reliable method to access complex molecules in an atom- and step-economical fashion.[1] To this end, rhodium(III) has been extensively studied[2] and numerous aromatic[3] and vinylic[4] C(sp2)–H bond activations have been reported by our group and others. Activation of C(sp3)–H bonds with transition metal catalysts and their subsequent elaboration represents a highly desirable yet largely elusive goal, with few Rh(III)-catalyzed examples having been reported. In 2010, Glorius described the formation of pyrroles by allylic C(sp3) –H activation of enamines (Scheme 1, eq 1).[5] Wang discovered that a reactive benzylic C–H bond of 8-methylquinolines is susceptible to activation followed by alkenylation (Scheme 1, eq 2).[6a] We were particularly intrigued by a recent report by Cossy and coworkers who demonstrated that unsaturated sulfonamides afford vinylpyrrolidines by allylic C-H bond amination (Scheme 1, eq 3).[7] This latter reaction presumably proceeds via a π-allyl Rh intermediate and we hypothesized that we could intercept this intermediate to access different structures. Herein we report the coupling of unsaturated sulfonamides and alkynes via an allylic C(sp3)-H activation/electrocyclization sequence. The reaction affords stereochemically complex valuable azabicyclic products[8] with complete control of diastereoselectivity (Scheme 1, eq 4).
Scheme 1.

Rh(III)-Catalyzed C(sp3)-H Activation
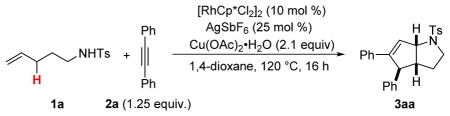
We first investigated the reactivity of unsaturated N-tosylsulfonamide 1a and diphenylacetylene 2a with [RhCp*Cl2]2 (5 mol %), AgSbF6 (25 mol %) and Cu(OAc)2•H2O (2.1 equiv) in 1,4-dioxane (c 0.1M, 120 °C, 16 h). We were pleased to observe formation of 1-azabicyclo[3.3.0]octane 3aa with complete control of the relative stereochemistry of the three contiguous stereocenters formed during the reaction (Table 1, entry 1).
Table 1.
Optimization of the conditions for the formation of 3aa
| ||
|---|---|---|
| Entry | Variation from the optimized conditions | Yield (%) |
| 1 | none | 70 |
| 2 | 5 mol % [RhCp*Cl2]2 | 26 |
| 3 | 5 mol % [IrCp*Cl2]2 | trace |
| 4 | 5 mol % [Ru(p-cymene)Cl2]2 | 0 |
| 5 | no [RhCp*Cl2]2 | 0 |
| 6 | no AgSbF6 | 0 |
| 7 | no Cu(OAc)2•H2O | 0 |
| 8 | 80 °C instead of 120 °C | 0 |
Single crystal X-ray analysis of this compound confirmed our structural assignment and revealed that the phenyl group is located on the exo face of the molecule.[9]
Although 3aa was obtained in low yield (26%), a higher catalyst loading drastically improves the yield to 70% (Table 1, entry 1). The most important conclusions resulting from our optimization studies are: (a) other potential catalysts for C–H activation such as Ir(III)[10] or Ru(II)[11] complexes did not give satisfactory conversions (Table 1, entries 3 and 4), (b) control experiments demonstrated that the presence of Rh(III), Ag(I) and Cu(II) species are all necessary for the reaction to take place (Table 1, entries 5–7) and (c) lowering the temperature to 80 °C completely shuts down the reactivity (Table 1, entry 8).[12]
With these optimized conditions in hand, we examined the tolerance of the reaction of various sulfonamide substrates with diphenylacetylene 2a. We were pleased to discover that N-tosylsulfonamide 1b could be converted into 3ba with good yield and complete diastereoselectivity (Table 2, entry 1). It is worth noting that the presence of the alkyne completely shuts down the potential C(sp3)-H amination pathway since no trace of pyrrolidine (Scheme 1, eq 3) is observed during this transformation.[7]
Table 2.
