

Abstract
The focus of this study was on the bastadin class of bromotyrosine derivatives, commonly isolated from Ianthella marine sponges, and is the first report on the secondary metabolites from Ianthella cf. reticulata. Two new bastadins were isolated, (E,Z)-bastadin 19 (1b), a diastereoisomer of the known (E,E)-bastadin 19 (1a), and dioxepine bastadin 3 (2), an unusual dibenzo-1,3-dioxepine. A bastadin NMR database was created and assisted in the structure determination of 1b, 2 and the rapid dereplication of ten other known compounds including bastadins 2–9 (3-10), 13 (11) and 19 (1a). The geometry of the 2-(hydroxyimino)-N-alkylamide chains, a chemical feature present in all bastadins, was further probed and new insights regarding the natural oxime configuration are discussed. Bastadins possessing (E,Z), (Z,E) or (E,E)-dioxime configurations could be artifacts of isolation or storage in solution. Therefore, this point was explored by photochemical and thermal isomerization studies, as well as molecular mechanics calculations. Bastadins 13 (11) and 19 (1a) exhibited moderate inhibition against Trypanosoma brucei and bastadin 4 (5) was cytotoxic to HCT-116 colon cancer cells.
Sponges of the genus Ianthella (order Verongida, family Ianthellidae) produce almost exclusively brominated secondary metabolites. The only exception to this pattern is represented by the bastaxanthin carotenoids.1 All of the brominated compounds can be divided into two groups: bromobenzofurans represented by the iantherans 2–4 and bromotyrosine derivatives such as aeroplysin,5 ianthelline,6 ianthesine,7 and the bastadins. The latter group represents the most ubiquitous substances isolated from the genus Ianthella. The first examples reported (1980–81) were from the Verongid sponge Ianthella basta and included bastadins 1 and 2 reported by Kazlauskas and coworkers.8, 9 Subsequently, there have been copious studies on Ianthella, resulting in the discovery of some 28 bastadins. Many exhibit cytotoxic activity against cancer cells and some are still under evaluation as potential therapeutic leads.9–16 Somewhat unusual are the additional reports of bastadins from two other sponges including a Verongid of the genus Psammaplysilla 15 and the Dendroceratid Dendrilla.16
A signature structural feature present in all bastadins reported to date is the (E)-2-(hydroxyimino)-N-alkylamide functional group (HI). The moiety, imbedded in a matrix of bromophenol rings, is also a key residue present in the psammaplin family of bromotyrosines, extensively studied from the Verongida sponge, Pseudoceratina (a.k.a. Psammaplysilla or Druinella) purpurea.17 Decades ago, Schmitz reported that a psammaplin A diastereomer containing both an (E)- and (Z)-2-(hydroxyimino)-N-alkylamide was transformed on standing to the E,E isomer and suggested the Z,Z isomer thereof, was the natural metabolite that isomerized to E,E upon extraction and work-up.18 This significant finding has been overlooked in all subsequent research on bastadin and psammaplin metabolites reported to date.19
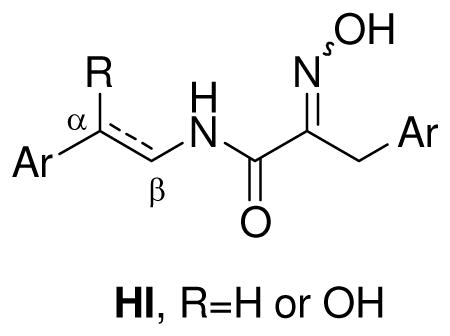
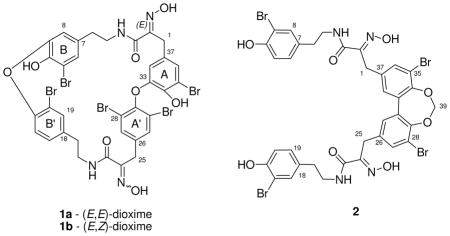
The availability of a recent collection of I. cf. reticulata prompted us to reinvestigate the structure of several known bastadins. In this study, we report an isomer of (E,E)-bastadin 19 (1a) that possesses both the (E)- and (Z)-2-(hydroxyimino)-N-alkylamides, which we have named (E,Z)-bastadin 19 (1b), in addition to an unusual dioxepine bastadin 3 (2). The characterization of compound 2, whose central core, like all bastadins, possessed an H/C ratio < 1,20 was challenging when based on analysis of the NMR data. In order to assist in the structure determination of new analogs and the rapid dereplication of known bastadins, we created a bastadin NMR database that provided benchmark chemical shift values for each aromatic and HI substructure found in the bastidins. This database assisted in the structure elucidation of 2, which contained a dibenzo-1,3-dioxepine group21 sandwiched between the two 2-(hydroxyimino)-N-alkylamide arrays. Finally, using the bastadin NMR database, we were able to propose 1H NMR data revisions for bastadin 23.16
Results and Discussion
We examined the methanol extract from I. cf. reticulata obtained from Papua New Guinea (coll. no. 05435) by LCMS and concluded it contained a complex mixture of several bastadins. In order to efficiently deal with the vast prior work, a biogenetically inspired22 outline was assembled, as shown in Figure 1, to include all of the previously reported bastadins. This sketch provided the basis for an immediate dereplication matrix similar to that we previously employed in analyzing sponge-derived oxy-polyhalogenated diphenyl ethers (O-PHDEs).20 The known bastadins, divided into four structure types, I – IV, based on their aryl-aryl linkages with annotations accompanying each structure, include: (1) prebastadin (preBAS, I), (2) dimer of hemibastadin (dihemBAS, II), (3) bastadin (BAS, III), and (4) isobastadin (isoBAS, IV).
Figure 1.


Sponge-derived bastadin structural families consisting of preBas, dihemBAS, BAS and isoBAS.
