

Abstract
Purpose
IPI-504 is a novel, water-soluble, potent inhibitor of heat-shock protein 90 (Hsp90). Its potential anticancer activity has been validated in preclinical in vitro and in vivo models. We studied the activity of IPI-504 after epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) therapy in patients with advanced, molecularly defined non–small-cell lung cancer (NSCLC).
Patients and Methods
Patients with advanced NSCLC, prior treatment with EGFR TKIs, and tumor tissue available for molecular genotyping were enrolled in this prospective, nonrandomized, multicenter, phase II study of IPI-504 monotherapy. The primary outcome was objective response rate (ORR). Secondary aims included safety, progression-free survival (PFS), and analysis of activity by molecular subtypes.
Results
Seventy-six patients were enrolled between December 2007 and May 2009 from 10 United States cancer centers. An ORR of 7% (five of 76) was observed in the overall study population, 10% (four of 40) in patients who were EGFR wild-type, and 4% (one of 28) in those with EGFR mutations. Although both EGFR groups were below the target ORR of 20%, among the three patients with an ALK gene rearrangement, two had partial responses and the third had prolonged stable disease (7.2 months, 24% reduction in tumor size). The most common adverse events included grades 1 and 2 fatigue, nausea, and diarrhea. Grade 3 or higher liver function abnormalities were observed in nine patients (11.8%).
Conclusion
IPI-504 has clinical activity in patients with NSCLC, particularly among patients with ALK rearrangements.
INTRODUCTION
Heat-shock protein 90 (Hsp90) is integral in protein homeostasis and regulates the stability of key proteins involved in oncogenesis, proliferation, and survival through its role as a protein chaperone.1 Hsp90 is an emerging focus of cancer therapy by virtue of its ability to inhibit multiple vital signaling pathways simultaneously.2,3 Furthermore, mutated oncoproteins, including epidermal growth factor receptor (EGFR), may preferentially rely on Hsp90 chaperones more than their wild-type counterparts, further increasing the appeal of Hsp90 as a therapeutic target for cancers defined by such mutations.4–7
Non–small-cell lung cancer (NSCLC) is a heterogeneous disease that can be subclassified based on driver mutations, specific oncogene alterations that lead to biologic dependence on the driver's signaling pathway, or oncogene addiction. The most common driver mutations in NSCLC appear to involve the genes for KRAS, EGFR, and anaplastic lymphoma kinase (ALK).8–10 When potent and specific inhibitors are used to block the signal from the driver oncogene, treatment can be extremely effective, as demonstrated in the case of EGFR tyrosine kinase inhibitors (TKIs) in EGFR-mutant NSCLC.11–13 This success may be mirrored with ALK TKI therapy in ALK-rearranged NSCLC.14
Retaspimycin hydrochloride (IPI-504) is a novel, water-soluble, potent inhibitor of Hsp90. An analog of 17-allylamino-17-demethoxygeldanamycin (17-AAG), IPI-504's potential anticancer activity has been validated in in vitro and in vivo models.15,16 A phase I/II study of IPI-504 in patients with NSCLC was conducted. In the phase I dose-escalation portion of the study, IPI-504 monotherapy in patients with NSCLC demonstrated a favorable adverse effect profile and evidence of clinical benefit.17 We therefore conducted this multicenter phase II portion of the study to prospectively assess the efficacy of IPI-504 after EGFR TKI therapy in patients with advanced NSCLC. EGFR genotype was mandatory so that differences in activity by mutation status could be observed. We retrospectively assessed other biomarkers to identify groups with differential responses to therapy.
PATIENTS AND METHODS
Study Design and Patients
This was a nonrandomized two-armed phase II clinical trial to assess the objective response rate (ORR) by RECIST (Response Evaluation Criteria in Solid Tumors) 1.0 to IPI-504 monotherapy in patients with advanced NSCLC who either had an activating EGFR mutation or were EGFR wild-type.18 Each genotype-defined arm of the trial functioned as a Simon two-stage study with planned interim evaluation after 10 patients and expanded enrollment of an additional 19 patients if there was at least one partial response (PR) or stable disease lasting ≥ 3 months, which was achieved for both arms. While available tissue for EGFR analysis was mandatory, completed EGFR genotype was not required at study entry, thus the trial remained open until both cohorts fully enrolled, which led to overenrollment of the wild-type arm. Secondary aims included describing the safety and progression-free survival (PFS) of the regimen, and examining molecular markers associated with response.
Patients were recruited between December 2007 and May 2009 from 10 United States cancer centers. To be eligible, patients had to have stage IIIB (with pleural effusion), or stage IV NSCLC with progression on EGFR TKI therapy at some point in their history; adequate renal, hepatic, and bone marrow function; Eastern Cooperative Oncology Group performance status of 0 to 2; measurable disease by RECIST 1.0; no active or untreated CNS metastases; no significant cardiac conduction abnormalities (based on findings from similar compounds) or ongoing keratoconjunctivitis (based on nonclinical findings with an oral IPI-504 formulation); and either previously defined EGFR genotype or sufficient tumor tissue to undergo genotype assessment.18,19 There was no limit on prior therapies. All patients signed written informed consent and the study was monitored by all local institutional review boards. Funding for the trial was provided by Infinity Pharmaceuticals Inc.
