

Abstract
Objective:
To investigate the clinical spectrum and distinguishing features of adenylate cyclase 5 (ADCY5)–related dyskinesia and genotype–phenotype relationship.
Methods:
We analyzed ADCY5 in patients with choreiform or dystonic movements by exome or targeted sequencing. Suspected mosaicism was confirmed by allele-specific amplification. We evaluated clinical features in our 50 new and previously reported cases.
Results:
We identified 3 new families and 12 new sporadic cases with ADCY5 mutations. These mutations cause a mixed hyperkinetic disorder that includes dystonia, chorea, and myoclonus, often with facial involvement. The movements are sometimes painful and show episodic worsening on a fluctuating background. Many patients have axial hypotonia. In 2 unrelated families, a p.A726T mutation in the first cytoplasmic domain (C1) causes a relatively mild disorder of prominent facial and hand dystonia and chorea. Mutations p.R418W or p.R418Q in C1, de novo in 13 individuals and inherited in 1, produce a moderate to severe disorder with axial hypotonia, limb hypertonia, paroxysmal nocturnal or diurnal dyskinesia, chorea, myoclonus, and intermittent facial dyskinesia. Somatic mosaicism is usually associated with a less severe phenotype. In one family, a p.M1029K mutation in the C2 domain causes severe dystonia, hypotonia, and chorea. The progenitor, whose childhood-onset episodic movement disorder almost disappeared in adulthood, was mosaic for the mutation.
Conclusions:
ADCY5-related dyskinesia is a childhood-onset disorder with a wide range of hyperkinetic abnormal movements. Genotype-specific correlations and mosaicism play important roles in the phenotypic variability. Recurrent mutations suggest particular functional importance of residues 418 and 726 in disease pathogenesis.
We previously reported that a p.A726T mutation in adenylate cyclase 5 (ADCY5) in one family caused autosomal dominant familial dyskinesia with facial myokymia (FDFM; OMIM 600293) because of the distinctive features of perioral and periorbital twitches, choreiform and dystonic movements of the neck and arms, and increased reflexes.1 Subsequently, we identified a different ADCY5 missense mutation, p.R418W, in 2 sporadic cases with axial hypotonia, chorea, myoclonus, dystonia, and occasional facial movements, but not obvious myokymia.2 Adenylate cyclases convert adenosine-5ʹ-triphosphate (ATP) to 3′,5′-cyclic adenosine monophosphate (cAMP), a second messenger in signal transduction.3 Specificity and modulation of ADCY activity is related to tissue expression patterns and stimulators and inhibitors for each member of the gene family. ADCY5 is highly expressed in striatum, a region involved in modulating movement, and ADCY5 knock-out mice display a parkinsonian phenotype.4 Overexpression of either mutated protein in HEK293 cells increased cAMP accumulation,2 suggesting a gain-of-function effect.
The variability in adventitious movements, periodicity, and severity in 3 families prompted us to evaluate ADCY5 in other cases with similar features. To clarify the clinical spectrum and define possible genotype–phenotype relationships, we identified multiple new familial and sporadic cases and show that ADCY5-related dyskinesia is more common and constitutes a broader category than previously thought.
METHODS
Standard protocol approvals, registrations, and patient consents.
Blood samples and skin biopsy were obtained under approved Institutional Review Board protocols.
Patients and samples.
Patients with ADCY5 mutations were identified at multiple sites utilizing different strategies. Some patients had clinical exome sequencing and others had targeted ADCY5 sequencing based on characteristic phenotypes. Videos and medical records were reviewed and patients were evaluated in Neurology or Genetics Clinics at the University of Washington, Seattle (ID027, Ch4, FDFM, essential hereditary chorea [EHC] family, and chorea-dystonia [ChDys] family); Rady Children's Hospital, University of California San Diego (ID1 and ID032); Johns Hopkins All Children's Hospital, St. Petersburg, Florida (ID016); Lucile Packard Children's Hospital, Stanford University, Palo Alto, California (ID028); Phoenix Children's Hospital, Arizona (ID029); Hôpital de la Pitié Salpêtrière, Paris (ID018, ID019, ID019B, ID035); Hôpital Robert Debré, Paris (ID020); Hôpital Trousseau, Paris (ID021, ID024); and Hôpital Civil de Strasbourg, France (ID023, ID033).
Mutation detection and analysis.
For the EHC family, all 21 ADCY5 exons were Sanger sequenced (table e-1 on the Neurology® Web site at Neurology.org). Exome sequences for the ChDys family were obtained as previously described1 using SeqCap EZ Human Exome Library v3.0 (Roche NimbleGen; Madison, WI) capture and an Illumina (San Diego, CA) HiSeq 1000 system with paired-end 100-base reads. Clinical exome sequencing was performed at Baylor College of Medicine (Houston, TX) (ID016 and ID029), GeneDx (Gaithersburg, MD) (ID028), and ICM (Paris, France) (ID033 and ID035). Mutations in ID032 and case Ch4 were detected by molecular inversion probe (MIP) capture5 (table e-1) and sequencing on an Illumina MiSeq system. For the remaining sporadic cases, we sequenced exons 2, 10, and 18.