Scope of the Unsaturated Sulfonamidea
| Entry | Alkenyl sulfonamide | Product | Yield (%)b |
|---|---|---|---|
| 1 |
1b |
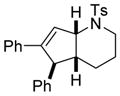 3ba |
72 |
| 2 |
1c |
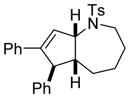 3ca |
66 |
| 3 |
1d |
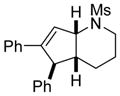 3da |
70 |
| 4 |
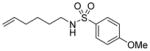 1e |
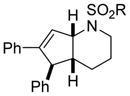 3ea (R = p-OMe-C6H4) |
70 |
| 5 |
1f |
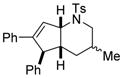 3fa/3′fa(dr = 1:1) |
42 |
Reactions were conducted with 1.0 equiv of 1 and 1.25 equiv of 2a, in the presence of 10 mol % [RhCp*Cl2]2, 25 mol % AgSbF6, 2.1 equiv Cu(OAc)2•H2O in dioxane at 120 °C.
Isolated yield.
Interestingly, larger bicyclic compounds can also be accessed; for example, the azabicycle 3ca bearing a seven-membered ring was obtained as a single diastereomer from sulfonamide 1c (Table 2, entry 2). Different electron-rich sulfonamides have successfully been involved in this transformation where methanesulfonamide 1d and p-methoxybenzenesulfonamide 1e lead to azabicyclic products 3da and 3ea, respectively (Table 2, entries 3 and 4). The very high diastereoselectivity observed during these transformations prompted us to investigate the reactivity of branched unsaturated N-tosylamide 1f. Upon treatment with the optimized conditions, the expected 1-azabicyclo[4.3.0]nonane was generated as a 1:1 mixture of diastereomers 3fa and 3′fa (Table 2, entry 5).
An array of diversely substituted alkynes were treated with N-tosylamides 1a or 1b under the optimized conditions (Table 3). Symmetrical tolanes 2b–f bearing an electron-donating alkyl or methoxy substituent in the para position are well tolerated and afford the corresponding azabicycles 3ab–3ae and 3bf as single diastereomers. Gratifyingly, heteroaryl moieties are also tolerated and azabicyclo[3.3.0]octane 3ag is obtained from reaction of alkyne 2g.
Table 3.
Scope of the Alkyne Substratea

|
See footnotes Table 2.
This transformation appears to be sensitive to the electronics of the alkyne as electron-withdrawing substituted tolane 2h only affords trace amounts of the corresponding bicycle 3bh. Steric hindrance also seems to play a crucial role since no reaction occurs with electron-rich symmetrical alkyne 2i bearing methoxy groups at the ortho positions. We were pleased to observe that the transformation proceeds smoothly with dienyne 2j to form azabicycle 3aj as a single diastereomer (Table 3).
We next investigated the regioselectivity of this transformation by examining the behavior of unsymmetrical alkynes. Tolane 2k was treated under the usual conditions with N-toluenesulfonamide 1b and the C(sp3)-H activation/cyclization sequence readily takes place. Although the expected bicyclic compound was synthesized in good yield, the electronic properties of the alkyne did not seem to influence the selectivity of the transformation as a mixture of 3bk and 3′bk was obtained in a 1:1 ratio (Scheme 2).
Scheme 2.

An Unsymmetrical Alkyne
In an effort to shed light on the mechanism of the reaction that generates these unexpected products, we conducted a series of deuterium labeling experiments. The use of dideuterated substrate [D2]-1a with the labels at the reactive allylic position furnished exclusively the monodeuterated product [D2]-3aa. (Scheme 3, eq 5). Of more interest is the reaction between 2a and tosylamides (E)-[D]-1a and (Z)-[D]-1a deuterated at the terminal position of the olefin (position 6). In both cases, analysis of the 1H NMR spectra of the resulting products [D]-3aa shows deuteration at position 6 and at position 8, providing evidence that a 1,3 shift could be involved during the transformation (Scheme 3, eqs 6 and 7).
Scheme 3.

Deuterium Labeling Experiments
Based on these observations, we propose a mechanism for this transformation (Scheme 4). First, the active Rh(III) catalyst A activates the allylic C(sp3)-H bond of 1a to provide a η3 π-allyl complex B in equilibrium with its haptotropic η1 isomer C. Complexation with alkyne 2a followed by migratory insertion affords the vinylrhodium(III) complex D which undergoes a direct vinyl-to-allyl 1,3-Rh migration to produce the bis(allyl)rhodium(III) species E.[13] A 4π-electrocyclization through intermediate F leads to π-allylrhodium(III) complex G. This diastereo-determining step proceeds via a conrotatory mechanism where the torquoselectivity is governed by steric factors highlighted in an empirical model (Figure 1). A counter-clockwise cyclization (red pathway) creates a destabilizing steric repulsion on the bottom face of the pentadienyl moiety between the rhodium catalyst and the bulky phenyl group while a more favorable clockwise cyclization (blue pathway) occurs with a steric repulsion between the rhodium and the less bulky alkyl chain. After N-metalation, the rhoda(III)azacyclohexane H is obtained and generates the azabicyclic compound 3aa by reductive elimination. This late N-cyclization step is in agreement with the absence of diastereocontrol with branched sulfonamide 1f. A final Cu mediated oxidation of the resulting Rh(I) complex I closes the catalytic cycle.