Treatment and Evaluation
Treatment consisted of a 30-minute infusion of intravenous IPI-504 on days 1, 4, 8, and 11 of a 21-day cycle. Therapy continued until progressive disease, intolerable adverse effects, or elective withdrawal. A total of 76 patients were enrolled. The starting dose was 400 mg/m2 for 75 patients. In April 2009, the dose for patients who were on study (n = 19) was lowered to 225 mg/m2, due to hepatotoxicities observed at the 400 mg/m2 dose in a separate trial of IPI-504 in patients with GI stromal tumors,20 and the last enrolled patient started at a dose of 225 mg/m2.
All patients were assessed for safety by history, physical examination, blood chemistries, liver function tests, and blood counts, at baseline and before each infusion. Amylase and lipase were assessed if there were symptoms of pancreatitis. Adverse events were graded using the National Cancer Institute Common Toxicity Criteria version 3.0. Slit lamp eye examinations were performed during screening and ECGs were obtained during screening and before and after the first infusion, to evaluate for keratitis and QTc prolongation, respectively. Radiographic evaluation of tumor response by computed tomography scan was performed after every two cycles. All films were reviewed and independently assessed by a central radiology core laboratory.
Patient Tumor Molecular Analyses
Tumor tissue specimens from all patients were assessed for EGFR mutations via direct sequencing of exons 18 to 21, using standard methods, at participating institutions' CLIA-certified internal laboratories or else at Genzyme Corporation (Cambridge, MA). A subset of patients also underwent EGFR (n = 25), KRAS (n = 30), and BRAF (n = 5) genotyping analyses with allele-specific amplification-refractory mutation system assays (DxS, United Kingdom) at Infinity Pharmaceuticals, Inc (Cambridge, MA). Patients who underwent successful EGFR testing via both methods were classified using the result of the more sensitive assay (allele-specific amplification-refractory mutation system).
Posthoc analyses of other molecular markers of interest were performed in all patients for whom sufficient tissue was available. The primary analyses were performed in a CLIA-certified laboratory at Massachusetts General Hospital and consisted of the SNaPshot assay (Applied Biosystems, Foster City, CA), adapted to detect key oncogenic mutations in EGFR, KRAS, PIK3CA, BRAF, PTEN, AKT, TP53, NRAS, CTNNB1 (beta-catenin), APC, KIT, JAK2, NOTCH1, and FLT3 (n = 21); and the fluorescent in situ hybridization break-apart assay for detection of ALK gene rearrangements, using methods previously described (n = 15).10,21 Other analyses included genotyping by Oncomap analysis (Dana-Farber Cancer Institute, Boston, MA) covering 1,155 mutations in 114 cancer genes (n = 10). In addition, coding exons for 11 genes (ALK exons 20-29, BRAF exon 15, EGFR, ERBB2, HSP90AA1, HSP90AB1, KRAS, MET, NF1, PTEN, and STK11) were sequenced by the Sanger method at Functional Biosciences Inc (Madison, WI; n = 12). Ten potential mutations were then validated by the Sanger method at Genewiz (South Plainfield, NJ).
Western Blot Analyses
H1975 (EGFR L858R/T790M), HCC827 (EGFR del 19), H3122 (EML4-ALK), and MGH006 (EML4-ALK derived from a patient who was sensitive to the ALK inhibitor PF-02341066) cells were treated with increasing doses of 17-AAG for 24 hours. Western blotting was performed using previously described methods.22 Membranes were probed with antibodies against P-ALK (Cell Signaling Technology, Beverly, MA), ALK (Cell Signaling Technology), P-EGFR (Biosource, Carlsbad, CA), and EGFR (Santa Cruz Biotechnology Inc, Santa Cruz, CA). Chemiluminescence was detected using the Syngene G:Box camera (Synoptics, Cambridge, UK).
Cell Survival Assays
Cells were seeded at 2,000 cells per well of a 96-well plate. After overnight incubation, the cells were treated in sextuplet with serial dilutions of 17-AAG for 72 hours. Viable cell titer relative to untreated cells was determined using Syto60 assays as previously described.22
Statistical Considerations
The primary end point of the study was ORR, calculated as the sum of patients with confirmed CR or PR divided by the number of treated patients. Each arm (EGFR mutant and wild-type) was analyzed independently. The study was powered assuming a null ORR of 5% and a target ORR of 20%.
Summary statistics were used to describe safety and included all patients treated with IPI-504. PFS was defined as the time from enrollment to progressive disease or death, censored at the last known follow-up, and was calculated with the Kaplan-Meier method, following the intent-to-treat principle.
RESULTS
Patients
Seventy-six patients were enrolled. EGFR genotype analysis did not need to be completed before initiation of treatment; consequently eight patients (10%) with indeterminate genotype were not assigned to either the EGFR mutant or wild-type arms. The median age was 64 years (range, 31 to 82 years) and was similar between the genotypes (Table 1). The entire study population had an over-representation of women (63%) and never-smokers (45%), which was even more pronounced among EGFR mutants (71% women, 61% never-smokers). The study cohort was also heavily pretreated with a median of four prior systemic regimens and a median time since diagnosis of 27.5 months. Prior EGFR TKI therapy had yielded a 54% response rate and lasted a median of 10.5 months among EGFR mutation–positive patients.
Table 1.