To investigate shared ancestry in FDFM and EHC, we determined haplotypes surrounding ADCY5. We genotyped short tandem repeat markers (STRPs) as described6 and sequenced portions of the introns flanking exons 5, 6, and 14, containing informative single nucleotide polymorphisms (SNPs) (figure 1A).
Figure 1. Organization of ADCY5 and locations of mutations.

(A) Schematic of ADCY5 exons shows the number of families with each mutation (in parentheses) and the positions of some intragenic short tandem repeat markers (STRPs) and single nucleotide polymorphisms included in the haplotype analysis of the EHC and FDFM families. Markers in green rectangles define distinct haplotypes not shared by the 2 families. Genotypes of flanking STRPs D31271, D3S1278, D3S3606, D3S1292, and D3S1569 were also used (not shown). Mutations seen in multiple affected persons are in red and those identified only in a single individual are in green. In Chen et al.,2 p.R418 was placed near the end of the sixth helix of the first transmembrane domain; the current consensus is that this residue is in the C1a domain. (B) ADCY5 protein contains 2 membrane-spanning and 2 major bipartite cytoplasmic domains. Binding of dopamine to the excitatory dopamine 1 receptor (D1R) activates the striatal stimulatory heterotrimeric G protein GαOLF that in turn activates ADCY5. On activation, the C1a and C2a regions of ADCY5 form a catalytic pocket in which conversion of adenosine-5ʹ-triphosphate (ATP) binding to 3′,5′-cyclic adenosine monophosphate (cAMP) occurs. EHC = essential hereditary chorea; FDFM = familial dyskinesia with facial myokymia. Modified from Chen et al.2 with permission (copyright © 2014 American Neurological Association).
To investigate mosaicism for ADCY5 mutations, we performed allele-specific PCR amplification with primers containing either the reference sequence or the mutant nucleotide in the 3′-position and a mismatched nucleotide in the preceding position. A common reverse primer was used for both alleles. PCR products were visualized and sequenced.
RESULTS
Selected case descriptions.
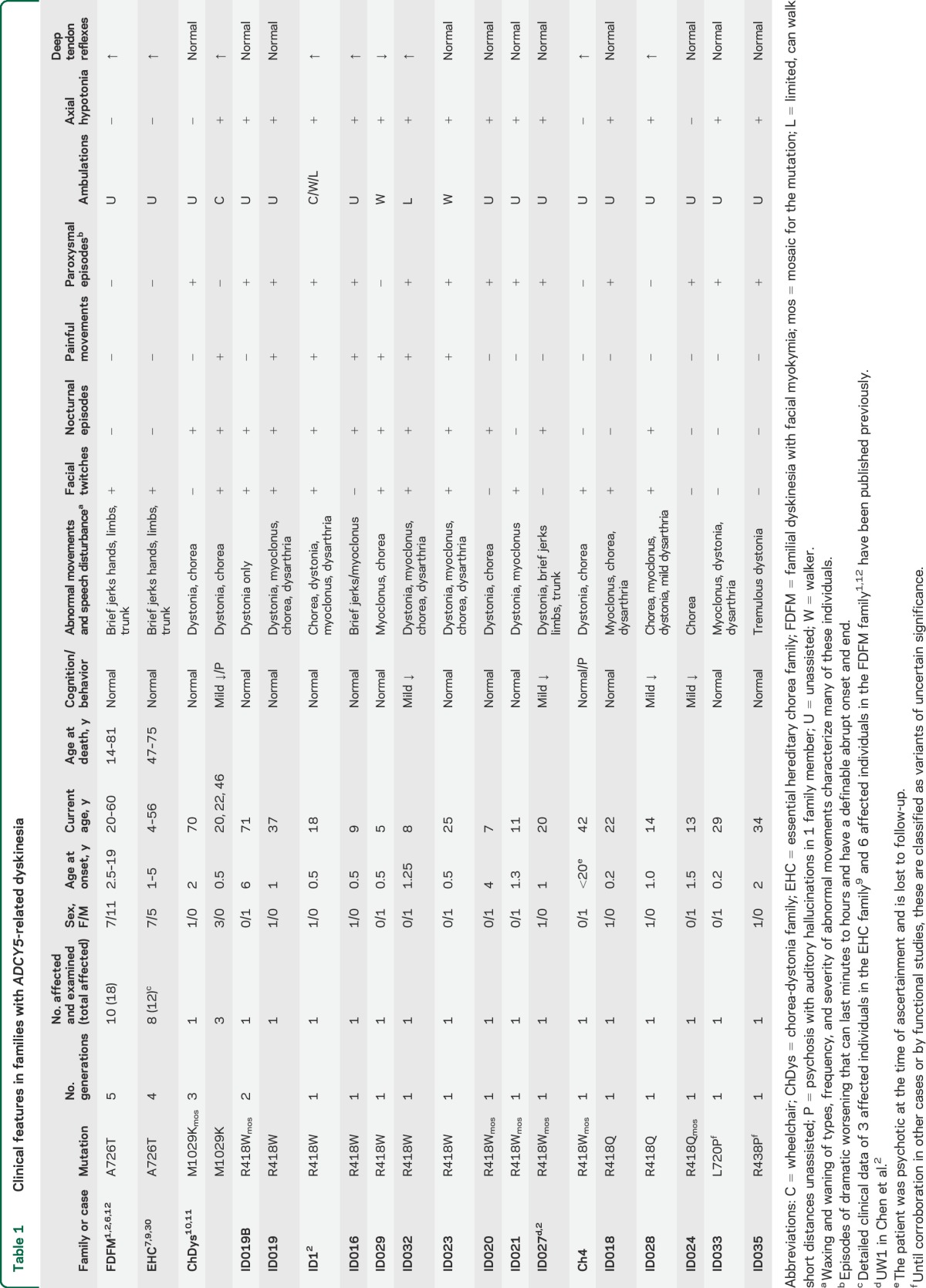
All cases are summarized in table 1.