To employ the information of Figure 1 in dereplicating and recognizing new structures it is important to note that the bastadin structure is comprised of two classes of constitutional elements. One substructural region contains the four aromatic rings A, B, A′, and B′ (shown in I of Figure 1, with rings coded as 1 to 8 in Table 1), and the other consists of the two HI arrays (coded as chains 9 to 12 in Table 1). Easily recognized from Table 1 are that the aryl ring protons exhibit either four distinct ABX systems (first row) or four distinct two-spin systems (second row). Pinpointing the connectivity in the HI moiety is straightforward and involves matching data collected to that of either a saturated chain, an unsaturated chain, or two possible saturated chains with an attached OH group. After the rapid identification of the eight possible substructures, collection and analysis of 2D NMR data may be required. Employing the dereplication matrix of Table 1 allowed the characterization of the known bastadins including: 2 (3),8, 9 3 (4),9 4 (5),9, 23 5 (6),9 6 (7),9, 14 7 (8),9, 24 8 (9),10, 23 9 (10),10, 23 13 (11)25 and 19 (1a).26
Table 1.
NMR (1H and 13C in DMSO-d6) Database Values at Protonated Positions of the Rings and (E)-hydroxyimino-N-alkyl Amide Chains from Known Marine Sponge-derived Bastadins

|
values for the presence of two bromines in the aryl at the ortho position of the aryl-ether linkage
values do not include the NMR chemical shifts of bastadin-23
values for the linkage in α position for R = 10
values for the linkage in α position for R = 11
values for double bond conjugated with a phenol ring
values for NMR data reported in CD3CN
The first of the two new metabolites to be analyzed was compound 1b of formula C34H27Br5N4O8, as established by HRESIMS. This formula provided a match to four previously reported bastadins: 5 (6),9, 14, 15, 22, 24, 26, 27 15,16, 28 1629 and 19 (1a), 24, 26 while also ruling out the presence of benzene rings coded as 3 and 6 in Table 1. The constitutions of the four aryl rings, containing nine protons as three distinct two-spin systems and one ABX pattern, were established using the Table 1 database plus gCOSY NMR data. The specific data (correlated to ring types) included: (a) 2H singlet at δH 7.55 = ring 7, (b) meta-coupled protons at δH 7.15/6.33 = ring 5, (c) ABX spin system at δH 7.50/7.08/6.65 = ring 4, and (d) meta-coupled system at δH 6.75/6.01 which did not match NMR data of Table 1 but eventually assigned by 2D NMR data = ring 5. The requirement of such aryl rings ruled out bastadin 16 (comprised of rings = 1, 5, 7, 7). The presence of two non-identical HI residues coded as 9, was approached based on resonances including δH 2.61 (t)/2.59 (t); δH 3.24 (m)/.30 (m); 3.61 (s)/δH 3.28. The two aryl A-HI-aryl B and aryl A′-HI-aryl B′ connectivity’s determined by gHMBC experiments indicated the HI chains were linked as follows, A5-HI-B5 and A′7-HI-B′4. The mismatch of these arrangements with those of bastadin 5 (6) [A5-HI-B4 and A′7-HI-B′5] and bastadin 15 [A5-HI-B7 and A′4-HI-B′5] (Figure S2, Supporting Information) ruled out each of these structures. While (E,E)-bastidin 19 (1a) was consistent with this analysis, it did not fit the C-25 carbon chemical shift (δC 36.0) in the southern HI group. Schmitz’s discussions18 were the basis for assigning the structure as 1b, named (E,Z)-bastadin 19. This final conclusion was based on using the diagnostic 13C chemical shifts identified for the HI oxime allylic methylene: δC ≈ 28 for E configuration vs. δC ≈ 36 for Z configuration.18 The relevant 13C NMR data for 1a are δC 27.7/28.3; whereas 1b exhibits δC 27.8/36.0.
At this stage we confirmed that HI geometry differences previously observed for two psammaplin A diastereomers were replicated in the bastadin 19 diastereomers 1a and 1b. Based on this parallel pattern we considered the possibility that all bastadin natural products actually contain HI moieties with an initial all Z configuration. This intimates that the (E,Z), (Z,E), or (E,E) structures may not be the initial biosynthetic products, but could arise during the isolation steps or during extract storage. Interconversion reactions shown in Figure 2 were conducted to validate this point. First, (E,E)-bastadin 19 (1a) was subjected to photoisomerization using an incident light of 310–330 nm with acetophenone as triplet sensitizer.30 A mixture of recovered starting material plus new diastereoisomers was obtained consisting of: 1a [E,E, 36%], 1b [E,Z, 25%], (1c) [Z,E, 20%], and (1d) [Z,Z, 19%]. Their geometries were confirmed based on the diagnostic δC shifts for C1/C25 discussed above and shown in Table 2. Second, the semi-synthetic sample of (Z,Z)-bastadin 19 (1d) was subjected to isomerization in an NMR tube employing heat through brief sonication (30 min). The mixture of diastereoisomers plus starting material obtained consisted of: 1a [E,E, 9%], 1b [E,Z, 19%], 1c [Z,E, 24%], and 1d [Z/Z, 48%]. Finally, this mixture of compounds 1a-1d, on standing in solvent for prolonged periods (≈ 720 h) at room temperature, was completely transformed into 1a.
Figure 2.

Photochemical and thermal interconversions of bastadin 19 (1a-d) diastereoisomers.
Table 2.