Demographics by EGFR, KRAS, and ALK Genotype
| Demographic | Total |
EGFR Status (n = 68) |
KRASStatus (n = 38) |
ALK Status (n = 15) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Wild Type |
Mutant |
Wild Type |
Mutant |
Wild Type |
Rearranged |
|||||||||
| No. | % | No. | % | No. | % | No. | % | No. | % | No. | % | No. | % | |
| No. of patients | 76 | 40 | 28 | 26 | 12 | 12 | 3 | |||||||
| Median age, years | 64.0 | 63.0 | 66.0 | 61.0 | 65.0 | 65.5 | 48.0 | |||||||
| Range | 31-82 | 31-79 | 44-82 | 31-81 | 52-76 | 48-76 | 31-58 | |||||||
| Sex | ||||||||||||||
| Female | 48 | 63 | 22 | 55 | 20 | 71 | 17 | 65 | 7 | 58 | 9 | 75 | 1 | 33 |
| Male | 28 | 37 | 18 | 45 | 8 | 29 | 9 | 35 | 5 | 42 | 3 | 25 | 2 | 67 |
| Race | ||||||||||||||
| Asian | 11 | 14 | 6 | 15 | 5 | 18 | 4 | 15 | 0 | 2 | 17 | 1 | 33 | |
| Black or African American | 4 | 5 | 2 | 5 | 2 | 7 | 2 | 8 | 0 | 0 | 0 | |||
| White | 61 | 80 | 32 | 80 | 21 | 75 | 20 | 77 | 12 | 100 | 10 | 83 | 2 | 67 |
| Smoking status | ||||||||||||||
| Current smoker | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |||||||
| Never-smoker | 34 | 45 | 13 | 33 | 17 | 61 | 13 | 50 | 0 | 3 | 25 | 3 | 100 | |
| Previous smoker | 42 | 55 | 27 | 68 | 11 | 39 | 13 | 50 | 12 | 100 | 9 | 75 | 0 | |
| Median time since diagnosis, months | 27.5 | 24.6 | 37.2 | 25.7 | 20.6 | 28.5 | 29.7 | |||||||
| Range | 8-120 | 8-120 | 11-108 | 10-120 | 11-71 | 11-71 | 10-120 | |||||||
| Histology | ||||||||||||||
| Adenocarcinoma | 59 | 78 | 31 | 78 | 23 | 82 | 21 | 81 | 10 | 83 | 11 | 92 | 3 | 100 |
| Bronchioloalveolar | 4 | 5 | 2 | 5 | 2 | 7 | 0 | 1 | 8 | 0 | 0 | |||
| Large cell | 2 | 3 | 2 | 5 | 0 | 1 | 4 | 1 | 8 | 0 | 0 | |||
| Squamous | 6 | 8 | 4 | 10 | 1 | 4 | 3 | 12 | 0 | 1 | 8 | 0 | ||
| Unspecified NSCLC | 5 | 7 | 1 | 3 | 2 | 7 | 1 | 4 | 0 | 0 | 0 | |||
| Median No. of prior treatment regimens for NSCLC | 4.0 | 4.0 | 3.0 | 3.0 | 3.5 | 4.0 | 3.0 | |||||||
| Range | 1-11 | 1-7 | 1-11 | 1-6 | 2-7 | 2-7 | 3-5 | |||||||
| Best prior response to EGFR TKI treatment | ||||||||||||||
| CR | 1 | 1 | 0 | 1 | 4 | 1 | 4 | 0 | 0 | 0 | ||||
| PR | 18 | 24 | 2 | 5 | 14 | 50 | 3 | 12 | 1 | 8 | 1 | 8 | 0 | |
| Total time on EFGR TKI prior to study, months | ||||||||||||||
| Median | 1.8 | 1.5 | 10.5 | 1.7 | 1.2 | 1.9 | 0.0 | |||||||
| Range | 0-61 | 0-25 | 0-61 | 0-61 | 0-16 | 0-16 | 0-1 | |||||||
NOTE. Patients may be counted in more than one column dependent upon molecular analysis.
Abbreviations: EGFR, epidermal growth factor receptor; NSCLC, non–small-cell lung cancer; TKI, tyrosine kinase inhibitor; CR, complete response; PR, partial response.
Toxicity
IPI-504 was generally well-tolerated. Most adverse events were grades 1 or 2; nine patients (12%) had dose reductions for toxicity, while 11 (14%) discontinued therapy for adverse events. The most commonly reported adverse events (regardless of relationship to drug) were fatigue, nausea, diarrhea, vomiting, cough, anorexia, and joint/muscle aches (Table 2). About one third of patients had transient, nontoxic purple-colored urine due to renal clearance of an IPI-504 chromometabolite. In terms of laboratory abnormalities, AST, ALT, and alkaline phosphatase elevations were common (49%, 41%, and 62%, respectively), but grade 3 or greater elevations were infrequent (9%, 7%, and 5%, respectively). No grade 3 or 4 bilirubin elevation was noted. Three patients died while on study. Two patients died of complications from pneumonia, including sepsis and respiratory distress, which were considered possibly related to study drug; both had renal failure and/or elevations in ALT and AST. The third patient died of respiratory failure, which was considered unrelated to study drug.
Table 2.
