

Abstract
Objective:
To assess the role of CHCHD2 variants in patients with Parkinson disease (PD) and Lewy body disease (LBD) in Caucasian populations.
Methods:
All exons of the CHCHD2 gene were sequenced in a US Caucasian patient-control series (878 PD, 610 LBD, and 717 controls). Subsequently, exons 1 and 2 were sequenced in an Irish series (355 PD and 365 controls) and a Polish series (394 PD and 350 controls). Immunohistochemistry and immunofluorescence studies were performed on pathologic LBD cases with rare CHCHD2 variants.
Results:
We identified 9 rare exonic variants of unknown significance. These variants were more frequent in the combined group of PD and LBD patients compared to controls (0.6% vs 0.1%, p = 0.013). In addition, the presence of any rare variant was more common in patients with LBD (2.5% vs 1.0%, p = 0.050) compared to controls. Eight of these 9 variants were located within the gene's mitochondrial targeting sequence.
Conclusions:
Although the role of variants of the CHCHD2 gene in PD and LBD remains to be further elucidated, the rare variants in the mitochondrial targeting sequence may be a risk factor for Lewy body disorders, which may link CHCHD2 to other genetic forms of parkinsonism with mitochondrial dysfunction.
Parkinson disease (PD) is the second most common neurodegenerative disorder, with an estimated prevalence of 1%–2% among individuals older than 60 years.1 Approximately 5%–10% of patients have a positive family history of PD.2 Recently, heterozygous mutations in the coiled-coil-helix-coiled-coil-helix domain containing 2 (CHCHD2) gene were identified in 4 of 341 independent Japanese families with an autosomal dominant pattern of inheritance of PD.3 CHCHD2 protein contains an N-terminal mitochondrial targeting sequence (MTS) and a twin Cx9C motif in a C-terminal coiled-coil-helix-coiled-coil-helix (CHCH) domain4 and it is localized to the mitochondria.5 To date, functional analyses have elucidated several important roles for CHCHD2; it regulates cytochrome c oxidase (COX) activity, and acts as a transcription factor to regulate COX expression, thereby facilitating mitochondrial electron transport chain flux under low oxygen conditions. CHCHD2 also inhibits mitochondria-mediated apoptosis.5 Mitochondrial dysfunction has been implicated in both early-onset genetic forms of PD (via the PINK1 and PARKIN mediated mitophagy pathway) and late-onset sporadic disease.6,7 Interestingly, it was also reported that specific variants in the CHCHD2 gene may affect susceptibility to sporadic PD.3 The role of variation in the CHCHD2 gene in Caucasian patients with PD and in pathologically confirmed Lewy body disease (LBD) is unclear. The aim of this study was to evaluate the association of CHCHD2 variants with risk of clinically diagnosed PD and pathologically diagnosed LBD.
METHODS
Participants.
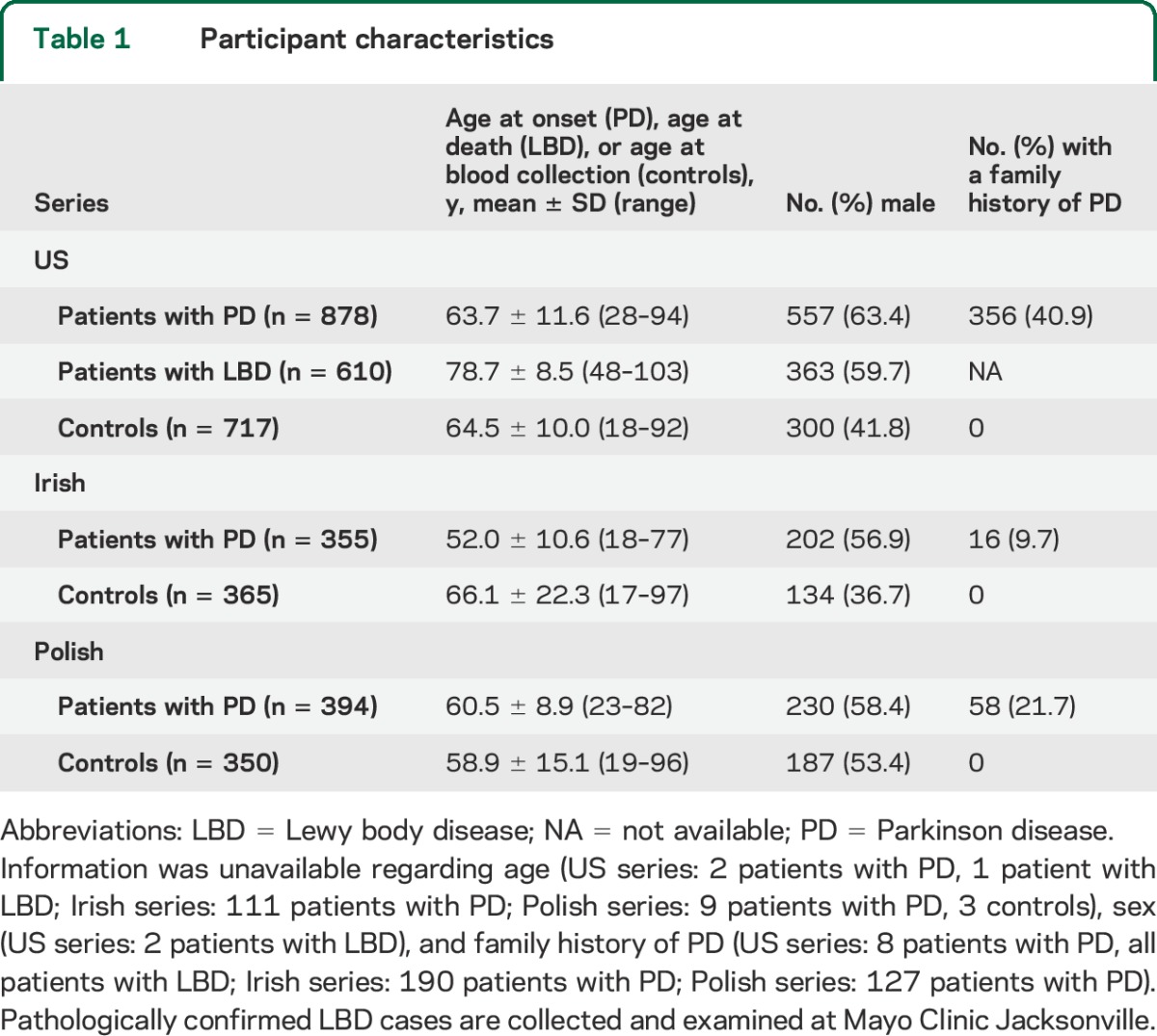
A total of 3,669 participants, including 1,627 patients with clinical PD, 610 patients with pathologically confirmed LBD, and 1,432 controls, were included in this study (see details in table 1), and collected between the years 1998 and 2014. The clinical and pathologic diagnoses were established according to the consensus criteria for PD8 and LBD.9,10 Controls were individuals free of PD or a related movement disorder at the time of examination. All participants were non-Hispanic Caucasian and were unrelated within and between disease groups. There was no overlap between the PD and LBD groups. Information was collected regarding age (age at onset in patients with PD, age at death in the patients with LBD from the US series, age at blood sample collection in controls) and sex. Family history of PD in first or second degree was also collected for patients with PD and controls.