Scheme 4.

Proposed Mechanism
Figure 1.
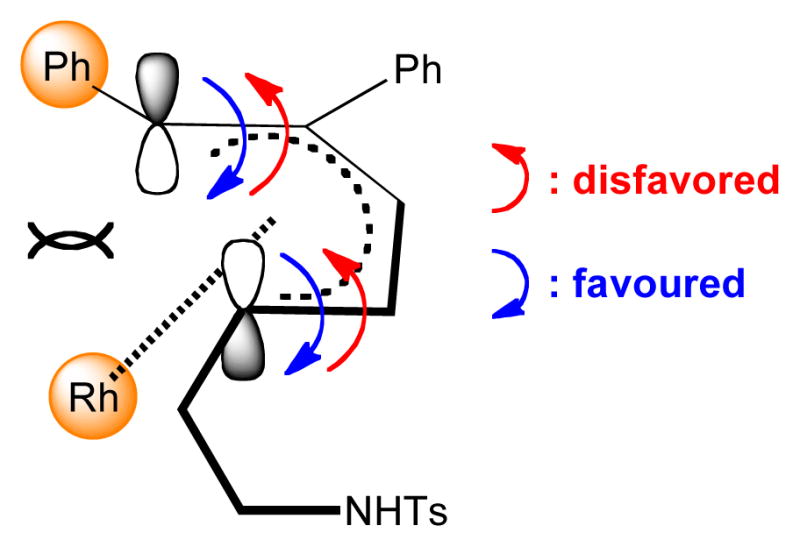
Model to rationalize diastereoselectivity
To conclude, we have discovered a new Rh(III)-catalyzed allylic C(sp3)-H activation/4π-electrocyclization sequence of unsaturated sulfonamides and alkynes which generates 1-azabicycles with complete control of the three newly formed stereocenters.[14] Deuterium labeling experiments reveal a rare direct vinyl-to-allyl 1,3-Rh migration. Studies to provide insight in this new reactivity and expand it to other useful synthetic applications are ongoing.
Experimental Section
In a 1.5 dram vial were added N-tosylamide 1a (47.9 mg, 0.200 mmol), diphenylacetylene 2a (44.6 mg, 0.250 mmol, 1.25 equiv), Cu(OAc)2•H2O (83.9 mg, 0.420 mmol, 2.1 equiv), AgSbF6 (17.2 mg, 0.050 mmol, 25 mol %) and [RhCp*Cl2]2 (12.3 mg, 0.020 mmol, 10 mol %). After addition of 1,4-dioxane (2 mL, 0.1M), the vial was sealed and heated at 120 °C for 16 h. The resulting blue mixture was filtrated through a short plug of silica and Celite (hexanes/EtOAc: 30:70) and concentrated under reduced pressure. Analysis of the crude material by 1H NMR and 13C NMR revealed the presence of a single diastereomer (d. r. > 96:4). Purification by flash chromatography (hexanes/EtOAc: 95:5 to 80:20) gave 58.3 mg (70%) of 3aa as a colorless oil.
Supplementary Material
Footnotes
We thank NIGMS for support (GM80442), and Johnson Matthey for a generous loan of rhodium salts. We thank Natthawat Semakul (CSU) for solving the structure of 3aa.