Table 1.
Clinical features in families with ADCY5-related dyskinesia
Simplex cases.
ID016 (p.R418W).
Extreme irritability and exaggerated Moro reflex were noted at 1 week and continued throughout the first year. Hypotonia was apparent by 6 months and motor milestones were significantly delayed. The patient sat at 18 months and walked at 3 years. At 1 year, sudden severe limb movements during sleep developed and subsequently increased in frequency and severity. Similar daytime movements, occasionally painful, sometimes were severe enough to cause falls from her stroller. Minor stresses such as viral infections provoked multiweek episodes. Examination at age 8 years showed normal cognition, marked dysarthria, and moderately severe axial hypotonia especially of the neck. Gait was slow, uncoordinated, and jerky, with a tendency to veer to the side and walk in a circle. She fell frequently. At rest, there were almost continuous sudden myoclonic-like jerks of limbs or trunk. Tendon reflexes were brisk, with normal or occasionally upgoing plantar reflexes.
Several brain MRIs had normal results. Video EEG showed bifrontal irregular bursts of 3–4 Hz delta activity sometimes associated with spike and polyspike activity that did not clearly correlate with muscle jerks and dystonic movements, but one episode of rhythmic head bobbing corresponding with EEG evidence of a seizure was recorded. Comprehensive metabolic evaluation showed only borderline pyruvate and complexes I and III OXPHOS enzyme activities. Despite normal POLG1 sequence (Medical Neurogenetics, Atlanta, GA) and absence of mitochondrial DNA mutations, a diagnosis of possible myoclonic epilepsy related to mitochondrial OXPHOS disorder was given. Numerous medications were trialed, including CoQ10, carnitine, amantadine, carbamazepine and levodopa-carbidopa. Current medications are levetiracetam, levocarnitine, clonazepam, and melatonin, with moderate control of abnormal movements.
ID027 (p.R418W mosaic).
A partial description of this case (denoted UW1) is published.2 Early motor milestones were delayed. Involuntary trunk and limb movements developed at age 5 years. Bouts of movements lasted days or weeks, followed by long quiescent periods. For example, on examination at age 19 years, there were frequent spontaneous dystonic arm, leg, and trunk movements, sustained dystonic posturing of the trunk and legs while walking, and moderately severe dysarthria (video 1). Video EEG documented the movements but showed normal EEG activity. Two months later, gait and speech were normal, there were no adventitious movements, and the examination elicited only occasional slight hesitation in rapid alternating movements and finger-to-nose testing. Tendon reflexes and brain MRI were normal. The patient is one of 5 cases in this study with cognitive deficits. During early childhood, her IQ was estimated at 60, and at 18 she scored 19/30 on the Montreal Cognitive Assessment.
Familial cases.
The EHC family (p.A726T).
This family (figure 2A) was first reported in 1976 as essential benign chorea.7 Clinical screening of the BCH1 gene TITF/NKX2-18 was negative. Manifestations are consistent and detailed descriptions of some affected individuals in the EHC family, including IV-5, have been published.7,9 Episodic jerky movements of the face, upper extremities, and feet began between ages 1 and 5 years. Tendon reflexes were hyperactive with flexor plantar responses, but sometimes normalized in adulthood. Muscle tone was normal. Movements worsened with stress, were socially embarrassing, and were not considered benign by the family. Manifestations were nonprogressive and in at least one individual, III-4, diminished substantially with age. Chlordiazepoxide, phenobarbital, or clonazepam had no benefit. Recent EMG in individual IV-5 showed neither facial myokymia nor peripheral neuropathy, but random generation of motor unit action potentials corresponding to the facial muscle twitches. Video 2 shows EHC IV-5 at 44 and 57 years.
Figure 2. Pedigrees of 2 families with mutations in ADCY5.