Table 1.
Participant characteristics
Standard protocol approvals, registrations, and patient consents.
Written informed consent was obtained from all participants and the study was approved by all institutional review boards from the participating centers. Informed consent for pathologically confirmed cases was obtained from the next of kin.
Genetic analysis.
For direct sequence analysis, each exon was amplified by polymerase chain reaction using published primers for CHCHD2.3 DNA was extracted from frozen brain tissue and whole blood samples using standard protocols. Genomic sequences were analyzed with SeqScape version 2.5 using 3730XL DNA Analyzer (ABI; Applied Biosystems, Foster City, CA). RefSeq accession NM_016139.2 was used to number all variants within the CHCHD2 gene and protein. In the discovery stage, we sequenced all 4 exons of CHCHD2 in a US series including PD, LBD, and controls. We found 9 exonic variants located in exons 1 and 2 and none in exons 3 and 4. In the second stage, we sequenced exons 1 and 2 in the Irish and Polish series. The 3 variants in intron 3 and the 6 variants in the 3′UTR were not assessed in the Irish or Polish series. There was no evidence of a departure from Hardy-Weinberg equilibrium in controls in any of the series (all p > 0.01).
Statistical analysis.
For common variants (defined as those with a minor allele frequency [MAF] of 1% or greater), associations with disease were evaluated using logistic regression models. Associations were examined for the separate disease outcomes of PD, LBD, and PD or LBD. Models in the US series and Polish series were adjusted for age (age at PD onset in patients with PD, age at death in patients with LBD, age at blood collection in controls) and sex, models in the Irish series were adjusted for sex (age was not adjusted for due to missing data), and models in the combined series were adjusted for sex and series (age was not adjusted for due to missing data in the Irish series). Each variant was examined under an additive model (i.e., effect of each additional minor allele). Odds ratios and 95% confidence intervals were estimated.
For rare variants (defined as those with a MAF of less than 1%), we did not perform any single-variant analysis owing to the low power such analysis would have. Instead, separately in each series and in the combined series, we performed gene burden tests by collapsing across rare variants and comparing the frequency of the presence of any rare variant in controls to that of PD, LBD, and the combined PD and LBD patient groups using Fisher exact test.11 Since variants in intron 3 and 3′UTR were not assessed in the Irish and Polish series, we did not examine the presence of rare variants in the combined series (US, Irish, and Polish). However, we did examine the presence of rare exonic variants found in exons 1 and 2 in the combined series. p Values of 0.05 or lower were considered statistically significant. Statistical analysis was performed using R Statistical Software (version 2.14.0; R Foundation for Statistical Computing, Vienna, Austria).
In silico analyses.
For prediction of the functional consequences of coding variants on CHCHD2 protein sequence, we used software programs available on the Internet: PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/),12 SIFT (http://sift.jcvi.org/),13 and Mutation Taster (http://www.mutationtaster.org/).14 The results are shown in table e-1 on the Neurology® Web site at Neurology.org. Splice site prediction of exonic variants identified in the CHCHD2 gene was calculated by using the online bioinformatics tool Human Splicing Finder (version 2.4.1; http://www.umd.be/HSF/HSF.html).15
Neuropathologic analysis.
All 610 cases in the LBD series underwent a standardized neuropathologic assessment for Alzheimer-type and Lewy-related pathologies by a single pathologist (D.W.D.) as reported previously.16 Braak neurofibrillary tangle stage17 and Thal amyloid phase18 were assigned to each case based upon thioflavin S fluorescent microscopy.19 Immunohistochemistry for α-synuclein (non-Aβ component precursor; 1:3,000; Mayo Clinic, Jacksonville, FL)20 was used to establish neuropathologic diagnosis of LBD,10,21 with deparaffinized and rehydrated sections pretreated with 95% formic acid for 30 minutes and then steamed in distilled water for 30 minutes. Lewy-related pathology was assessed in cortex, amygdala, basal forebrain, and brainstem and classified as brainstem, transitional, or diffuse Lewy body disease.9 Five brain samples with CHCHD2 variants (p.P2L, p.G4R, p.A37V, and p.A93V) were available for further analysis. We performed immunohistochemistry with polyclonal CHCHD2 antibody (1:800; Proteintech, Chicago, IL) on temporal lobe and midbrain sections. The deparaffinized and rehydrated sections were steamed in pH6 citrate buffer for 30 minutes. We also performed immunofluorescence double staining with polyclonal CHCHD2 antibody (1:400, Proteintech) and monoclonal Tom20 antibody (1:500, Santa Cruz Biotechnology, Dallas, TX) on hippocampal sections from participants with CHCHD2 variants (n = 5) as well as LBD cases without CHCHD2 variants (n = 3) and control brains (n = 2).