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Crabtree RH. J Chem Soc Dalton Trans. 2001:2437–2450. [Google Scholar]; b) Godula K, Sames D. Science. 2006;312:67–72. doi: 10.1126/science.1114731. [DOI] [PubMed] [Google Scholar]; c) Lyons TW, Sanford MS. Chem Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Ackermann L. Chem Rev. 2011;111:1315–1345. doi: 10.1021/cr100412j. [DOI] [PubMed] [Google Scholar]; e) Yeung CS, Dong VM. Chem Rev. 2011;111:1215–1292. doi: 10.1021/cr100280d. [DOI] [PubMed] [Google Scholar]; f) Delord-Wencel J, Dröge T, Liu F, Glorius F. Chem Soc Rev. 2011;40:4740–4761. doi: 10.1039/c1cs15083a. [DOI] [PubMed] [Google Scholar]; g) Arockiam PB, Bruneau C, Dixneuf PH. Chem Rev. 2012;112:5879–5918. doi: 10.1021/cr300153j. [DOI] [PubMed] [Google Scholar]; h) Kuhl N, Hopkinson MN, Wencel-Delord J, Glorius F. Angew Chem. 2012;124:10382–10401. doi: 10.1002/anie.201203269. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2012;51:10236–10254. doi: 10.1002/anie.201203269. [DOI] [PubMed] [Google Scholar]; i) Beller M, Bolm C, editors. Transitions Metals for Organic Chemistry. 2. Wiley-VCH; Weinheim: 2004. [Google Scholar]; j) Yu J-Q, Shi Z-J. C-H Activation. Springer; Berlin, Germany: 2010. [Google Scholar]; k) Chen X, Engle KM, Wang DH, Yu JQ. Angew Chem. 2009;121:5196–5215. [Google Scholar]; Angew Chem Int Ed. 2009;48:5094–5115. doi: 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Engle KM, Mei TS, Wasa M, Yu JQ. Acc Chem Res. 2012;45:788–802. doi: 10.1021/ar200185g. [DOI] [PMC free article] [PubMed] [Google Scholar]; m) Rouquet G, Chatani N. Angew Chem. 2013;125:11942–11959. doi: 10.1002/anie.201301451. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2013;52:11726–11743. doi: 10.1002/anie.201301451. [DOI] [PubMed] [Google Scholar]
- 2.For selected reviews, see: Colby DA, Bergman RG, Ellman JA. Chem Rev. 2010;110:624–655. doi: 10.1021/cr900005n.Satoh T, Miura M. Chem Eur J. 2010;16:11212–11222. doi: 10.1002/chem.201001363.Patureau FW, Wencel-Delord J, Glorius F. Aldrichim Acta. 2012;45:31–41.Song G, Wang F, Li X. Chem Soc Rev. 2012;41:3651–3678. doi: 10.1039/c2cs15281a.Colby DA, Tsai AS, Bergman RG, Ellman JA. Acc Chem Res. 2012;45:814–825. doi: 10.1021/ar200190g.Kuhl N, Shröder N, Glorius F. Adv Synth Catal. 2014;356:1443–1460.
- 3.For selected examples of Rh(III)-catalyzed aromatic C(sp2)-H activation, see: Ueura K, Satoh T, Miura M. Org Lett. 2007;9:1407–1409. doi: 10.1021/ol070406h.Stuart DR, Bertrand-Laperle M, Burgess KMN, Fagnou K. J Am Chem Soc. 2008;130:16474–16475. doi: 10.1021/ja806955s.Hyster TK, Rovis T. J Am Chem Soc. 2010;132:10565–10569. doi: 10.1021/ja103776u.Guimond N, Gouliaras C, Fagnou K. J Am Chem Soc. 2010;132:6908–6909. doi: 10.1021/ja102571b.Ryu J, Shin K, Park SH, Kim JY, Chang S. Angew Chem. 2012;124:10042–10046.Angew Chem Int Ed. 2012;51:9904–9908. doi: 10.1002/anie.201205723.Zhao D, Shi Z, Glorius F. Angew Chem. 2013;125:12652–12656.Angew Chem Int Ed. 2013;52:12426–12430.Liu G, Shen Y, Zhou Z, Lu X. Angew Chem. 2013;125:6149–6153.Angew Chem Int Ed. 2013;52:6033–6037. doi: 10.1002/anie.201300881.Hyster TK, Ruhl KE, Rovis T. J Am Chem Soc. 2013;135:5364–5367. doi: 10.1021/ja402274g.Hyster TK, Rovis T. Synlett. 2013:1842–1844. doi: 10.1055/s-0033-1339510.Hu F, Xia Y, Ye F, Liu Z, Ma C, Zhang Y, Wang J. Angew Chem. 2014;126:1388–1391. doi: 10.1002/anie.201309650.Angew Chem, Int Ed. 2014;53:1364–1367. doi: 10.1002/anie.201309650.