NMR Data for (E,E)-Bastadin 19 (1a), (E,Z)-Bastadin 19 (1b), (Z,E)-Bastadin 19 (1c) and (Z,Z)-Bastadin 19 (1d) in DMSO-d6
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| 1a | 1b | 1c | 1d | |||||
|
| ||||||||
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 27.7 | 3.38, s | 27.8 | 3.28, s | 35.9 | 3.26, s | 35.8 | 3.16, s |
| 2 | 151.3 | 150.8 | 151.2 | 150.6 | ||||
| 3 | 162.9 | 162.9 | 161.8 | 161.5 | ||||
| 4 | - | 8.20, t (6.0) | 7.75, t (6.8) | 8.13, t (6.0) | 8.15, brs | |||
| 5 | 40.3 | 3.17, m | 39.6a | 3.24, m | 39.9a | 3.13, m | 39.9a | 3.18, m |
| 6 | 33.3 | 2.52, t (7.0) | 32.7 | 2.61, t (6.6) | 33.5 | 2.42, t (6.9) | 33.1 | 2.53, nd |
| 7 | 131.9 | 132.1 | 131.6 | 131.8 | ||||
| 8 | 117.6 | 6.34, d (2.0) | 118.4 | 6.33, d (1.9) | 117.7 | 6.44, d (1.8) | 117.8 | 6.40, brs |
| 9 | 145.2 | 144.5 | 144.9 | 144.8 | ||||
| 10 | 143.5 | 143.9 | 143.7 | 143.6 | ||||
| 11 | 110.9 | 110.0 | 110.9 | 110.9 | ||||
| 12 | 127.5 | 7.11, d (2.0) | 128.0 | 7.15, d (1.9) | 127.5 | 7.10, d (1.8) | 127.7 | 7.14, d (1.8) |
| 14 | 113.4 | 112.9 | 113.2 | 113.0 | ||||
| 15 | 151.4 | 151.6 | 151.2 | 151.3 | ||||
| 16 | 120.1 | 6.78, d (8.0) | 119.1 | 6.65, d (8.7) | 119.6 | 6.83, d (8.4) | 119.5 | 6.79, d (8.4) |
| 17 | 129.7 | 7.10, dd (2.0, 8.0) | 129.5 | 7.08, dd (1.9, 8.7) | 129.6 | 7.13, dd (1.8, 8.4) | 129.5 | 7.10, dd (1.8, 8.4) |
| 18 | 137.0 | 136.1 | 137.0 | 136.5 | ||||
| 19 | 133.5 | 7.51, d (2.0) | 133.5 | 7.50, d (1.9) | 133.2 | 7.53, d (1.8) | 133.3 | 7.50, d (1.8) |
| 20 | 34.0 | 2.69, t (6.7) | 33.9 | 2.59, t (6.6) | 33.1 | 2.83, t (6.0) | 33.4 | 2.65, t (6.9) |
| 21 | 39.5a | 3.37, m | 39.5a | 3.30, m | 39.4a | 3.48, m | 39.4a | 3.36, m |
| 22 | 7.79, t (6.0) | 8.33, t (6.6) | 8.18, t (6.6) | 8.32, brs | ||||
| 23 | 163.2 | 162.1 | 163.3 | 162.4 | ||||
| 24 | 151.0 | 151.5 | 150.9 | 151.3 | ||||
| 25 | 28.3 | 3.73, s | 36.0 | 3.61, s | 28.2 | 3.78, s | 35.8 | 3.57, s |
| 26 | 137.9 | 137.3 | 137.7 | 137.4 | ||||
| 27 | 133.1 | 7.47, s | 133.8 | 7.55, s | 133.0 | 7.47, s | 133.7 | 7.53, s |
| 28 | 117.3 | 117.4 | 117.2 | 117.2 | ||||
| 29 | 146.2 | 146.6 | 146.2 | 146.6 | ||||
| 30 | 117.3 | 117.4 | 117.2 | 117.2 | ||||
| 31 | 133.1 | 7.47, s | 133.8 | 7.55, s | 133.0 | 7.47, s | 133.7 | 7.53, s |
| 33 | 144.6 | 144.6 | 143.7 | 144.5 | ||||
| 34 | 141.9 | 141.8 | 142.2 | 142.1 | ||||
| 35 | 110.1 | 110.1 | 110.3 | 110.3 | ||||
| 36 | 125.9 | 6.90, d (2.0) | 125.1 | 6.75, d (1.9) | 126.8 | 6.98, d (1.8) | 126.5 | 6.98, d (1.8) |
| 37 | 128.4 | 128.4 | 128.2 | 128.8 | ||||
| 38 | 113.0 | 5.96, d (2.0) | 113.2 | 6.01, d (1.9) | 113.2 | 6.00, d (1.8) | 113.0 | 6.03, d (1.8) |
| 2-NOH | 11.95, brs | 11.64, brs | 12.02, brs | 11.58, brs | ||||
| 24-NOH | 11.65, brs | 11.44, brs | 11.44, brs | 11.45, brs | ||||
| 10-OH | 9.96, brs | 9.77, brs | 9.81,b brs | 9.80, brs | ||||
| 34-OH | 9.80, brs | 9.96, brs | 10.00,b brs | 9.98, brs | ||||
determined by HMBC correlations
interchangeable signals
The configurational lability of (E)- and (Z)-oximes has been previously explored. Oximes in the solid state display a high degree of configurational stability, however equilibrium between the E and Z isomers occurs in solution and the thermodynamically favored isomer predominates.31 In order to gain additional insights for the configurational stability of the HI residue in solution, molecular mechanics calculations were carried out on (E)- and (Z)-2-(hydroxyimino)-N-methylpropanamide as a simplified model. We wanted to compare the calculated results with our solution observations described above. Molecular mechanics calculations began with a conformation search in Spartan ‘06, resulting in four global minima being predicted: s-trans-Z, s-cis-Z, s-trans-E, and s-cis-E, whose structures are shown in Scheme 1. Two possible conformations, s-trans and s-cis, exist for each E and Z configurational isomer. The minimized energies, with and without H-bonding, for each of the four isomers were calculated with the MMX force field (dielectric constant = 1.5) in PCMODEL and are shown in Scheme 1. Both sets of calculations with and without H-bonding predict that the s-cis conformations are at least 15 kJ/mol higher than the corresponding s-trans, indicating a negligible population of any s-cis conformer at room temperature in solution. The lowest energy among the s-trans isomers with H-bonding was calculated for s-trans-Z (−2.52 kJ/mol), which has a H-bond between the hydrogen of the amide N-H and oxygen of the oxime (1.72 Å). However, without this intramolecular H-bond the s-trans-E calculated energy is lower than s-trans-Z by 4.0 kJ/mol and the former would predominate in solution. The latter case more closely reflects the results we observed, where intramolecular H-bonding is minimized due to solvation, and 1b, 1c, and 1d undergo thermal isomerization to 1a in solution.