RESULTS
We identified a total of 23 variants in our US series (PD, LBD, and controls; total 2,205 samples), Irish series (PD and controls; total 720 samples), and Polish series (PD and controls; total 744 samples); 4 are common variants and 19 are rare variants of which MAF is less than 1%. Nine were exonic variants within exons 1 and 2 (table 2 and figure 1). Of the 4 common variants (rs816406, rs816407, rs10043, rs8406), 3 of them (rs816407, rs10043, rs8406) were in strong linkage disequilibrium (r2 > 0.96). The frequency of each CHCHD2 variant is displayed in table 2 for the US series, Irish series, Polish series, and combined series, while associations with disease for common variants and gene burden tests for rare variants are shown in table e-2. None of the 4 common CHCHD2 variants were associated with disease in any of the series (all p ≥ 0.15).
Table 2.
Frequency of CHCHD2 variants in patients with PD, patients with LBD, and controls
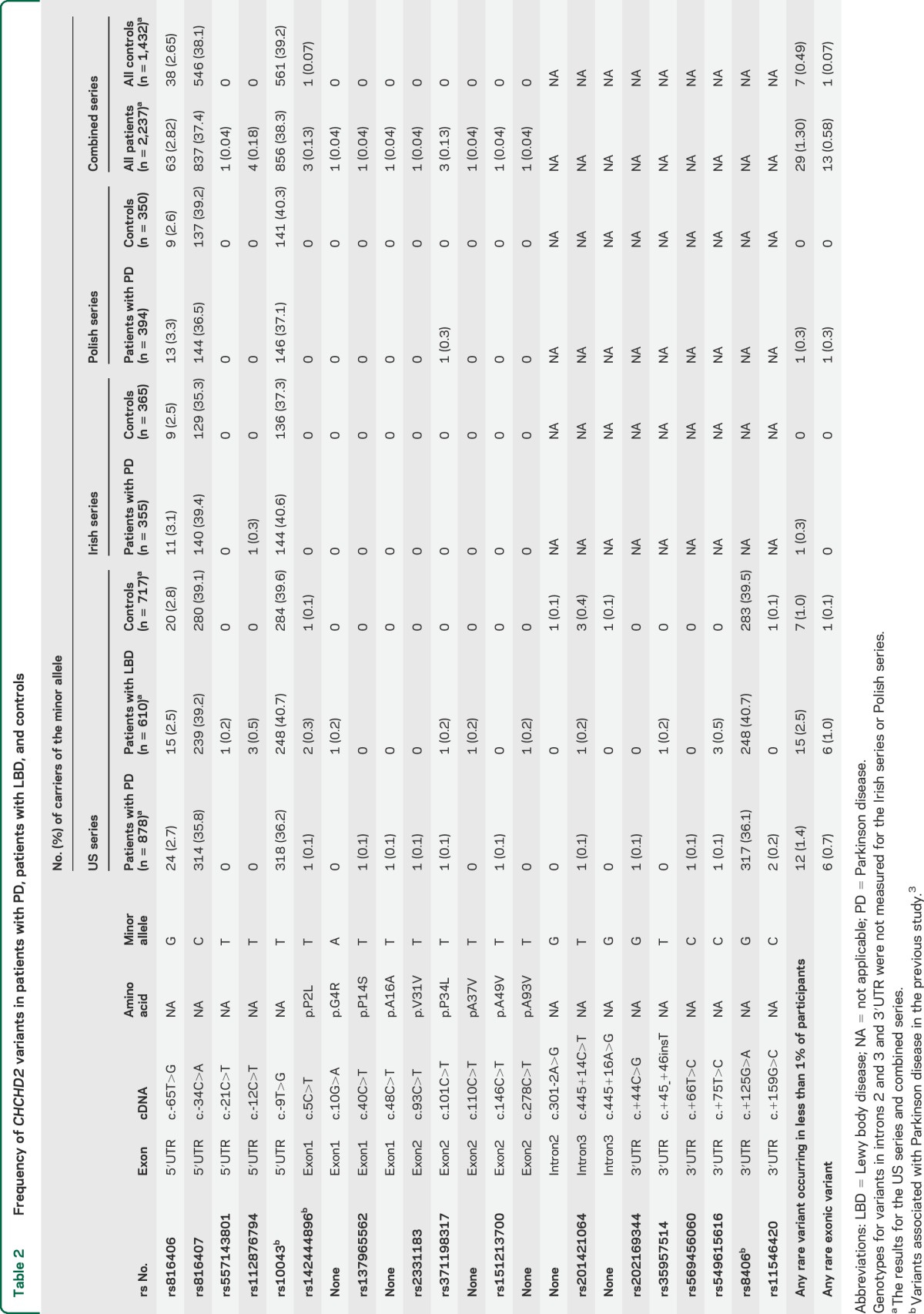
Figure 1. CHCHD2 variants found in Parkinson disease and Lewy body disease.