Most Frequent Adverse Events Regardless of Causality
| Parameter | Patients With |
|||||
|---|---|---|---|---|---|---|
| Any Event |
Grade 1 or 2 Event |
≥ Grade 3 Event |
||||
| No. | % | No. | % | No. | % | |
| MedDRA preferred term | ||||||
| Fatigue | 44 | 57.9 | 41 | 53.9 | 6 | 7.9 |
| Nausea | 43 | 56.6 | 41 | 53.9 | 6 | 7.9 |
| Diarrhea | 40 | 52.6 | 37 | 48.7 | 8 | 10.5 |
| Vomiting | 28 | 36.8 | 25 | 32.9 | 6 | 7.9 |
| Cough | 24 | 31.6 | 24 | 31.6 | 2 | 2.6 |
| Urine color abnormal | 22 | 28.9 | 22 | 28.9 | 0 | 0.0 |
| Anorexia | 19 | 25.0 | 18 | 23.7 | 4 | 5.3 |
| Arthralgia | 19 | 25.0 | 17 | 22.4 | 2 | 2.6 |
| Myalgia | 19 | 25.0 | 18 | 23.7 | 1 | 1.3 |
| Headache | 19 | 25.0 | 19 | 25.0 | 0 | 0.0 |
| Abdominal pain | 18 | 23.7 | 18 | 23.7 | 1 | 1.3 |
| Constipation | 18 | 23.7 | 18 | 23.7 | 2 | 2.6 |
| Dyspnea | 18 | 23.7 | 15 | 19.7 | 6 | 7.9 |
| Back pain | 16 | 21.1 | 16 | 21.1 | 0 | 0.0 |
| Infusion site pain | 15 | 19.7 | 15 | 19.7 | 0 | 0.0 |
| Dehydration | 14 | 18.4 | 11 | 14.5 | 3 | 3.9 |
| Musculoskeletal chest pain | 13 | 17.1 | 11 | 14.5 | 3 | 3.9 |
| Pyrexia | 12 | 15.8 | 12 | 15.8 | 0 | 0.0 |
| Vision blurred | 12 | 15.8 | 12 | 15.8 | 0 | 0.0 |
| Insomnia | 12 | 15.8 | 12 | 15.8 | 0 | 0.0 |
| Dizziness | 12 | 15.8 | 12 | 15.8 | 0 | 0.0 |
| Liver function tests* | ||||||
| Any abnormality† | 52 | 68.4 | 43 | 56.6 | 9 | 11.8 |
| Alkaline phosphatase | 47 | 61.8 | 43 | 56.6 | 4 | 5.3 |
| AST | 37 | 48.7 | 30 | 39.5 | 7 | 9.2 |
| ALT | 31 | 40.8 | 26 | 34.2 | 5 | 6.6 |
| Total bilirubin | 3 | 3.9 | 3 | 3.9 | 0 | 0.0 |
Maximum post-baseline grade, based on laboratory values.
Any abnormality includes abnormalities in alkaline phosphatase, AST, ALT, and/or total bilirubin.
Response and Molecular Analyses
Sixty-eight patients (89%) had successful EGFR genotype analyses, with 28 patients (37% of the 76 enrolled) assigned to the EGFR mutation–positive arm and 40 (53%) to the wild-type arm. Of the EGFR mutant patients, 17 (61%) had exon 19 deletions, six (21%) had the exon 21 L858R point mutation, one (4%) had exon 20 insertions, and four (14%) had two mutations each (two patients with exon 18 G719S and exon 21 L861Q mutations; two with exon 19 deletion and exon 20 T790M mutations). Note that both patients with a T790M mutation were genotyped from biopsies obtained after EGFR TKI treatment. Reasons for indeterminate EGFR genotype included insufficient tumor tissue or poor quality of available tissue. Thirty-eight patients (50%) underwent KRAS mutation testing and 12 (16%) had a mutation; 15 (20%) underwent ALK rearrangement testing and three (4%) were positive (Appendix Fig A1, online only). Demographics of the KRAS mutation–positive patients were notable for a positive smoking history, and those of the ALK-rearranged patients were notable for young age, male predominance, and never-smoking (Table 1).
The ORR to IPI-504 was 7% (five of 76) overall, 10% (four of 40) in EGFR wild-type patients and 4% (one of 28) in EGFR mutants. The median duration of response was 120 days. Four of the five responses were confirmed with a follow-up scan. The EGFR mutation–positive patient with a RECIST PR had an L858R mutation, had previously had a PR to the combination of erlotinib and enzastaurin lasting approximately 8 months, and transitioned directly from erlotinib to IPI-504. Responses were also seen in three (12%) of the 26 KRAS wild-type patients, in one (8%) of the 12 patients known to be ALK wild-type, and in two (67%) of the three patients with an ALK rearrangement (Table 3, Figs 1, 2A). Note that two of the three KRAS wild-type responders had ALK rearrangement, but the third was confirmed ALK wild-type. The single patient that started treatment at 225 mg/m2 had 5% tumor shrinkage.
Table 3.
Efficacy
| Parameter | Total |
EGFR Status (n = 68) |
KRAS Status (n = 38) |
ALK Status (n = 15) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Wild Type |
Mutant |
Wild Type |
Mutant |
Wild Type |
Rearranged |
|||||||||
| No. | % | No. | % | No. | % | No. | % | No. | % | No. | % | No. | % | |
| No. of patients | 76 | 40 | 53 | 28 | 37 | 26 | 34 | 12 | 16 | 12 | 16 | 3 | 4 | |
| Objective response rate (all PRs) | 5 | 7 | 4 | 10 | 1 | 4 | 3* | 12 | 0 | 0 | 1 | 8 | 2 | 67 |
| RECIST stable disease or better for at least 3 months | 18 | 24 | 10 | 25 | 6 | 21 | 4 | 15 | 5 | 42 | 3 | 25 | 3 | 100 |
| Median PFS, months | 2.86 | 2.86 | 2.76 | 2.86 | 3.91 | 2.43 | ||||||||
| 95% CI | 2.43-4.18 | 1.18-5.33 | 2.40-3.91 | 1.22-10.20 | 1.12-4.18 | 1.13-5.33 | Unable to determine | |||||||
Abbreviations: PR, partial response; RECIST, Response Evaluation Criteria in Solid Tumors; PFS, progression-free survival.