Scheme 1.

Calculated global energy minima of (E)-and (Z)-2-(hydroxyimino)-N-methylpropanamides
The occurrence of the (Z)-oxime functional group is rare in natural products. In plants, the tyrosine derived-glucosinolate and cyanogenic glucoside dhurrin involves the transformation of L-tyrosine to (Z)-p-hydroxyphenylacetaldehyde oxime.32 Nocardicin A, a novel β-lactam from the actinomycete Nocardia uniformis, contains a 2-(hydroxyimino)-N-alkylamide substructure strikingly similar to those found in bastadins. Biosynthetic studies have suggested that the oxidation of the amine to Z-oxime in nocardicin A may proceed through a similar mechanism as proposed for dhurrin.33 An intramolecular hydrogen bonding mechanism was proposed to account for the predominant formation of Z-oxime (nocardicin A) relative to E-oxime (nocardicin B).34 This mechanism aptly accounts for the biosynthesis of Z-oximes in bastadin 1b and psammaplins (Scheme 2), as well as the preferential formation of (E)-p-hydroxyphenylacetaldehyde oxime over the (Z)-oxime in dhurrin. Two successive oxidations of the amine leads to a N,N-dihydroxy-HI intermediate in which one of the hydroxyls is hydrogen bonded to the neighboring amide, fixing the position of the two oxygen atoms and precluding the free rotation about the C-N bond. This intermediate is supported by observations in dhurrin biosynthetic studies that have shown the N-hydroxylation reactions are catalyzed by a multifunctional monooxygenase with a single catalytic site where the second oxygen incorporated in the N-hydroxytyrosine is quantitatively retained in the p-hydroxyphenylacetaldehyde oxime (the second oxygen is denoted as bold in Scheme 2).35 The neighboring amide facilitates dehydration to form the nitroso-HI intermediate and then tautomerization leading to the Z-oxime. Based on these biosynthetic studies, Schmitz’s E-Z isomerization results, and our current work, we are tempted to speculate that all bastadin and psammaplin metabolites are produced with the (Z)-oxime configuration and subsequently isomerize to the more thermodynamically favored E isomer in solution.
Scheme 2.

Extension of Townsend’s Z-oxime biosynthetic proposal34 to rationalize the initial biosynthesis of Z-HI in bastidins and psammaplins followed by Z-E isomerization in solution.
The next substance analyzed, compound (2), displayed a HREIMS m/z at [M−H]− 952.8664 for the formula C35H29Br4N4O8 and is unique in the bastadin family. A symmetrical dimeric biaryl-containing structure, analogous to that of bastadin 3 (4) [C34H30Br4N4O8],9, 23 was intimated from the count of H15 and C18 observed by NMR. These two compounds had parallel NMR properties as shown in Table 3. The aryl and aliphatic groups present were pinpointed from 1H and HMQC NMR data including: (a) an ABX spin system, δH 7.28/6.83/6.96 = ring 2 for two aryl groups (Table 1); (b) a meta-coupled constellation at δH 7.51/7.40, for two aryl rings not represented in Table 1; (c) HI arrays identified as chain 9 by the methylene protons at δH 3.30; δH 2.64 and δH 3.83 (Table 1); and (d) an one isolated dioxy-methylene at δH 5.66 (2H, s), δC 101.3. The gHMBC correlations from H-27/H-36 and H-31/H-38 to C-1/C-25 provided conclusive evidence that the two identical side chains were attached to the biaryl ring at C-26/C-37 of 2. This connection was also supported by NOESY correlations from δH 3.83 (H-1/H-25) to δH 7.40 (H-31/H-38) and to δH 7.51 (H-27/H-36). Finally, gHMBC correlations from H2-39 at δH 5.66 to δC 147.7 (C-29/C-34), and also H-27/36 and H-31/38 to C-29/C-34 linked the biaryl system through the isolated methylene at C-39. These 2D NMR data sets required embedding a 1,3-dioxepine ring into the biaryl system to complete the structure, which we named dioxepine bastadin 3 (2). The dibenzo-1,3-dioxepine scaffold is rare in natural products chemistry and is found only in the cercosporins isolated from Cercospora spp., a terrestrial fungus.36 We do not consider this compound to be an artifact generated during the isolation process from possible diol condensation with methylene chloride or other solvents.
Table 3.