(A) The upper panel in blue represents the protein coding transcripts of the CHCHD2 gene and the exons are numbered (CHCHD2; RefSeq accession number NM_016139.2). The lower panel represents the protein domains and motifs that are referred to MitoProt II (http://ihg.gsf.de/ihg/mitoprot.html)31 and Pfam database (http://pfam.xfam.org/).32 Exonic variants shown in black were identified in patients with Parkinson disease or pathologically confirmed Lewy body disease in our study. Mutations in red were identified in Japanese patients with autosomal dominant Parkinson disease.3 CHCH = coiled coil 1-helix 1-coiled coil 2-helix 2; MTS = mitochondrial targeting sequence. (B) Nonsynonymous CHCHD2 variants found in this study and sequence alignment with various species. Amino acid position of mutation is highlighted in black. RefSeq accession numbers are as follows; Homo sapiens, NP_057223.1; Pan troglodytes, XP_001161277.1; Macaca mulatta, XP_001089512.1; Canis lupus, XP_536830.2; Bos Taurus, NP_001029918.1; Mus musculus, NP_077128.2; Rattus norvegicus, NP_001015019.1; Gallus gallus, NP_001006218.1; Danio rerio, NP_957061.1.
Rare exonic CHCHD2 variants were more common in the combined group of patients with PD and LBD compared to controls (0.6% vs 0.1%, p = 0.013, table e-2). This finding was driven mostly by the US series, where although not significant, rare exonic variants were more common compared to controls for both patients with PD (0.7% vs 0.1%, p = 0.14, table e-2) and patients with LBD (1.0% vs 0.1%, p = 0.053, table e-2). In the US series, rare exonic variants were found in 6 patients with PD and 6 patients with LBD. Only one healthy control with a rare variant (p.P2L) was found and the age at examination was 55 years. No rare exonic variants were observed in the Irish series in either patients with PD or controls, and in the Polish series rare exonic variants were observed in only one patient with PD (0.3%) and no controls. Rare exonic variants were also more common in the combined group of patients with PD (not including the US series patients with LBD) compared to controls (0.4% vs 0.1%, p = 0.074, table e-2). Within the US series, the presence of any rare variant was more common for patients with LBD, although this was only borderline significant (2.5% vs 1.0%, p = 0.050, table e-2).
Characteristics of the variants found in this study are summarized in table e-1, including MAF of public databases (Exome Variant Server [http://evs.gs.washington.edu/EVS/], Exome Aggregation Consortium [http://exac.broadinstitute.org/], and 1000 Genomes [http://www.1000genomes.org/]) and in silico analyses. We identified 9 rare exonic variants with unknown significance (p.P2L, p.G4R, p.P14S, p.A16A, p.V31V, p.P34L, p.A37V, p.A49V, and p.A93V; figure 1) in patients with PD or LBD in this study. Eight of these (p.P2L, p.G4R, p.P14S, p.A16A, p.V31V, p.P34L, p.A37V, and