Asterisks denote individuals who provided DNA samples and all of these who were affected were examined. The family-specific mutation segregated completely with affectation status; no nonmanifesting carriers were identified. Age at death (d) or current age (c) is shown. (A) Family EHC with autosomal dominant essential chorea and p.A726T ADCY5 mutation, updated from the diagram published in 1976.7 Medical records of individuals II-2 and III-3 had been reviewed and individual IV-3 had been examined previously.7 Individual VI-8 was examined but not sampled. Clinical descriptions of III-4, IV-5, and V-5 were detailed previously (denoted II-3, III-5, and IV-2).9 (B) Family ChDys, first described in 1967, with severe chorea and dystonia and p.M1029K ADCY5 mutation. In individual II-2, the mutation was detectable only by allele-specific amplification. EHC = essential hereditary chorea; ChDys = chorea-dystonia.
The ChDys family (p.M1029K).
Individual II-2 (figure 2B) was first described at age 19 years in 1967 as sporadic paroxysmal choreoathetosis without EEG abnormality.10 Choreiform movements with dystonic trunk posturing began at 6 months, occurred as frequently as 20 times per day, and could last for hours and be so severe that the patient was unable to stand. Anxiety or sudden activity but not passive motion precipitated episodes. However, she also had asymptomatic periods of several days. Chlordiazepoxide decreased attack frequency, but diphenylhydantoin, phenobarbital, reserpine, ethosuximide, imipramine, and haloperidol were unhelpful and diphenhydramine worsened the movements. In the 3rd decade, gradual improvement in frequency and severity of episodes occurred.11 Neurologic examination at age 30 years had normal results except for mild jerks of trunk, hands, and face. By age 56, the patient had only subtle truncal jerks, and by age 68, her neurologic examination was essentially normal.
The proband's daughter, III-6, had hypotonia without pathologic reflexes. Motor milestones were delayed. She crawled at 2 years and walked at 2.5 years. By age 4 years, she manifested dysarthria and marked chorea of all extremities, face, and tongue.11 Movements were continuous while awake and absent during sleep. In contrast to her mother, disease progressed, and by age 8 years, she had severe movements, poor head control, and contractures of knees and ankles, and required a wheelchair. Tendon reflexes were brisk with ankle clonus. In her 20s, triggers such as stress, hunger, and heat precipitated painful episodes of arm flexion and internal rotation and hip flexion, with limited responsiveness for up to 30 minutes despite remaining alert. Unpredictable episodes of severe jerks and spasms continued, as did frequent dystonic tongue thrusting and severe dysarthria. In her 40s, she was hospitalized with a psychotic depression, and she has had several recurrences of psychosis with delusions and auditory hallucinations. She graduated from high school in special education, but at age 50 years had a Mini-Mental State Examination score of 16/26. Brain MRI was normal.
The granddaughters (IV-4 and IV-5) are now 22 and 20 years old. They were hypotonic from infancy, with delayed motor milestones. IV-4 never walked. IV-5 at age 8 had bilateral ankle-foot orthotics and used a wheelchair at school. At 8 and 9 years, respectively, they had marked dystonia and jerky movements of the trunk and all limbs. Muscle tone was decreased but episodically increased with movements. Upper extremity tendon reflexes were normal but there were unsustained ankle clonus and variable Babinski responses. In IV-4, brain MRI at age 1 year was normal, as were muscle and skin biopsies at age 3. Their intellectual abilities were considered normal and they graduated from high school in special education primarily because of their movement disorder. Video 3 shows patient IV-4 at 8 and 20 years.
FDFM (p.A726T).
Clinical descriptions of this family, with essentially the same manifestations as EHC, were published, with detailed descriptions of 7 affected people.6,12 In 2014, repeat hand and face EMG of IV-46 showed frequent involuntary bursts of motor units provoked by needle insertion and volition, similar to those in EHC IV-5. These bursts were not typical of myokymic discharges because they were often stimulus provoked, highly variable in duration, and not rhythmic. Most EEGs in this family have been normal but spike and polyspike activities were recorded in 2 individuals, one treated with mysoline.12 Various anxiolytics and antiepileptics have produced no improvement (e.g., chlordiazepoxide, primidone, amitriptyline, trifluoperazine, diphenylhydantoin). Acetazolamide and propranolol have been of modest anecdotal benefit in a few persons.
Detection of ADCY5 mutations and investigation of mosaicism.
In EHC, Sanger sequencing revealed the c.2176G>A, p.A726T mutation previously found in FDFM. To investigate the possibility of a shared ancestor in FDFM, of German ancestry, and EHC, of English ancestry, we genotyped one intragenic and 5 flanking STRPs and 5 intronic SNPs. Distinct haplotypes span a 28-kb region containing ADCY5, demonstrating that the mutations arose independently (figure 1A).
In the ChDys family, we sequenced 6 exomes (figure 2B) and prioritized rare nonsynonymous variants carried by all 4 affected individuals. ADCY5 was absent from this list of genes. However, direct review revealed a missense mutation (c.3086T>A, p.M1029K) in 3 affected individuals, but not II-2. Coverage of this position was poor (range 5–9 reads) and marked by the GATK strand-bias filter. The mutation affects a highly conserved residue (PhastCons13 1.0 and GERP14 4.53), has a nonconservative Grantham score15 of 95, is not present in the Exome Variant Server or the 1000 Genomes database, and is classified as possibly damaging by PolyPhen216,17 (0.576) and deleterious by SIFT18 (0.02). Given the clinical history of II-2, we considered the possibility of somatic mosaicism. Sanger sequencing of exon 18 in DNA from uncultured peripheral blood and skin again detected only the wild-type allele. Allele-specific amplification followed by sequencing revealed the mutant allele in blood, skin, and cultured fibroblasts, confirming that she is mosaic (figure 3A).