NMR Data for Bastadin 3 (4) and Dioxepine Bastadin 3 (2) in DMSO-d6
|
|
||||
|---|---|---|---|---|
| 4 | 2 | |||
|
| ||||
| position | δC | δH (J in Hz) | δC | δH (J in Hz) |
| 1, 25 | 27.6 | 3.71, s | 28.3 | 3.83, s |
| 2, 24 | 151.7 | 151.3 | ||
| 3, 23 | 162.9 | 163.0 | ||
| 4, 22 | - | 7.93, t (6.6) | - | 8.02, t (6.6) |
| 5, 21 | 40.4 | 3.29, m | 40.3 | 3.30, m |
| 6, 20 | 33.6 | 2.63, t (7.4) | 33.5 | 2.64, t (6.6) |
| 7, 18 | 131.4 | 131.5 | ||
| 8, 17 | 132.5 | 7.28, d (1.9) | 132.6 | 7.28, d (1.9) |
| 9, 16 | 108.9 | 109.0 | ||
| 10, 15 | 152.2 | 152.3 | ||
| 11, 14 | 116.2 | 6.82, d (8.3) | 116.2 | 6.83, d (8.7) |
| 12, 19 | 128.7 | 6.95, dd (1.9, 8.3) | 128.8 | 6.96, dd (1.9, 8.7) |
| 26, 37 | 128.9 | 135.8 | ||
| 27, 36 | 132.0 | 7.30, d (1.9) | 132.8 | 7.51, d (1.9) |
| 28, 35 | 111.2 | 115.4 | ||
| 29, 34 | 149.6 | 147.7 | ||
| 30, 33 | 127.6 | 132.6 | ||
| 31, 38 | 131.0 | 6.94, d (1.9) | 128.5 | 7.40, d (1.9) |
| 39 | - | - | 147.7 | 5.66, (s) |
| 2/24-NOH | 11.79, (brs) | 11.97, (brs) | ||
| 9/15-OH | 10.01, (brs) | 9.99, (brs) | ||
| 29/34-OH | 8.95, (brs) | |||
During the dereplication process we discovered that the aryl 1H NMR data reported in the literature for bastadin 23 (12)16 did not match the patterns expected based on the trends shown in Table 1. Of further concern was that its structure was based solely on 1D 1H NMR analysis and 2D NOESY correlations. The literature values shown in Figure 3, especially for δH 6.58 (H-27) are not in accord with an aryl H-27 proton ortho to the Br and meta to an OAr, which is the substitution pattern of ring A′ (Figure S3, Supporting Information) of structure 12. There are two possibilities for revisions to rectify this ambiguity (Figure 3). One involves the amended assignments shown for structure 12 and it incorporates the assumption that literature J values are misreported. The other requires a revised structure 12a whose ring A′ provides an acceptable rationalization of the observed 1H NMR data. We preferred the first option because the substitution pattern of ring A′ in 12a is inconsistent both with all prior bastadin structures and the overview of the biogenetic pathways21 leading to its members.
Figure 3.

The literature 1H NMR assignment16 for the A′ ring in bastadin 23 (12) – possible revisions.
The bioassay component of this project included evaluation of both extract fractions and pure compounds. The MeOH extract (coded XFM) showed 80 % inhibition at 1.25 μg/mL against T. brucei, the causative agent of human African trypanosomiasis (sleeping sickness) designated as a neglected tropical disease (NTD) by the National Institutes of Health.37 Several compounds including 1a, 1b, 2, 3 and 5-11 were evaluated against the parasite. This follow-up screening revealed that only the isoBAS type structure with its ether linkages C9-O-C15 and C29-O-C33 (Figure 1) such as bastadin 13 (11) [IC50 = 1.77 μM] and bastadin 19 (1a) [IC50 = 1.31 μM] exhibited significant inhibition against T. brucei, although the testing set did not include the dioxepine bastadin 3 (2), due to its scarcity, nor the three isobastadins (1b-1d) due to their instability. Additional screening involved evaluation of 1a, 1b, 2 and 4-7 against the colon carcinoma cell line HCT-116. Among this set, only bastadin 4 (5) [IC50 = 1.28 μM] showed significant activity.
Conclusions
The most consistent sources of the bastadins are sponges from the genera Ianthella (order Verongida, family Ianthellidae). The results reported in our isolation work underscore this point and the sponge Ianthella cf. reticulata represents, as shown in Table 4, a fourth such species containing these heteroatom-rich structures. The literature also clearly shows (Table 4) that the bastadins can be isolated from other sponges identified as Psammaplysilla 15 (order Verongida, family Aplysinellidae) and Dendrilla 16 (order Dendroceratida, family Darwinellidae). However, for each of the two non-Ianthella sponges there is only one report of bastadins (D. cactos and P. purpurea). Consequently, these results seem inconsistent and could be explained by errors in the establishing their identity. It is tempting to envision that the bastadin bromotyrosine family represents a chemical signature for the genus Ianthella. Our isolation of (E,Z)-bastadin 19 (1b) is significant and points to the difficulty of obtaining bastadin analogs of all Z geometry. Based on our photochemical and thermal isomerization results, molecular mechanics calculations of a HI analog, and reported biosynthetic studies, we propose the bastidins and psammaplins initially contain the (Z)-oxime configuration and subsequently isomerize to the more thermodynamically stable E isomer in solution during extraction and/or work-up. Another unexpected finding of this study was the isolation of 2, a new dibenzo-1,3-dioxepine derivative of the known bastadin 3 (4). The bastadin database assembled here will facilitate future studies of bastadin-containing sponges, whose structures are challenging to establish because a core element has an H/C ratio of less than one.
Table 4.
Distribution of Bastadins Reported in this Study within Marine Sponges
| Bas. No. | Cmpd. No. | Ianthella basta | Ianthella quadrangulata | Ianthella flabelliformis | Dendrilla cactos | Psammaplysilla purpurea |
|---|---|---|---|---|---|---|
| 2 | 3 | ✓8, 9 | ✓22 | |||
| 3 | 4 | ✓9, 24 | ||||
| 4 | 5 | ✓8, 9, 24, 26 | ✓14 | |||
| 5 | 6 | ✓9, 22, 24, 26, 27 | ✓14 | ✓22, 38* | ✓15 | |
| 6 | 7 | ✓9, 22, 24 | ✓14 | ✓38* | ✓16 | |
| 7 | 8 | ✓9, 24, 26 | ✓14 | ✓16 | ✓15 | |
| 8 | 9 | ✓10, 23 | ||||
| 9 | 10 | ✓10, 23 | ✓38* | |||
| 13 | 11 | ✓25 | ✓14 | ✓38* | ||
| 19 | 1a | ✓26 | ✓38** | ✓16 |
Comparison of exact mass measurements with literature data indicated the presence of bastadins 5, 6, 9, 10, and 11, or corresponding isomeric bastadins 12, 13, 15, 16, 18 and 20.38
This reference failed to mention that bastadin 19 can not easily be distinguished using exact mass measurements from bastadins 5, 15, and 16.38
Experimental Section
General Experimental Procedures
The NMR spectra were recorded in DMSO-d6, acetone-d6 and CD3OD on a Varian 500 and 600 MHz for 1H and at 125 MHz for 13C NMR. High and low-resolution mass measurements were obtained respectively from a FAB and an ESI-TOF mass spectrometer. LCMS were performed with a Phenomenex Luna C18 RP 5μm column (150 × 4.6 mm) using an ESITOFMS, a Waters 996 photodiode array detector, and a Sedex55 evaporative light scattering (ELS) detector. Semi-preparative HPLC was performed using a Phenomenex 5μm Luna C18 RP column (250 × 10 mm) by using a single wavelength (λ = 254 nm) for compound detection.