p.A49V) were located in the MTS of CHCHD2 (figure 1). Seven variants (p.P2L, p.G4R, p.P14S, p.A16A, p.V31V, p.A49V, and p.A93V) were predicted as damaging by at least one of the prediction tools. Two variants (p.A37V and p.A93V) were extremely rare even in large public databases. We found 2 synonymous variants (p.A16A; c.48C>T and p.V31V; c.93C>T) not seen in our control series and the MAF in the public databases are exceedingly low. They were predicted as disease-causing by Mutation Taster due to splicing site changes; however, Human Splicing Finder found no potential new sites for p.A16A and p.V31V variants. Human Splicing Finder did, however, predict one new enhancer motif at c.44 in p.A16A and 2 sites broken (at c.88 and c.89) and one new enhancer motif (at c.93) in p.V31V.
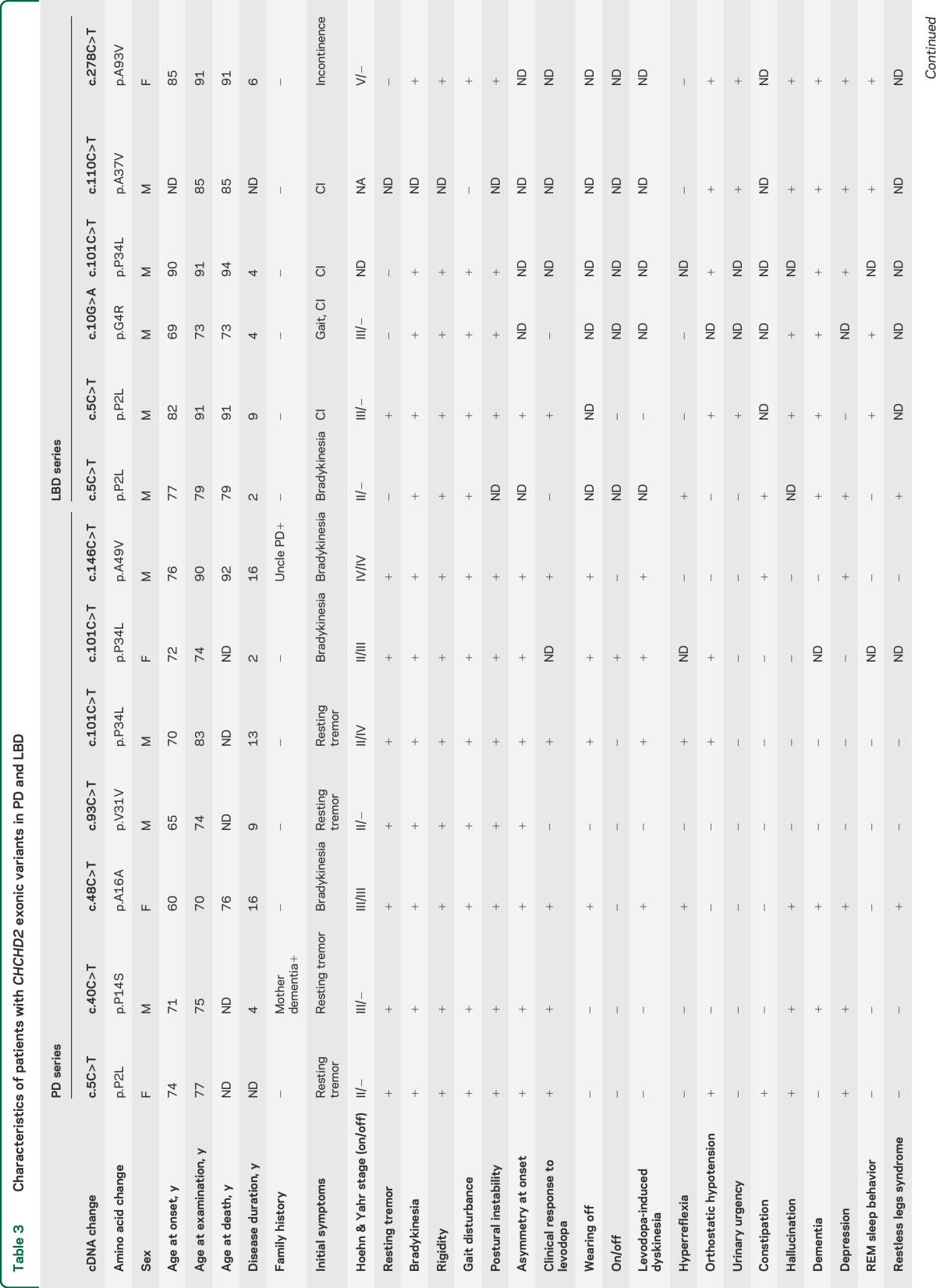
The clinical information of patients with rare exonic variants is shown in table 3. Seven patients had clinically diagnosed PD and 6 pathologically diagnosed LBD. Mean age at onset was 72.8 ± 6.8 years (range 60–90), mean age at death was 85.1 ± 7.6 years (range 73–94), and disease duration was 8.0 ± 5.5 years (range 2–16). Two patients (p.A49V and p.P14S) have a family history of PD or dementia, but DNA from their affected relatives was not available. Parkinsonism was the most common feature (12/13) among carriers although one individual with p.A37V was reported to have presented with cognitive impairment without significant parkinsonism. Dementia and hallucinations were frequently observed (7/13), as were depression (8/13) and orthostatic hypotension (7/13) in CHCHD2 variant carriers.
Table 3.
Characteristics of patients with CHCHD2 exonic variants in PD and LBD
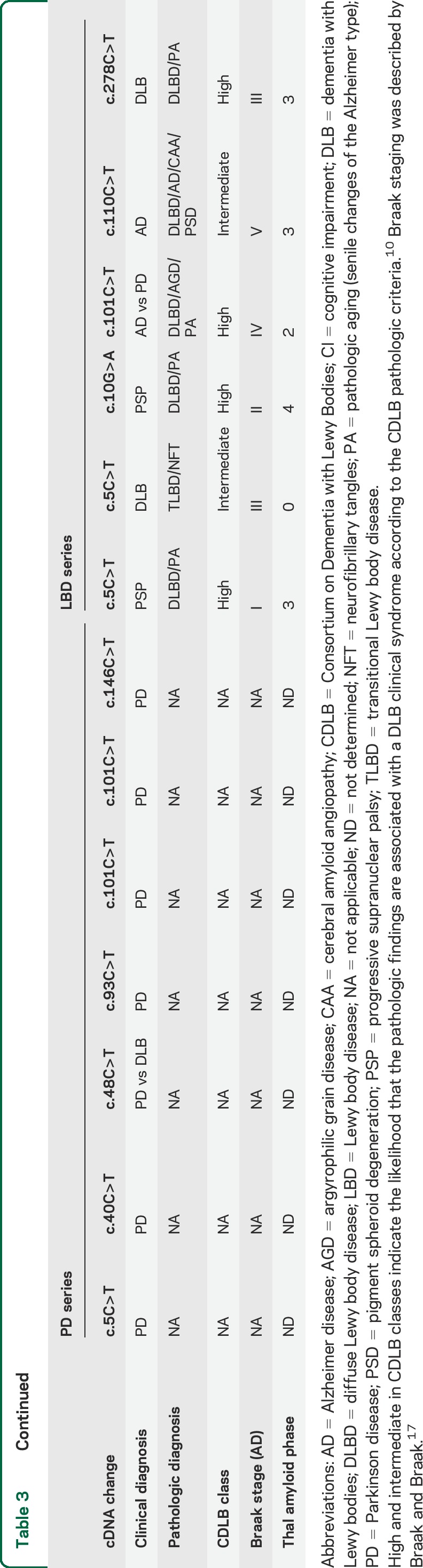
Neuropathologic data are available for 6 LBD cases with exonic variants and is presented in table 3. Five patients were classified as diffuse type of Lewy body pathology and one as a transitional type.9,22 To assess the expression of CHCHD2 at protein level, we performed immunohistochemistry for CHCHD2 in patients with LBD with exonic variants (p.P2L, p.G4R, p.A37V, and p.A93V; table 3). The granular staining pattern of CHCHD2 was observed in neuronal and glial cells in the substantia nigra and temporal cortex (figure 2C). Almost all Lewy bodies and pale bodies were negative for CHCHD2 (figure 2D). However, only a few pale bodies (p.G4R) and the surroundings of Lewy bodies (p.A93V) were slightly stained in a granular pattern (figure 2, E and F). Immunofluorescence staining suggested the expression level of CHCHD2 was decreased in patients with LBD with variants compared to age-matched controls (n = 2) and patients with LBD without variants (n = 3; figure 2, G–R). Interestingly, the case with the CHCHD2 p.A93V substitution, which lies outside the MTS region, showed normal protein expression in a subset of neurons (figure 2O).