Two of the three KRASwild-type responders were ALK rearranged; the third was confirmed ALK wild-type.
Fig 1.
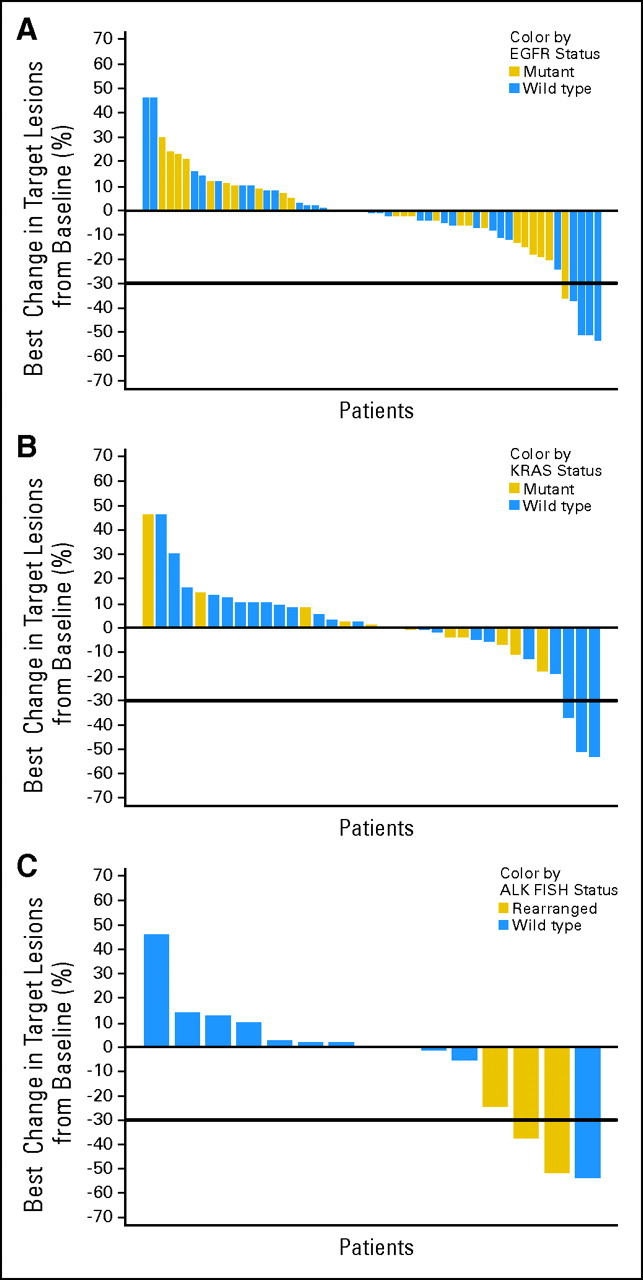
Best percent change in size of target lesions on study. Percent change in measurable tumor at best response is displayed by genotype. (A) Epidermal growth factor receptor (EGFR) mutation status, (B) KRAS mutation status, and (C) ALK rearrangement status.
Fig 2.
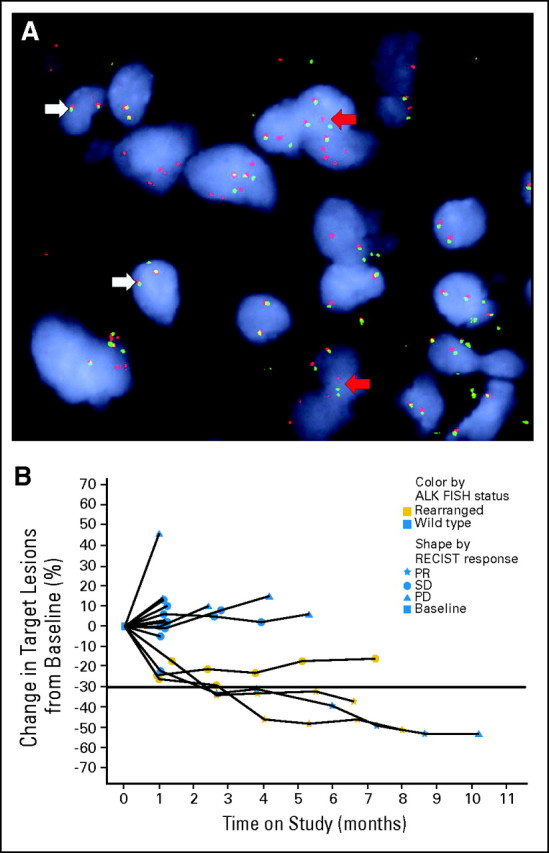
Patients with ALK rearrangements on study. (A) Example of a positive fluorescent in situ hybridization (FISH) break-apart assay in a patient with ALK rearrangement. White arrows indicate the wild-type allele with close proximity of the red and green probes yielding a yellow signal. Red arrows indicate the ALK rearrangement with separated red and green probes. (B) Change in size of target lesions over time for patients tested for ALK rearrangement. RECIST, Response Evaluation Criteria in Solid Tumors; PR, partial response; SD, stable disease; PD, progressive disease.