NMR Database and Calculations
The bastidin NMR database was generated by averaging chemical shift values within each substructure type from reported literature values; see Supporting Information, Figures S1–S4 for individual chemical shifts. Chemical shifts were averaged and the error was reported to three standard deviations. Molecular modeling was carried out with Spartan ’06, version 1.1.2, Wavefunction Inc., June 15, 2007. Global minima were calculated using the Conformer Distribution program in Spartan ’06 utilizing the SYBYL force field. Energy minimization was carried out with PCMODEL, version 7.5 (Serena Software) using the MMX force field, Steepest Descent minimization method, dielectric constant = 1.5, with and without H-bonds.
Animal Material
The sponge (coll. no. 05435, 500 g wet weight) was collected in November 2005 by SCUBA in Milne Bay (10°37.005′ S: 150°95.620′ E), Papua New Guinea. Collection depth was approximately 30 ft. The sponge was identified taxonomically by Dr. Nicole J. de Voogd as I. cf. reticulata, and a voucher specimen was deposited at the Naturalis, National Museum of Natural History, Leiden, The Netherlands, under the registration number RMNH Por 5392.
Extraction and Isolation
Samples were preserved in the field according to our standard laboratory procedures39 and stored at 4 °C until extraction was performed. The sponges were extracted with hexanes (XFH), CH2Cl2 (XFD), and MeOH (XFM) using an accelerated solvent extractor (ASE). The MeOH extract (05435XFM, > 1 g) was submitted to a H2O/n-BuOH partitioning procedure. The resultant n-BuOH soluble extract (05435XFMWBS, 850 mg) was fractionated using several rounds of semi preparative reversed-phase HPLC (flow rate: 2 mL/min). Isocratic conditions were employed with CH3CN and water, each containing 0.1% formic acid. The fractionation (54:46 CH3CN-H2O) afforded 23 fractions. Fractions H4 (55.3 mg, 6.1×10−2 % dry weight), H6 (106.0 mg, 1.2×10−1 % dry weight), H8 (28.7 mg, 3.2×10−2 % dry weight), H16 (120.0 mg, 1.3×10−1 % dry weight) and H18 (18.2 mg, 2.0×10−2 % dry weight) provided 4, 11, 9, 1a and 8 respectively. Fractions H7 (23.0 mg), H9 (23.0 mg), H12 (4.6 mg), H20 (23.0 mg used of 37.2 mg) and H22 (11.7 mg) were each purified using 45:55 CH3CN-H2O and gave, respectively, 3 (4.0 mg, 4.0×10−3 % dry weight), 10 (2.4 mg, 3.0×10−3 % dry weight), 1b (2.0 mg, 2.0×10−3 % dry weight), 5 (11.0 mg, 1.2×10−2 % dry weight) accompanied by 6 (3.5 mg, 4.0×10−3 % dry weight) and 7 (2.0 mg, 2.0×10−3 % dry weight) accompanied by 2 (1.1 mg, 1.0×10−3 % dry weight).
Sensitized Photoisomerization
A degassed solution containing acetophenone (triplet sensitizer) and 1a (30 nM) in 1 mL of acetonitrile/methanol (3/1) was irradiated through one Pyrex culture tube at 310–330 nm for 2 h. The resultant residue was fractionated using semi-preparative reversed-phase HPLC with acetonitrile and water (52:48 CH3CN-H2O), each containing 0.1% formic acid. Fractions H2 (4.5 mg, 19 % yield), H4 (6.6 mg, 20 % yield), H6 (8.6 mg, 25 % yield), H8 (11.3 mg, 36% yield) provided 1d, 1c, 1b, and 1a, respectively.
Thermal Isomerization
A solution of 1d in DMSO-d6 contained in a NMR tube was sonicated in a beaker of MeOH for 30 min. The resultant 1H NMR spectrum is available as Figure S23 (Supporting Information).
Room Temperature Isomerization
The mixture of 1a-1d obtained from the thermal isomerization of 1d (see above) in DMSO-d6 was kept at room temperature for 720 h.
Cytotoxicity against HCT-116
IC50 values were determined as previously described.40
Trypanosoma brucei Assay
IC50 values were determined as previously described.37
(E,E)-Bastadin 19 (1a): white powder; 1H NMR and 13C NMR in DMSO-d6 (see Table 2); ESIMS [M − H]− m/z 1012.8 (11), 1014.8 (56), 1016.8 (100), 1018.8 (99), 1020.8 (56), 1022.8 (13).
(E,Z)-Bastadin 19 (1b): white powder; 1H NMR and 13C NMR in DMSO-d6 (see Table 2); ESIMS [M − H]− m/z 1012.5 (6), 1014.5 (55), 1016.5 (99), 1018.5 (100), 1020.5 (38), 1022.5 (3); HRESIMS [M−H]− m/z 1012.7677 (calcd for C35H2979Br4N4O8 1012.7668).
(Z,E)-Bastadin 19 (1c): white powder; 1H NMR and 13C NMR in DMSO-d6 (see Table 2); ESIMS [M − H]− m/z 1012.5 (6), 1014.5 (55), 1016.5 (99), 1018.5 (100), 1020.5 (38), 1022.5 (3).