Figure 2. Neuropathologic assessment in Lewy body disease.

(A, B) Hematoxylin & eosin (H&E) and α-synuclein (non-Aβ component precursor [NACP]) staining reveal Lewy bodies in substantia nigra. Immunohistochemistry for CHCHD2 shows the granular staining pattern in neuron and glial cells in temporal cortex (C) and substantia nigra. Almost all Lewy bodies and pale bodies were negative for CHCHD2 (D), but the rim of Lewy bodies are partially stained in some neurons (E). (F) Some pale bodies are also stained in granular pattern. (G–R) Immunofluorescence double-staining for CHCHD2 (red) and Tom20 (green) in hippocampal CA3 regions. CHCHD2 and Tom20 are expressed in mitochondria in control (G–I) and Lewy body disease (LBD) without CHCHD2 variants (J–L). (M–O) The expression level of CHCHD2 is decreased in patients with LBD with p.A93V variant but intact in some neurons. (P–R) In patients with p.P2L variant, which is located in mitochondrial targeting sequence, CHCHD2 expression is decreased. Bars = 50 μm.
DISCUSSION
In our Caucasian multicenter analysis, we did not find any definite pathogenic CHCHD2 mutations that cosegregated with disease in multi-incident pedigrees such as p.T61I, which was found in 2 large Japanese pedigrees with PD.3 The association observed in the Japanese population with the common variants, rs10043 and rs8406 (both single nucleotide polymorphisms are in high linkage disequilibrium), was not replicated in the Caucasian series; this could in part be due to ethnic-specific allele frequencies. Interestingly, the MAF of rs10043 and rs8406 was much lower in the Japanese population (∼4%) compared to that observed in our Caucasian series (∼23%). This may suggest a different haplotype structure and the potential for Japanese (Asian)-specific variant causing the reported association. One rare variant reported to influence susceptibility in PD in the Japanese study, p.P2L (rs142444896), was also rare in the Caucasian series but was not significant when compared to controls (1 PD, 2 LBD vs 1 control).3
We found a total of 9 rare exonic variants of unknown significance. Given the lack of multi-incident families or additional carriers, functional studies are needed to elucidate the nature of each variant. In the combined series, our gene burden test found an association between PD-LBD series and the number of rare exonic CHCHD2 variants within exons 1 and 2 (p = 0.013). In addition, we identified an association between LBD and any rare variant (p = 0.050). The exonic variants (p.P2L, p.G4R, p.P14S, p.A16A p.V31V, p.P34L. p.A37V, and p.A49V) are located in the MTS domain (figure 1). The MTS is critical for the CHCHD2 protein to be localized in mitochondrial intermembrane space.3 Those variants in the MTS might have an alternate pathomechanism compared to other exonic variants (e.g., p.A93V); notably, the reported pathogenic mutations found in the original Japanese study are located outside of the MTS domain (p.T61I, c.300+5G>A and p.R145Q; figure 1).3 The clinical data available for carriers suggests the age at onset is variable and most present with sporadic PD (table 3). In the original autosomal dominant pedigrees with pathogenic mutations, the mean age at onset was relatively late (56.2 ± 8.1 years).3 Given the sporadic nature of the disease, the rare variants we observed (p.P2L, p.G4R, p.P14S, p.A16A, p.V31V, p.P34L, p.A37V, p.A49V, and p.A93V) in this study might have lower penetrance than the pathogenic mutations found in Japan (p.T61I, c.300+5G>A and p.R145Q).3
Proteins similar to CHCHD2 with a twin Cx9C motif are involved in the production of energy by oxidative phosphorylation (OxPhos).23 In fact, CHCHD2 was first recognized as one of the regulators of OxPhos in a computational screen.24 Knockdown of CHCHD2 decreases the mitochondrial membrane potential and increases the generation of reactive oxygen species.25 These data suggest that CHCHD2 is an important mitochondrial regulator that can influence the energy levels of cells. Considering that impaired mitochondrial biogenesis and quality control may contribute to PD,26 further studies will be necessary to assess the role of CHCHD2 for mitochondrial homeostasis. It will be interesting to see if CHCHD2 plays a role in the PINK1/Parkin-dependent mitochondrial quality control pathway. Recently, a mutation in the CHCHD10 gene that encodes the mitochondrial CHCHD10 protein (OMIM 615903) was identified as the cause of late-onset familial amyotrophic lateral sclerosis with frontotemporal dementia in a large French pedigree27; one of the affected patients also shows signs of parkinsonism. CHCHD10 is a close paralog of CHCHD2 and the genetic and protein structures of both are similar. Further studies examining the role of genetic variation in both genes across neurodegenerative disorders are warranted.4,28,29
Although the results of this study suggest that rare CHCHD2 variants may be associated with risk of PD and LBD, it should also be noted that different genetic associations may be observed for alternate Lewy body disorders, e.g., PD and dementia with Lewy bodies both associate with the SNCA locus although the association signals appear independent.30 Indeed, there are a number of caveats to the present study, e.g., the low power to detect the association of rare CHCHD2 variants with risk of PD or LBD. Even when collapsing across rare variants and utilizing gene burden tests, the frequencies of the resulting collapsed rare variant variables were low, and therefore the possibility of type II error (i.e., a false-negative association) is important to consider. We also only have a limited number of pathologic cases with CHCHD2 variants so our staining studies can only be considered exploratory and larger samples sizes are required to replicate both our genetic and pathologic findings. With that in mind, the field of genetics and particularly late-onset neurodegenerative disease is moving toward the discovery of rare variants with incomplete penetrance. Large consortia or meta-analytical approaches will be needed to better understand the role of rare CHCHD2 variants in these diseases. Functional studies elucidating the cellular pathways altered by mutant CHCHD2 will be warranted to clarify the role of this protein in disease.