At the time of analysis, 35 patients (46%) had a PFS event (progression or death), and 41 (54%) were censored. The estimated median PFS for all patients was 2.86 months (95% CI, 2.43 to 4.18 months), although the three patients with ALK rearrangements each received IPI-504 for approximately 7 months (Fig 2B). Additional genetic results from SNaPshot, Oncomap, DxS genotyping, and Sanger sequencing are summarized in Appendix Table A1.
Laboratory assessments of lung cancer models harboring EGFR mutations and ALK rearrangements confirmed that the EGFR mutant models were sensitive to Hsp90 inhibition with 17-AAG, as previously demonstrated, but also revealed that ALK-rearranged models were highly sensitive. Indeed, both the stability of the ALK-rearranged protein and the viability of the cancer were highly sensitive to Hsp90 inhibition (Fig 3).
Fig 3.

ALK rearranged cancer cell lines are sensitive to heat-shock protein 90 (Hsp90) inhibitors. (A) Cancer cell lines with epidermal growth factor receptor (EGFR) mutations (H1975, L858R/T790M, and HCC827 exon 19 deletion) or ALK gene rearrangement (H3122 and MGH-006) were treated with increasing doses of the Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin (17-AAG) for 24 hours. Protein lysates were probed with the indicated antibodies. (B) NCI-H1975, HCC827, MGH-006, and NCI-H3122 cells were treated with increasing doses of 17-AAG for 72 hours. Total cell viability was determined by Syto60 assay. Data are presented as the percent of viable cells relative to cells grown in the absence of drug.
DISCUSSION
To our knowledge, this is the first trial of an Hsp90 inhibitor in molecularly defined cohorts of patients with advanced NSCLC. We have demonstrated that IPI-504 is active in NSCLC, with a response rate of 7% (five of 76) in the overall study population, 10% (four of 40) in patients who were EGFR wild-type, 4% (one of 28) in EGFR mutants with acquired resistance to TKIs, 12% (three of 26) among KRAS wild-type patients, and 8% (one of 12) among ALK wild-type patients. The most intriguing finding is the posthoc analysis demonstrating that two of three patients known to have ALK rearrangements had a PR to IPI-504 and the third patient had stable disease (24% reduction) durable for 7.2 months. This is the first clinical demonstration of activity of an Hsp90 inhibitor in patients with ALK rearrangements.
ALK is a member of the insulin superfamily of receptor tyrosine kinases and was initially associated with anaplastic large-cell lymphoma, which commonly has ALK oncogenic signaling mediated by fusion between the ALK kinase domain and the partner protein nucleophosmin.23 More recently, EML4-ALK and other rearrangements involving the ALK locus have been described in NSCLC as transforming driver mutations conferring sensitivity to therapy with ALK TKIs.10,14,24 Preclinical models have demonstrated that nucleophosmin ALK is a client of Hsp90,25 and our data indicate that EML4-ALK is also a key client (Fig 3).
Overall, the study validates the hypothesis that an Hsp90 inhibitor, by virtue of the chaperone role of Hsp90 for multiple oncoproteins and its pervasive effect on key signaling pathways, has the potential to be an effective cancer therapy against multiple types of oncogene-addicted cancers, including those that have developed resistance to receptor-specific targeted treatments. TKIs that inhibit driver mutations in such cancers have been extremely effective, including imatinib in chronic myelogenous leukemia and gastrointestinal stromal tumors (targets BCR-ABL and CKIT, respectively), gefitinib and erlotinib in NSCLC (targets EGFR), and potentially PF-02341066 in NSCLC (targets ALK).11,14,26,27 Our study confirms that inhibition of a driver mutation need not be via a receptor-specific molecule in order to be effective.
It is notable that despite extensive preclinical evidence that Hsp90 inhibition, and specifically IPI-504 treatment, leads to effective cell killing and tumor regression in EGFR mutation–positive models, including those with acquired resistance to EGFR TKIs, we saw few responses in patients with EGFR mutations.28–30 There could be several reasons for this observation. Our population of patients was particularly atypical in that the median time from diagnosis among patients with EGFR mutations was 2 years and 54% had been treated with at least two prior EGFR TKI agents. Since their cancers had become resistant to EGFR TKIs, the biology of their tumors may have changed from being dependent on a single oncogene to a more heterogeneous state. The dose of IPI-504 could also have been a factor in the modest response rate among EGFR mutants. Our analyses of cancer cell lines suggest that lower concentrations of Hsp90 inhibitors may be required to downregulate expression of EML4-ALK compared with mutant EGFR. The dose-response curve for EGFR mutant cancers was modestly shifted to the right compared with the dose-response curve for ALK-rearranged cancers. This potentially wider therapeutic window may possibly have contributed to the higher response rate observed in the patients with ALK rearrangements. Importantly, the lack of acquired resistance to ALK-specific therapy among the patients with ALK rearrangements may imply a discrete molecular biology that was more susceptible to Hsp90 inhibition than the patients with EGFR mutations, all of whom had previously received and acquired resistance to EGFR TKIs. None of the patients on our trial (regardless of genotype) had previously been treated with ALK-specific inhibitors.