(Z,Z)-Bastadin 19 (1d): white powder; 1H NMR and 13C NMR in DMSO-d6 (see Table 2); ESIMS [M − H]− m/z 1012.5 (6), 1014.5 (55), 1016.5 (99), 1018.5 (100), 1020.5 (38), 1022.5 (3).
Dioxepine bastadin 3 (2): white powder; 1H NMR and 13C NMR in DMSO-d6 (see Table 3); ESIMS [M − H]− m/z 948.9 (7), 606.6 (46), 608.6 (100), 610.6 (89), 612.6 (51), 614.6 (11); HRESIMS [M−H]− m/z 948.8714 (calcd for C35H2979Br4N4O8 948.8724).
Bastadin 2 (3): white powder; NMR data are in accordance with the literature.9
Bastadin 3 (4): white powder; 1H NMR and 13C NMR in DMSO-d6 (see Table 3); 1H NMR (CD3OD, 600 MHz) δH 7.42 (2H, d, J =1.8 Hz, H-27/36), 7.30 (2H, J =1.8 Hz, H-8/17), 7.06 (2H, d, J =1.8 Hz, H-31/38), 6.96 (2H, dd, J = 1.8, 8.1 Hz, H-12/19), 6.78 (2H, d, J =8.1 Hz, H-11/14), 3.84 (4H, s, H-1/25), 3.38 (4H, t, 7.2, H-5/21), 2.67 (4H, t, 6.6, H-6/20); 13C NMR (CD3OD, 125 MHz) δC 165.9 (C-3/23), 153.9 (C-10/15), 153.2 (C-2/24), 150.9 (C-29/34), 134.4 (C-8/17), 134.1 (C-27/36), 133.2 (C-7/18), 132.8 (C-31/38), 131.3(C-26/37), 130.2 (C-12/19), 128.8 (C-30/33), 117.4 (C-11/14), 112.6 (C-28/35), 110.9 (C-9/16), 42.2 (C-5/21), 35.4 (C-6/20), 28.9 (C-1/25); ESIMS [M − H]− m/z 1014.7 (6), 1016.7 (55), 1018.7 (97), 1020.7 (100), 1022.7 (41), 1024.7 (11).
Bastadin 4 (5): white powder; NMR data are in accordance with the literature.23
Bastadin 5 (6): white powder; NMR data are in accordance with the literature.41
Bastadin 6 (7): white powder; NMR data are in accordance with the literature.9, 16
Bastadin 7 (8): white powder; NMR data are in accordance with the literature.24
Bastadin 8 (9): white powder; [α]27D= −2.8 (c=1, MeOH) NMR data are in accordance with the literature.25
Bastadin 9 (10): white powder; NMR data are in accordance with the literature.23
Bastadin 13 (11): white powder; NMR data are in accordance with the literature.25
Supplementary Material
Acknowledgments
This research was supported by NIH grant R01-CA047135 (P.C.) and NMR equipment grants from NSF CHE-0342912 and NIH S10-RR19918. We thank W. Boggess at the University of Notre Dame for providing HRESIMS measurements and R. Bogomolni at the University of California Santa Cruz for use of the lamp to perform the photoisomerization reaction. We would like to thank L. Matainaho, University of Papua New Guinea and the crew and skipper (C. DeWit) of the M/V Golden Dawn for assistance in specimen collection.
Footnotes
Dedicated to the late Dr. Richard E. Moore of the University of Hawaii at Manoa for his pioneering work on bioactive natural products.
Supporting Information Available: Tables and Figures are provided which include mass spectrometry and NMR spectroscopy studies for 1b-1d, 2, and 4. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Ramdahl T, Kazlauskas R, Bergquist P, Liaaenjensen S. Biochem Syst Ecol. 1981;9:211–213. [Google Scholar]
- 2.Okamoto Y, Ojika M, Sakagami Y. Tetrahedron Lett. 1999;40:507–510. [Google Scholar]
- 3.Okamoto Y, Ojika M, Suzuki S, Murakami M, Sakagami Y. Bioorg Med Chem. 2001;9:179–183. doi: 10.1016/s0968-0896(00)00234-0. [DOI] [PubMed] [Google Scholar]
- 4.Greve H, Meis S, Kassack MU, Kehraus S, Krick A, Wright AD, Konig GM. J Med Chem. 2007;50:5600–5607. doi: 10.1021/jm070043r. [DOI] [PubMed] [Google Scholar]
- 5.Minale L, Sodano G, Chan WR, Chen AM. J Chem Soc Chem Commun. 1972:674–675. [Google Scholar]
- 6.Litaudon M, Guyot M. Tetrahedron Lett. 1986;27:4455–4456. [Google Scholar]
- 7.Okamoto Y, Ojika M, Kato S, Sakagami Y. Tetrahedron. 2000;56:5813–5818. [Google Scholar]
- 8.Kazlauskas R, Lidgard RO, Murphy PT, Wells RJ. Tetrahedron Lett. 1980;21:2277–2280. [Google Scholar]
- 9.Kazlauskas R, Lidgard RO, Murphy PT, Wells RJ, Blount JF. Aust J Chem. 1981;34:765–786. [Google Scholar]
- 10.Miao S, Andersen RJ. J Nat Prod. 1990;53:1441–1446. doi: 10.1021/np50072a007. [DOI] [PubMed] [Google Scholar]
- 11.Pettit GR, Butler MS, Bass CG, Doubek DL, Williams MD, Schmidt JM, Pettit RK, Hooper JNA, Tackett LP, Filiatrault MJ. J Nat Prod. 1995;58:680–688. doi: 10.1021/np50119a005. [DOI] [PubMed] [Google Scholar]
- 12.Aoki S, Cho S, Hiramatsu A, Kotoku N, Kobayashi M. J Nat Med. 2006;60:231–235. doi: 10.1007/s11418-006-0045-3. [DOI] [PubMed] [Google Scholar]