Supplementary Material
ACKNOWLEDGMENT
The authors thank those who contributed to their research, particularly the patients and families who donated brain and DNA samples for this work.
GLOSSARY
- CHCHD2
coiled-coil-helix-coiled-coil-helix domain containing 2
- COX
cytochrome c oxidase
- LBD
Lewy body disease
- MAF
minor allele frequency
- MTS
mitochondrial targeting sequence
- OxPhos
oxidative phosphorylation
- PD
Parkinson disease
Footnotes
Supplemental data at Neurology.org
Editorial, page 2002
AUTHOR CONTRIBUTIONS
Kotaro Ogaki: drafting/revising the manuscript, study concept and design, interpretation of data. Shunsuke Koga: drafting/revising the manuscript, interpretation of data. Michael G. Heckman: drafting/revising the manuscript, interpretation of data. Fabienne C. Fiesel: revising the manuscript. Maya Ando: drafting/revising the manuscript. Catherine Labbé: revising the manuscript. Oswaldo Lorenzo-Betancor: revising the manuscript. Elisabeth L. Moussaud-Lamodière: revising the manuscript. Alexandra I. Soto-Ortolaza: revising the manuscript. Ronald L. Walton: revising the manuscript. Audrey J. Strongosky: revising the manuscript. Ryan J. Uitti: revising the manuscript, contribution of patients. Allan McCarthy: revising the manuscript, contribution of patients. Timothy Lynch: revising the manuscript, contribution of patients. Joanna Siuda: revising the manuscript, contribution of patients. Grzegorz Opala: revising the manuscript, contribution of patients. Monika Rudzinska: revising the manuscript, contribution of patients. Anna Krygowska-Wajs: revising the manuscript, contribution of patients. Maria Barcikowska: revising the manuscript, contribution of patients. Krzysztof Czyzewski: revising the manuscript, contribution of patients. Andreas Puschmann: revising the manuscript, contribution of patients. Kenya Nishioka: revising the manuscript. Manabu Funayama: revising the manuscript, study concept and design, interpretation of data. Nobutaka Hattori: revising the manuscript, study concept and design. Joseph E. Parisi: revising the manuscript, contribution of patients. Ronald C. Petersen: revising the manuscript. Neill R. Graff-Radford: revising the manuscript. Bradley F. Boeve: revising the manuscript, contribution of patients. Wolfdieter Springer: revising the manuscript. Zbigniew K. Wszolek: revising the manuscript, contribution of patients. Dennis W. Dickson: revising the manuscript, contribution of patients, interpretation of data. Owen A. Ross: drafting/revising the manuscript, study concept and design, interpretation of data.
STUDY FUNDING
R.J.U., O.A.R., Z.K.W., and D.W.D. are partially supported by the NIH/NINDS P50 NS072187. O.A.R. is supported by the National Institute of Neurological Disorders and Stroke R01 NS078086 and in part by the Michael J. Fox Foundation for Parkinson's Research. Z.K.W. is also partially supported by a gift from Carl Edward Bolch Jr., and Susan Bass Bolch. Samples included in this study were clinical patients or brain donors to the brain bank at Mayo Clinic in Jacksonville, which is supported by CurePSP/Society for Progressive Supranuclear Palsy and Udall Center for Excellence in Parkinson Research (P50 NS072187). O.A.R. is supported by The Little Family Foundation. W.S. is partially supported by NIH/NINDS R01NS085070, the Michael J. Fox Foundation for Parkinson's Research, and the Foundation for Mitochondrial Medicine, Mayo Clinic Foundation and the Center for Individualized Medicine, the Marriott Family Foundation, and a Gerstner Family Career Development Award. K.O. is supported by a research grant from the NAITO Foundation. F.C.F. is the recipient of an APDA Fellowship. C.L. is the recipient of a FRSQ postdoctoral fellowship and is a 2015 Younkin Scholar supported by the Mayo Clinic Alzheimer's Disease and Related Dementias Genetics program. The Mayo Clinic Florida is a Morris K. Udall Parkinson's Disease Research Center of Excellence (National Institute of Neurological Disorders and Stroke P50 NS072187) and an Alzheimer's disease Research Center (NIA P50 AG16574). Mayo Clinic is supported by the Mangurian Foundation for Lewy body research.
DISCLOSURE
K. Ogaki and S. Koga report no disclosures relevant to the manuscript. M. Heckman is a member of the editorial board of Parkinsonism and Related Disorders. F. Fiesel, M. Ando, C. Labbé, O. Lorenzo-Betancor, E. Moussaud-Lamodière, A. Soto-Ortolaza, R. Walton, and A. Strongosky report no disclosures relevant to the manuscript. R. Uitti receives compensation from the American Academy of Neurology for his work as associate editor for Neurology®. He is a member of the editorial board of Parkinsonism and Related Disorders. A. McCarthy, T. Lynch, J. Siuda, G. Opala, M. Rudzinska, A. Krygowska-Wajs, M. Barcikowska, and K. Czyzewski report no disclosures relevant to the manuscript. A. Puschmann received a honorarium for speaking and sponsoring for travel from Lundbeck AB, Sweden, is a member of the editorial board of Parkinsonism and Related Disorders, and serves as consultant to the Swedish National Board of Health and Welfare (Socialstyrelsen) for the preparation of national guidelines for the treatment of Parkinson's Disease; and receives research funding from The Swedish Parkinson Foundation (Parkinsonfonden) and governmental funding for clinical research within the Swedish National Health Services (ALF-YF) and from Bundy Academy, Sweden. K. Nishioka and M. Funayama report no disclosures relevant to the manuscript. N. Hattori is a member of the editorial board of Journal of Neurology, Neurosurgery, and Psychiatry, Neuroscience Research, Journal of Parkinson's Disease, and Journal of Neural Transmission. J. Parisi, R. Petersen, N. Graff-Radford, B. Boeve, and W. Springer report no disclosures relevant to the manuscript. Z. Wszolek is funded by NIH NS072187, is the Co-Editor in Chief of Parkinsonism and Related Disorders, and is the Associate Editor of the European Journal of Neurology. D. Dickson receives research support from the NIH (P50 AG016574; P50 NS072187; P01 AG003949) and CurePSP/Society for Progressive Supranuclear Palsy; is an editorial board member of American Journal of Pathology, Annals of Neurology, Journal of Neuropathology and Experimental Neurology, and Brain Pathology; and is the editor-in-chief of American Journal of Neurodegenerative Disease and International Journal of Clinical and Experimental Pathology. O. Ross is a member of the editorial board of PLoS ONE, American Journal of Neurodegenerative Disease, Molecular Neurodegeneration, and Parkinsonism and Related Disorders, and is funded by NIH grants NS078086 and NS072187 and the Michael J. Fox Foundation. Go to Neurology.org for full disclosures.