Figure 3. Allele-specific amplification documents mosaicism of ADCY5 mutations in representative cases.

Wild-type (wt) alleles (top panels) were amplified using wt-primer and mutant alleles (lower panels) amplified using primers containing the mutant nucleotide in the 3′ position and one mismatched nucleotide in the preceding position. (A) Sequence chromatograms for 2 members of the ChDys family. The mutant nucleotide A is detected in patient III-6 but not in her affected mother II-2. Allele-specific PCR in II-2 amplified the mutant allele from blood, skin, and fibroblast cells. The analysis is not quantitative, but the very faint mutant band in fibroblasts suggests a selective advantage for the wt allele during culture. (B) On the sequence chromatograms, in contrast to C and T peaks of equal intensity in ID016, much smaller signals are present for the mutant T allele in ID020, ID021, and ID027. Presence of the mutant allele was confirmed for all by allele-specific amplification. (C) Mosaicism for the mutant A nucleotide is shown for ID024, in contrast to the peaks of equal height for the wt and mutant alleles in ID018. In all panels, for the mosaics, the faint gel signals are consistent with the small mutant allele peaks in the chromatograms. Absence of a mutant allele in a normal control (NC) demonstrates the specificity of the allele-specific amplification. ChDys = chorea-dystonia.
We previously reported a de novo ADCY5 missense mutation, c.1252C>T, p.R418W, in ID1 and ID027.2 Whereas the exomes of ID1, ID016, and ID029 contained the variant allele in approximately half the reads, strand bias (7 variants of 32 reads) in ID027 raised the possibility that the patient might be mosaic for the mutation. In the clinical laboratory, Sanger sequencing using 2 sets of primers ruled out uneven allele amplification due to primer placement. Sequence electropherograms of exon 2 documented the lower peak height of the variant T compared to that of wild-type C, whereas these were equal in ID016, who carries the same mutation (figure 3B). The mutation was not carried by the parents of ID016 or ID029. Targeted sequencing of the 3 exons that contained the known mutations showed that ID019, ID020, ID021, and ID023 also carry p.R418W. The very low signals for the variant nucleotide in ID020, ID021, and the more mildly affected father of ID019 were confirmed by allele-specific amplification to reflect somatic mosaicism (figure 3B). MIP capture and sequencing identified p.R418W in ID032 and reported 1 variant read of 18 in Ch4. A very low level mosaicism was confirmed in Ch4 by allele-specific amplification only with overexposure of the blot. His MRI showed decreased signal intensity in globus pallidus.
Three individuals carried a different de novo mutation in the same residue, p.R418Q. Exome sequencing of ID028 obtained 8 variant and 18 reference reads; however, by Sanger sequencing, the heights of the mutant and reference nucleotide peaks were equal at the clinical laboratory and in our laboratory. Targeted sequencing identified the mutation in ID018 and ID024. Allele-specific amplification demonstrated mosaicism in ID024 (figure 3C). By clinical exome sequencing in patients with dystonia, we identified 2 novel de novo ADCY5 variants, p.L720P (ID033) and p.R438P (ID035), predicted to be pathogenic by Sift and PolyPhen and not found in the ExAC database. Figure 1B shows locations of mutations and variants in protein domains.
DISCUSSION
We show that ADCY5 mutations in the cytoplasmic domains cause a childhood-onset mixed hyperkinetic movement disorder with no or slow progression. Prior to molecular characterization, the varied character of the movement semiology, which may include chorea, dystonia, or myoclonus, sometimes affecting the face, led to diagnoses such as benign hereditary chorea, paroxysmal dyskinesia, cerebral palsy, or mitochondrial disease. Clues to the diagnosis are as follows: (1) axial hypotonia, sometimes accompanied by weakness; (2) facial chorea or dystonia; (3) nocturnal paroxysmal dyskinesia, often pronounced; (4) movement-related pain; (5) dramatic fluctuations in frequency and severity of movements; (6) no or mild cognitive impairment; (7) normal MRI; and (8) little or no progression. Some patients have paroxysms that can last for minutes to weeks, with or without provocation by movement. Other triggering factors have been prolonged periods of inactivity, fatigue, intercurrent illness, or other stressor. The waxing-waning pattern makes it difficult to attribute exacerbations or remissions to environmental factors or medications. However, clonazepam markedly reduced the nighttime sleep-disrupting myoclonic episodes in several patients and levetiracetam reduced movements in a few.