- 13.Aoki S, Cho S, Ono M, Kuwano T, Nakao S, Kuwano M, Nakagawa S, Gao JQ, Mayumi T, Shibuya M, Kobayashi M. Anticancer Drugs. 2006;17:269–278. doi: 10.1097/00001813-200603000-00005. [DOI] [PubMed] [Google Scholar]
- 14.Greve H, Kehraus S, Krick A, Keller G, Maier A, Fiebig HH, Wright AD, Konig GM. J Nat Prod. 2008;71:309–312. doi: 10.1021/np070373e. [DOI] [PubMed] [Google Scholar]
- 15.Carney JR, Scheuer PJ, Kelly-Borges M. J Nat Prod. 1993;56:153–157. doi: 10.1021/np50091a025. [DOI] [PubMed] [Google Scholar]
- 16.Reddy AV, Ravinder K, Narasimhulu M, Sridevi A, Satyanarayana N, Kondapi AK, Venkateswarlu Y. Bioorg Med Chem. 2006;14:4452–4457. doi: 10.1016/j.bmc.2006.02.033. [DOI] [PubMed] [Google Scholar]
- 17.Piña IC, Gautschi JT, Wang GY, Sanders ML, Schmitz FJ, France D, Cornell-Kennon S, Sambucetti LC, Remiszewski SW, Perez LB, Bair KW, Crews P. J Org Chem. 2003;68:3866–3873. doi: 10.1021/jo034248t. [DOI] [PubMed] [Google Scholar]
- 18.Arabshahi L, Schmitz FJ. J Org Chem. 1987;52:3584–3586. [Google Scholar]
- 19.Fusetani N, Masuda Y, Nakao Y, Matsunaga S, van Soest RWM. Tetrahedron. 2001;57:7507–7511. Tokaradines A and B were reported as the (Z)-oxime isomer, however we believe the authors incorrectly assigned the configuration. The E geometry would more aptly account for the syn-methylene carbon chemical shift of 30.7 ppm. [Google Scholar]
- 20.Calcul L, Chow R, Oliver AG, Tenney K, White KN, Wood AW, Fiorilla C, Crews P. J Nat Prod. 2009;72:443–449. doi: 10.1021/np800737z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keana JFW, Morse RH. Tetrahedron Lett. 1976:2113–2114. [Google Scholar]
- 22.Jaspars M, Rali T, Laney M, Schatzman RC, Diaz MC, Schmitz FJ, Pordesimo EO, Crews P. Tetrahedron. 1994;50:7367–7374. [Google Scholar]
- 23.Pordesimo EO, Schmitz FJ. J Org Chem. 1990;55:4704–4709. [Google Scholar]
- 24.Franklin MA, Penn SG, Lebrilla CB, Lam TH, Pessah IN, Molinski TF. J Nat Prod. 1996;59:1121–1127. doi: 10.1021/np960507g. [DOI] [PubMed] [Google Scholar]
- 25.Butler MS, Lim TK, Capon RJ, Hammond LS. Aust J Chem. 1991;44:287–296. [Google Scholar]
- 26.Mack MM, Molinski TF, Buck ED, Pessah IN. J Biol Chem. 1994;269:23236–23249. [PubMed] [Google Scholar]
- 27.Masuno MNH, AC, Pessah IN, Molinski TF. Mar Drugs. 2004;2:176–184. [Google Scholar]
- 28.Dexter AF, Garson MJ, Hemling ME. J Nat Prod. 1993;56:782–786. [Google Scholar]
- 29.Sun Ku P, Jurek J, Carney JR, Scheuer PJ. J Nat Prod. 1994;57:407–410. [Google Scholar]
- 30.Padwa A, Albrecht F. J Am Chem Soc. 1974;96:4849–4857. [Google Scholar]
- 31.Tennant G. In: Comprehensive Organic Chemistry, The Synthesis and Reaction of Organic Compounds. Part 8. Barton D, Ollis WD, editors. Vol. 2. Pergamon Press; Oxford; New York: 1979. pp. 383–590. [Google Scholar]
- 32.Sibbesen O, Koch B, Halkier BA, Moller BL. J Biol Chem. 1995;270:3506–3511. doi: 10.1074/jbc.270.8.3506. [DOI] [PubMed] [Google Scholar]
- 33.Kelly WL, Townsend CA. J Am Chem Soc. 2002;124:8186–8187. doi: 10.1021/ja025926g. [DOI] [PubMed] [Google Scholar]
- 34.Kelly WL, Townsend CA. J Bacteriol. 2005:739–746. doi: 10.1128/JB.187.2.739-746.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Halkier BA, Lykkesfeldt J, Moller BL. Proc Natl Acad Sci USA. 1991;88:487–491. doi: 10.1073/pnas.88.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lousberg RJJCh, Weiss U, Salemink CA, Amone A, Merlini L, Nasini G. Chem Commun. 1971:1463–1464. [Google Scholar]
- 37.Rubio BK, Tenney K, Ang KH, Abdulla M, Arkin M, McKerrow JH, Crews P. J Nat Prod. 2009;72:218–222. doi: 10.1021/np800711a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Motti CA, Freckelton ML, Tapiolas DM, Willis RH. J Nat Prod. 2009;72:290–294. doi: 10.1021/np800562m. [DOI] [PubMed] [Google Scholar]
- 39.Rodriguez J, Nieto RM, Crews P. J Nat Prod. 1993;56:2034–2040. doi: 10.1021/np50102a002. [DOI] [PubMed] [Google Scholar]
- 40.Robinson SJ, Tenney K, Yee DF, Martinez L, Media JE, Valeriote FA, van Soest RWM, Crews P. J Nat Prod. 2007;70:1002–1009. doi: 10.1021/np070171i. [DOI] [PubMed] [Google Scholar]
- 41.Couladouros EA, Pitsinos EN, Moutsos VI, Sarakinos G. Chem Eur J. 2001;11:406–421. doi: 10.1002/chem.200400904. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.