REFERENCES
- 1.de Rijk MC, Launer LJ, Berger K, et al. Prevalence of Parkinson's disease in Europe: a collaborative study of population-based cohorts: Neurologic Diseases in the Elderly Research Group. Neurology 2000;54:S21–S23. [PubMed] [Google Scholar]
- 2.Lesage S, Brice A. Parkinson's disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet 2009;18:R48–R59. [DOI] [PubMed] [Google Scholar]
- 3.Funayama M, Ohe K, Amo T, et al. CHCHD2 mutations in autosomal dominant late-onset Parkinson's disease: a genome-wide linkage and sequencing study. Lancet Neurol 2015;14:274–282. [DOI] [PubMed] [Google Scholar]
- 4.Longen S, Bien M, Bihlmaier K, et al. Systematic analysis of the twin cx(9)c protein family. J Mol Biol 2009;393:356–368. [DOI] [PubMed] [Google Scholar]
- 5.Liu Y, Clegg HV, Leslie PL, et al. CHCHD2 inhibits apoptosis by interacting with Bcl-x L to regulate Bax activation. Cell Death Differ 2015;22:1035–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron 2015;85:257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haelterman NA, Yoon WH, Sandoval H, Jaiswal M, Shulman JM, Bellen HJ. A mitocentric view of Parkinson's disease. Annu Rev Neurosci 2014;37:137–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kosaka K, Yoshimura M, Ikeda K, Budka H. Diffuse type of Lewy body disease: progressive dementia with abundant cortical Lewy bodies and senile changes of varying degree: a new disease? Clin Neuropathol 1984;3:185–192. [PubMed] [Google Scholar]
- 10.McKeith IG. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. J Alzheimers Dis 2006;9:417–423. [DOI] [PubMed] [Google Scholar]
- 11.Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet 2008;83:311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 2003;31:3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 2014;11:361–362. [DOI] [PubMed] [Google Scholar]
- 15.Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 2009;37:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wider C, Ross OA, Nishioka K, et al. An evaluation of the impact of MAPT, SNCA and APOE on the burden of Alzheimer's and Lewy body pathology. J Neurol Neurosurg Psychiatry 2012;83:424–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 18.Thal DR, Rub U, Orantes M, Braak H. Phases of a beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002;58:1791–1800. [DOI] [PubMed] [Google Scholar]
- 19.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 2012;123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beach TG, White CL, Hamilton RL, et al. Evaluation of alpha-synuclein immunohistochemical methods used by invited experts. Acta Neuropathol 2008;116:277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005;65:1863–1872. [DOI] [PubMed] [Google Scholar]
- 22.Uchikado H, Lin WL, DeLucia MW, Dickson DW. Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy. J Neuropath Exp Neur 2006;65:685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hell K. The Erv1-Mia40 disulfide relay system in the intermembrane space of mitochondria. Biochim Biophys Acta 2008;1783:601–609. [DOI] [PubMed] [Google Scholar]
- 24.Baughman JM, Nilsson R, Gohil VM, Arlow DH, Gauhar Z, Mootha VK. A computational screen for regulators of oxidative phosphorylation implicates SLIRP in mitochondrial RNA homeostasis. PLoS Genet 2009;5:e1000590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aras S, Bai M, Lee I, Springett R, Huttemann M, Grossman LI. MNRR1 (formerly CHCHD2) is a bi-organellar regulator of mitochondrial metabolism. Mitochondrion 2015;20:43–51. [DOI] [PubMed] [Google Scholar]
- 26.Scarffe LA, Stevens DA, Dawson VL, Dawson TM. Parkin and PINK1: much more than mitophagy. Trends Neurosci 2014;37:315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bannwarth S, Ait-El-Mkadem S, Chaussenot A, et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014;137:2329–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Banci L, Bertini I, Ciofi-Baffoni S, Tokatlidis K. The coiled coil-helix-coiled coil-helix proteins may be redox proteins. Febs Lett 2009;583:1699–1702. [DOI] [PubMed] [Google Scholar]
- 29.Zhang M, Xi Z, Zinman L, et al. Mutation analysis of CHCHD10 in different neurodegenerative diseases. Brain 2015;138:e300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bras J, Guerreiro R, Darwent L, et al. Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Hum Mol Genet 2014;23:6139–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Claros MG, Vincens P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur J Biochem 1996;241:779–786. [DOI] [PubMed] [Google Scholar]
- 32.Finn RD, Bateman A, Clements J, et al. Pfam: the protein families database. Nucleic Acids Res 2014;42:D222–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.