Although there is variability, a pattern of phenotypes associated with mutations in different domains emerged. C1b domain mutation A726T is associated with the mildest disease. The myokymia thought to characterize FDFM could not be confirmed on review of the original EMG tracings, nor was it observed on repeat study or in an individual in the EHC family with similar movements. These facial movements are likely chorea or dystonia. In the ChDys family with a p.M1029K mutation in C2a very near the catalytic domain, the 3 nonmosaic mutation carriers have had severe generalized unremitting dystonia and chorea, without myoclonic movements. Mutations in C1a and C2a cause moderate to severe disorders. It is of note that 13 of the 14 C1a mutations are de novo and 6 of these are mosaic. Mosaicism often results in milder disease that may remit for extended periods. Among the 14 cases with residue 418 mutations, 78% had onset in infancy, 71% have nocturnal movements, 79% have paroxysmal worsening, 86% have axial hypotonia, 43% have painful dystonia, 71% have facial movements, and 36% have increased reflexes.
Axial hypotonia is a common finding in patients with all the mutations, except those in C1b. The hypotonia is presumably central in origin, a process that has been reported to affect the axial muscles as well as the limbs, although strength is usually preserved.19 Study of more families will be needed to determine if the seizures documented in 3 individuals, the mild cognitive impairment in 5, the psychosis in 2, or the cardiomyopathy in 11 are part of the disease spectrum.
The range of disease severity related to mutation location may reflect the function of ADCY5 domains. On activation, C1a and C2a form the catalytic ATP–binding pocket. The C2b domain may affect this activity less directly. The mutational hot spot suggests arginine 418 has an important functional role.
Altered striatal dopamine signaling is one hypothesis for various forms of dystonia, including defects in the biosynthesis or transport of dopamine,20 DYT1 Torsin A-related dystonia,21,22 DYT25 GNAL-related dystonia,23,24 and adult-onset focal dystonia.25 ADCY5 is the principal adenylate cyclase integrating signals from multiple receptors including D1 and D2 striatal dopamine receptors.26 Based on increased cAMP accumulation in our functional studies of 2 ADCY5 mutations2 and the decrease in spontaneous movement in ADCY5-null mice,4 we suspected that increased adenylate cyclase activity might be a pathophysiologic factor in ADCY5-related dyskinesia. Recently, however, a splice site mutation in ADCY5 was identified in a father and son with chorea and dystonia.27 It is difficult to reconcile the similar manifestations caused by this loss-of-function mutation and the possible gain-of-function missense mutations in our patients. The missense mutations may have a different effect in vivo than what we observed in vitro. Functional studies in neuronal cells may shed light on this question.
To encompass the broader phenotype, we suggest changing the name from FDFM to ADCY5-related dyskinesia.28 ADCY5-related dyskinesia should be considered in patients without a diagnosis who manifest any of the cardinal features, whether episodic or constant, and with or without a positive family history. With the caveat that pathogenicity of the 2 novel variants requires corroboration, with 18 families described herein and 3 other published families,27,29 it appears this disorder is less rare than initially thought. As additional families are found, the full spectrum of mutations and their relationship to phenotype will be elucidated. Increased understanding of the pathogenesis of ADCY5-related dyskinesia will facilitate development of pharmacologic treatments for all forms of dystonia that involve the same or related pathways.
WEB-BASED RESOURCES
Exome Variant Server (EVS): evs.gs.washington.edu/
Exome Aggregation Consortium (ExAC): exac.broadinstitute.org/
PolyPhen-2 (Polymorphism Phenotyping v2): http://genetics.bwh.harvard.edu/pph2/
Sorting Intolerant From Tolerant: http://sift.jcvi.org/www/SIFT_intersect_coding_submit.html
Supplementary Material
ACKNOWLEDGMENT
The authors thank the families for their active participation in this research; John Wolff and Mark Matsushita, Division of Medical Genetics, University of Washington, for technical support; Colin Pritchard, MD, PhD, Department of Laboratory Medicine, University of Washington, for advice on investigation of mosaicism; and Shu Ching Hu, MD, PhD, Department of Neurology, University of Washington, for assessment of patient videos.