In our study, IPI-504 was generally safe and tolerable, with low rates of grade 3 or higher adverse events. The most common adverse events included nausea, fatigue, and diarrhea, and these were mostly grades 1 and 2. Grade 3 or higher liver function abnormalities were observed in nine patients (11.8%), and drug-related deaths were complicated by patients' underlying lung cancer.
Limitations of our study include its relatively small size and the atypical population studied, which may affect the generalizability of the results. Both the EGFR mutation–positive and wild-type cohorts had a long interval since diagnosis, a high number of prior therapies, and a low proportion of smokers. Furthermore, tumor tissue available for genetic analysis was primarily from diagnostic biopsies, before any targeted therapy or development of resistance that might have altered the genetic signature. However, the fact that tumor tissue for genotyping was collected from 100% of participants due to eligibility mandate was important. Not only did this allow us to make specific observations regarding response by EGFR genotype, we were able to carefully examine the minority of patients with robust responses, enabling the key observation of activity in ALK-rearranged NSCLC. We believe that all studies of targeted therapies should require tissue from all participants. It is not uncommon for studies of novel agents to show activity among only a minority of subjects, and our study effectively illustrates how posthoc molecular analysis of the best responding patients can provide direction for avenues of further research. Of note, we confirmed ALK rearrangement in our patients with the current standard break-apart fluorescent in situ hybridization assay that detects rearrangement in chromosome 2, but does not identify the specific variant of EML4-ALK present (EML4 has multiple break points at which it can partner with ALK).31 Therefore, we do not currently know if IPI-504 has a range of expected activity dependent on the oncogenic EML4-ALK variant.
In summary, although the predefined end point of 20% response was not observed in either EGFR-genotyped cohort, the novel Hsp90 inhibitor IPI-504 has activity in NSCLC, in particular among patients with ALK rearrangements. Additional study is required to prospectively evaluate the efficacy of Hsp90 inhibition in patients with ALK rearrangements and other oncogenic driver mutations.
Acknowledgment
We thank all of the patients who participated in the study, their families, and all the physicians and study staff involved; and Barbara Thomson for valuable editing and manuscript assistance.
Appendix
Fig A1.

Epidermal growth factor receptor (EGFR), KRAS, and ALK status for all samples. All patient genotyping for EGFR, KRAS, and ALK status including partial responses (PR) as noted. WT, wild-type; MUT, mutation; UNK, unknown.
Table A1.
Individual Patient Mutational Results
| Patient ID | EGFR Sequencing* | DxS EGFR† | DxS KRAS† | DxS BRAF† | Oncomap‡ | SNaPshot§ | Other Sanger Sequencing‖ | ALK FISH¶ |
|---|---|---|---|---|---|---|---|---|
| 1 | Mutant (D) | Mutant (D) | Wild type | EGFR and PIK3CA | NF1 | |||
| 2 | Mutant (D) | Mutant (D) | Wild type | EGFR | Wild type | |||
| 3 | Mutant (D) | Mutant (D) | ||||||
| 4 | Mutant (L) | Mutant (L) | ||||||
| 5 | Mutant (O) | Wild type | EGFR | EGFR and KRAS | Wild type | |||
| 6 | Mutant (D) | EGFR and TP53 | Wild type | |||||
| 7 | Mutant (D,O) | |||||||
| 8 | Mutant (D) | |||||||
| 9 | Mutant (L) | |||||||
| 10 | Mutant (D) | |||||||
| 11 | Mutant (D) | |||||||
| 12 | Mutant (D) | |||||||
| 13 | Mutant (D) | |||||||
| 14 | Mutant (D) | |||||||
| 15 | Mutant (L) | |||||||
| 16 | Mutant (L) | |||||||
| 17 | Mutant (D) | |||||||
| 18 | Mutant (D) | |||||||
| 19 | Mutant (D) | |||||||
| 20 | Mutant (D) | |||||||
| 21 | Mutant (L) | |||||||
| 22 | Mutant (D,O) | EGFR | ||||||
| 23 | Mutant (L) | |||||||
| 24 | Mutant (D) | |||||||
| 25 | Wild type | Mutant (O) | Wild type | Wild type | EGFR | |||
| 26 | Wild type | Mutant (D) | Wild type | |||||
| 27 | Wild type | Wild type | Mutant | Wild type | KRAS | Wild type | ||
| 28 | Wild type | Wild type | Mutant | |||||
| 29 | Wild type | Wild type | Mutant | |||||
| 30 | Wild type | Wild type | Wild type | Wild type | Wild type | Wild type | Wild type | Wild type |
| 31 | Wild type | Wild type | Wild type | Wild type | Wild type | TP53 | Wild type | Wild type |
| 32 | Wild type | Wild type | Wild type | Wild type | Wild type | Wild type | Wild type | Rearrangement detected |
| 33 | Wild type | Wild type | Wild type | Wild type | Wild type | CTNNB1 | Wild type | Rearrangement detected |
| 34 | Wild type | Wild type | Wild type | |||||
| 35 | Wild type | Wild type | Wild type | |||||
| 36 | Wild type | Wild type | Wild type | Wild type | Wild type | |||
| 37 | Wild type | Wild type | Wild type | |||||
| 38 | Wild type | Wild type | Wild type | |||||
| 39 | Wild type | Wild type | Wild type | |||||
| 40 | Wild type | Wild type | Wild type | |||||
| 41 | Wild type | Wild type | Wild type | Wild type | ||||
| 42 | Wild type | Wild type | Wild type | |||||
| 43 | Wild type | Wild type | ||||||
| 44 | Wild type | Mutant | KRAS | KRAS | Wild type | Wild type | ||
| 45 | Wild type | Mutant | KRAS | KRAS | Wild type | Wild type | ||
| 46 | Wild type | Mutant | KRAS | KRAS | Wild type | |||
| 47 | Wild type | Mutant | ||||||
| 48 | Wild type | Mutant | ||||||
| 49 | Wild type | Wild type | Wild type | |||||
| 50 | Wild type | Wild type | ||||||
| 51 | Wild type | Wild type | ||||||
| 52 | Wild type | Wild type | Wild type | |||||
| 53 | Wild type | Wild type | Wild type | |||||
| 54 | Wild type | KRAS | Wild type | |||||
| 55 | Wild type | KRAS | Wild type | |||||
| 56 | Wild type | KRAS | Wild type | |||||
| 57 | Wild type | Rearrangement detected | ||||||
| 58 | Wild type | |||||||
| 59 | Wild type | |||||||
| 60 | Wild type | |||||||
| 61 | Wild type | |||||||
| 62 | Wild type | |||||||
| 63 | Wild type | |||||||
| 64 | Wild type | |||||||
| 65 | Wild type | |||||||
| 66 | Wild type | |||||||
| 67 | Mutant (O) | Wild type | ||||||
| 68 | Mutant (D) | |||||||
| 69 | Wild type | Wild type | ||||||
| 70 | ||||||||
| 71 | ||||||||
| 72 | ||||||||
| 73 | ||||||||
| 74 | ||||||||
| 75 | ||||||||
| 76 |
NOTE. Blank cell indicates test was not performed or failed.