- 4.For selected examples of Rh(III)-catalyzed vinylic C(sp2)-H activation, see: Mochida S, Hirano K, Satoh T, Miura M. J Org Chem. 2009;74:6295–6298. doi: 10.1021/jo901077r.Hyster TK, Rovis T. Chem Commun. 2011;47:11846–11848. doi: 10.1039/c1cc15248c.Kuhl N, Shröder N, Glorius F. Org Lett. 2013;15:3860–3863. doi: 10.1021/ol4015915.Wang H, Beiring B, Yu DG, Collins KD, Glorius F. Angew Chem. 2013;125:12657–12661. doi: 10.1002/anie.201306754.Angew Chem Int Ed. 2013;52:12430–12434. doi: 10.1002/anie.201306754.Neely JM, Rovis T. J Am Chem Soc. 2014;136:2735–2738. doi: 10.1021/ja412444d.Piou T, Rovis T. J Am Chem Soc. 2014;136:11292–11295. doi: 10.1021/ja506579t.
- 5.Rakshit S, Patureau FW, Glorius F. J Am Chem Soc. 2010;132:9585–9587. doi: 10.1021/ja104305s. [DOI] [PubMed] [Google Scholar]
- 6.Liu B, Zhou T, Li B, Xu S, Song H, Wang B. Angew Chem. 2014;126:4275–4279.Angew Chem Int Ed. 2014;53:4191–4195. doi: 10.1002/anie.201310711.For a Rh(III)-catalyzed amidation of 8-methylquinolines, see: Wang N, Li R, Li L, Xu S, Song H, Wang B. J Org Chem. 2014;79:5379–5385. doi: 10.1021/jo5008515.
- 7.Cochet T, Bellosta V, Roche D, Ortholand J-Y, Greiner A, Cossy J. Chem Commun. 2012;48:10745–10747. doi: 10.1039/c2cc36067e. [DOI] [PubMed] [Google Scholar]
- 8.For examples of biologically active molecules bearing 1-azabicycle moieties, see: Linz W, Schindler U. 5,061,722. US Patent No. 1991Hermans H, Hubert KF, Knaeps GA, Willems JJM. 3,679,686. US Patent No. 1972Takano I, Yasuda I, Nishijima M, Hitotsuyanagi Y, Takeya K, Itokawa H. Bioorg Med Chem Lett. 1996;6:1689–1690.
- 9.See SI.
- 10.For selected examples of C-H activation catalyzed by [IrCp*Cl2]2, see: Kim H, Shin K, Chang S. J Am Chem Soc. 2014;136:5904–5907. doi: 10.1021/ja502270y.Kim J, Chang S. Angew Chem. 2014;126:2235–2239.Angew Chem Int Ed. 2014;53:2203–2207. doi: 10.1002/anie.201310544.Xie F, Qi Z, Yu S, Li X. J Am Chem Soc. 2014;136:4780–4787. doi: 10.1021/ja501910e.Kang T, Kim Y, Lee D, Wang Z, Chang S. J Am Chem Soc. 2014;136:4141–4144. doi: 10.1021/ja501014b.
- 11.For selected examples of C-H activation catalyzed by [Ru(p-cymene)Cl2]2, see: Kim J, Kim J, Chang S. Chem Eur J. 2013;19:7328–7333. doi: 10.1002/chem.201301025.Bhanuchandra M, Yadav MR, Rit RK, Kuram MR, Sahoo AK. Chem Commun. 2013;49:5225–5227. doi: 10.1039/c3cc41915k.
- 12.The use of a higher ratio of silver salt to Rh dimer (4:1) leads to lower isolated yields (18%). Furthermore, the use of cationic Rh (MeCN complex) as a precatalyst leads to very low conversions under these conditions. Whether this implicates a monocationic Rh intermediate as the active catalyst remains unclear.
- 13.Ikeda Y, Takano K, Kodama S, Ishii Y. Organometallics. 2014;33:3998–4004. [Google Scholar]
-
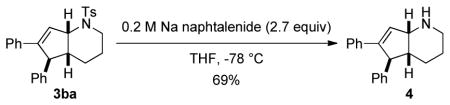
14.The protecting tosyl group of compound 3ba was removed by treatment with a solution of sodium naphtalenide in THF and the bicyclic secondary amine 4 was obtained in good yield. See SI for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.