GLOSSARY
- ADCY5
adenylate cyclase 5
- ATP
adenosine-5′-triphosphate
- cAMP
3′,5′-cyclic adenosine monophosphate
- ChDys
chorea-dystonia
- EHC
essential hereditary chorea
- FDFM
familial dyskinesia with facial myokymia
- MIP
molecular inversion probe
- SNP
single nucleotide polymorphism
- STRP
short tandem repeat marker
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Laura Amendola: acquisition of data, interpretation of data. Mathieu Anheim: acquisition of data, interpretation of data. Saunder Bernes: acquisition of data, interpretation of data. Thomas D. Bird: drafting/revising manuscript, study concept, analysis of data, obtaining funding, study coordination. Emily Bonkowski: acquisition of data, analysis of data, drafting/revising manuscript. Dong-Hui Chen: drafting/revising manuscript, acquisition, analysis and interpretation of data. Nathalie Damon-Perrière: acquisition of data, interpretation of data. Marie Y. Davis: acquisition of data, interpretation of data. Bertrand Degos: acquisition of data, interpretation of data, drafting/revising manuscript. Michael O. Dorschner: drafting/revising manuscript, acquisition of data, interpretation of data. Diane Doummar: acquisition of data, interpretation of data. Evan E. Eichler: acquisition of data, obtaining funding. Jennifer Friedman: drafting/revising manuscript, acquisition of data, study coordination. Alona Gad: analysis and interpretation of data. David Grabli: acquisition of data, interpretation of data, drafting/revising manuscript. Domitille Gras: acquisition of data, interpretation of data. Fuki Hisama: drafting/revising manuscript, acquisition of data. Olena Korvatska: drafting/revising manuscript, acquisition of data, interpretation of data. Katherine M. Mackenzie: acquisition of data, interpretation of data. Aurélie Méneret: drafting/revising manuscript, acquisition of data, analysis of data. Cyril Mignot: acquisition of data, interpretation of data. Wendy H. Raskind: drafting/revising manuscript, study concept, analysis of data, obtaining funding, study coordination. Emmanuel Roze: drafting/revising manuscript, acquisition of data, study coordination. Holly Stessman: acquisition of data, interpretation of data, drafting/revising manuscript. Philip D. Swanson: acquisition of data, interpretation of data. Ali Torkamani: acquisition of data, interpretation of data. Christine Tranchant: acquisition of data, interpretation of data. Oriane Trouillard: acquisition of data, interpretation of data. Marie Vidailhet: acquisition of data, interpretation of data, drafting/revising manuscript. Michael Weiss: acquisition of data, interpretation of data. Steven Winesett: acquisition of data, interpretation of data.
STUDY FUNDING
Supported in part by funds from the NIH (R01 NS069719) to W.H.R., R01 MH101221 to E.E.E., U01 HG006476 to A.T., Clinical and Translational Science Award (CTSA; UL1 TR001114-03) to A.T., Journées de Neurologie en Langue Française to A.M., INSERM (COSSEC) to E.R., AP-HP (DRC-PHRC) to E.R., Fondation pour la Recherche sur le Cerveau (FRC) to E.R., DHOS-INSERM to M.V., ANR (French National Institutes) to M.V., AMADYS to M.V., Alliance France Dystonie to A.M., and the Department of Veterans Affairs (T.D.B., W.H.R.).
DISCLOSURE
D. Chen reports no disclosures relevant to the manuscript. A. Méneret received a grant from JNLF (Journées de Neurologie en Langue Française). J. Friedman reports family financial interest in biotechnology. O. Korvatska, A. Gad, E. Bonkowski, H. Stessman, D. Doummar, and C. Mignot report no disclosures relevant to the manuscript. M. Anheim declares honoraria and travel grant from Novartis, Teva, Lundbeck, Abbvie, and Actelion. S. Bernes, M. Davis, and N. Perriere report no disclosures relevant to the manuscript. B. Degos received research support from INSERM (COSSEC) and patient's association France Parkinson, received travel funding from Novartis, Teva, Lundbeck, and Merz Pharma, received speech honorarium from Novartis and Medtronic, and served on scientific advisory boards for ORKYN. D. Grabli, D. Gras, F. Hisama, K. Mackenzie, P. Swanson, and C. Tranchant report no disclosures relevant to the manuscript. M. Vidailhet has been an invited speaker at MDS International meetings. She is on the scientific advisory board of Merz. She has received unrestricted research grants from DHOS-INSERM and ANR (French National Institutes) and from AMADYS and Alliance France Dystonie (patient associations). S. Winesett, T. Oriane, L. Amendola, and M. Dorschner report no disclosures relevant to the manuscript. M. Weiss received personal compensation for speaking from Walgreens and NuFactor and research support from ALS Therapy Alliance and the Northeast ALS Consortium. E. Eichler is an Investigator of the Howard Hughes Medical Foundation and receives funding from the National Institute of Mental Health (R01 MH101221). He is on the scientific advisory board (SAB) of DNAnexus, Inc. and is a consultant for Kunming University of Science and Technology (KUST) as part of the 1000 China Talent Program. A. Torkamani is the founder of and equity holder in Cypher Genomics. E. Roze is the recipient of a grant “poste d'accueil” AP-HP/CNRS. He received research support from INSERM (COSSEC), AP-HP (DRC-PHRC), Fondation pour la Recherche sur le Cerveau (FRC), Merz-Pharma, Orkyn, IP Santé, and Ultragenyx; served on scientific advisory boards for Orkyn, Ultragenyx, and Merz Pharma; received speech honorarium from Merz Pharma, Novartis, Ipsen-Pharma, and Orkyn; and received travel funding from Ipsen-Pharma, Teva, Abbvie, Merz-Pharma, Dystonia Europe, the Georgian Medical and Public Health Association, the International Federation of Clinical Neurophysiology, and the Movement Disorders Society. T. Bird is funded by the Department of Veterans Affairs. W. Raskind is funded by the National Institute of Neurological Diseases and Stroke (R01 NS069719) and the Department of Veterans Affairs. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Chen YZ, Matsushita MM, Robertson P, et al. Autosomal dominant familial dyskinesia and facial myokymia: single exome sequencing identifies a mutation in adenylyl cyclase 5. Arch Neurol 2012;69:630–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen YZ, Friedman JR, Chen DH, et al. Gain-of-function ADCY5 mutations in familial dyskinesia with facial myokymia. Ann Neurol 2014;75:542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanoune J, Pouille Y, Tzavara E, et al. Adenylyl cyclases: structure, regulation and function in an enzyme superfamily. Mol Cell Endocrinol 1997;128:179–194. [DOI] [PubMed] [Google Scholar]
- 4.Iwamoto T, Okumura S, Iwatsubo K, et al. Motor dysfunction in type 5 adenylyl cyclase-null mice. J Biol Chem 2003;278:16936–16940. [DOI] [PubMed] [Google Scholar]
- 5.O'Roak BJ, Stessman HA, Boyle EA, et al. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat Commun 2014;5:5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raskind WH, Matsushita M, Peter B, et al. Familial dyskinesia and facial myokymia (FDFM): follow-up of a large family and linkage to chromosome 3p21-3q21. Am J Med Genet B Neuropsychiatr Genet 2009;150B:570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bird TD, Carlson CB, Hall JG. Familial essential (“benign”) chorea. J Med Genet 1976;13:357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Breedveld GJ, van Dongen JW, Danesino C, et al. Mutations in TITF-1 are associated with benign hereditary chorea. Hum Mol Genet 2002;11:971–979. [DOI] [PubMed] [Google Scholar]
- 9.Fernandez M, Raskind W, Matsushita M, Wolff J, Lipe H, Bird T. Hereditary benign chorea: clinical and genetic features of a distinct disease. Neurology 2001;57:106–110. [DOI] [PubMed] [Google Scholar]
- 10.Perez-Borja C, Tassinari AC, Swanson AG. Paroxysmal choreoathetosis and seizures induced by movement (reflex epilepsy). Epilepsia 1967;8:260–270. [DOI] [PubMed] [Google Scholar]
- 11.Bird TD, Carlson CB, Horning M. Ten year follow-up of paroxysmal choreoathetosis: a sporadic case becomes familial. Epilepsia 1978;19:129–132. [DOI] [PubMed] [Google Scholar]
- 12.Fernandez M, Raskind W, Wolff J, et al. Familial dyskinesia and facial myokymia (FDFM): a novel movement disorder. Ann Neurol 2001;49:486–492. [PubMed] [Google Scholar]
- 13.Siepel A, Bejerano G, Pedersen JS, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 2005;15:1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res 2005;15:901–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grantham R. Amino acid difference formula to help explain protein evolution. Science 1974;185:862–864. [DOI] [PubMed] [Google Scholar]
- 16.Sunyaev S, Ramensky V, Koch I, Lathe W, III, Kondrashov AS, Bork P. Prediction of deleterious human alleles. Hum Mol Genet 2001;10:591–597. [DOI] [PubMed] [Google Scholar]
- 17.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res 2002;30:3894–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 2003;31:3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peredo DE, Hannibal MC. The floppy infant: evaluation of hypotonia. Pediatr Rev 2009;30:e66–e76. [DOI] [PubMed] [Google Scholar]
- 20.Marecos C, Ng J, Kurian MA. What is new for monoamine neurotransmitter disorders? J Inherit Metab Dis 2014;37:619–626. [DOI] [PubMed] [Google Scholar]
- 21.Asanuma K, Ma Y, Okulski J, et al. Decreased striatal D2 receptor binding in non-manifesting carriers of the DYT1 dystonia mutation. Neurology 2005;64:347–349. [DOI] [PubMed] [Google Scholar]
- 22.Zhang L, McCarthy DM, Sharma N, Bhide PG. Dopamine receptor and Galpha(olf) expression in DYT1 dystonia mouse models during postnatal development. PLoS One 2015;10:e0123104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fuchs T, Saunders-Pullman R, Masuho I, et al. Mutations in GNAL cause primary torsion dystonia. Nat Genet 2013;45:88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vemula SR, Puschmann A, Xiao J, et al. Role of Galpha(olf) in familial and sporadic adult-onset primary dystonia. Hum Mol Genet 2013;22:2510–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berman BD, Hallett M, Herscovitch P, Simonyan K. Striatal dopaminergic dysfunction at rest and during task performance in writer's cramp. Brain 2013;136:3645–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee KW, Hong JH, Choi IY, et al. Impaired D2 dopamine receptor function in mice lacking type 5 adenylyl cyclase. J Neurosci 2002;22:7931–7940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carapito R, Paul N, Untrau M, et al. A de novo ADCY5 mutation causes early-onset autosomal dominant chorea and dystonia. Mov Disord 2015;30:423–427. [DOI] [PubMed] [Google Scholar]
- 28.Shaw C, Hisama F, Friedman J, Bird TD. ADCY5-related dyskinesia. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews. Seattle, WA: 1993–2015. Available at: http://www.ncbi.nlm.nih.gov/books/NBK263441/. [Google Scholar]
- 29.Mencacci NE, Erro R, Wiethoff S, et al. ADCY5 mutations are another cause of benign hereditary chorea. Neurology 2015;85:80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bird TD, Hall JG. Additional information on familial essential (benign) chorea. Clin Genet 1978;14:271–272. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.