Abbreviations: EGFR, epidermal growth factor receptor; FISH, fluorescence in situ hybridization; Mutant (D), mutation based upon an exon 19 deletion; Mutant (L), L858R mutation; Mutant (O), other mutation; Mutant (D,O), mutation based upon an exon 19 deletion and another mutation.
EGFR sequencing was performed at participating institutions' CLIA-certified internal laboratories or at Genzyme.
EGFR, KRAS, and BRAF genotyping analysis with the allele-specific ARMS assay (DxS, United Kingdom) was performed at Infinity Pharmaceuticals, Inc (Cambridge, MA).
Oncomap analysis was performed at the Dana-Farber Cancer Institute (Boston, MA) covering 1,155 mutations in 114 cancer genes.
SNaPshot assay (Applied Biosystems, Foster City, CA), adapted to detect key oncogenic mutations in EGFR, KRAS, PIK3CA, BRAF, PTEN, AKT, TP53, NRAS, Beta-catenin, NOTCH, and FLT3 was performed at a CLIA-certified laboratory at Massachusetts General Hospital.
Sanger sequencing of ALK exons 20 to 29, BRAFexon 15, EGFR, ERBB2, HSP90AA1, HSP90AB1, KRAS, MET, NF1, PTEN, and STK11was performed at Functional Biosciences Inc (Madison, WI). Ten potential mutations were then validated by the Sanger method at Genewiz (South Plainfield, NJ).
FISH break-apart assay for detection of ALK gene rearrangements was performed at a CLIA-certified laboratory at Massachusetts General Hospital.
Footnotes
Supported by Infinity Pharmaceuticals Inc.
Presented in part in abstract format at the 45th Annual Meeting of the American Society of Clinical Oncology, May 29-June 2, 2009, Orlando, FL, and the 46th Annual Meeting of the American Society of Clinical Oncology, June 4-8, 2010, Chicago, IL.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
Clinical trial information can be found for the following: NCT00431015.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: Nafeeza Hafeez, Infinity Pharmacueticals (C); Jennifer Sweeney, Infinity Pharmaceuticals (C); John R. Walker, Infinity Pharmaceuticals (C); Christian Fritz, Infinity Pharmaceuticals (C); Robert W. Ross, Infinity Pharmaceuticals (C); David Grayzel, Infinity Pharmaceuticals (C) Consultant or Advisory Role: Pasi A. Jänne, Aveo Pharmaceuticals (U), Roche (U), AstraZeneca (U), Boehringer Ingelheim (C), Aveo Pharmaceuticals (C), Pfizer (U); Jeffrey A. Engelman, Novartis (C) Stock Ownership: Pasi A. Jänne, Gatekeeper Phamaceuticals; Nafeeza Hafeez, Infinity Pharmaceuticals; Jennifer Sweeney, Infinity Pharmaceuticals; John R. Walker, Infinity Pharmaceuticals; Christian Fritz, Infinity Pharmaceuticals; Robert W. Ross, Infinity Pharmacueticals; David Grayzel, Infinity Pharmaceuticals; Guillermo Paez, Infinity Pharmaceuticals Honoraria: Renato G. Martins, Eli Lilly, Genentech Research Funding: Lecia V. Sequist, Infinity Pharmaceuticals; Scott Gettinger, Infinity Pharmaceuticals; Renato G. Martins, OSI Pharmaceuticals, Eisai, Genentech, Novartis, Pfizer, Amgen, Bayer Pharmaceuticals, Eli Lilly; Pasi A. Jänne, Pfizer; Rogerio Lilenbaum, Infinity Pharmaceuticals; Ronald Natale, Amgen, Eli Lilly, Millennium Pharmaceuticals, Novartis Expert Testimony: None Other Remuneration: Pasi A. Jänne, Genzyme
AUTHOR CONTRIBUTIONS
Conception and design: Lecia V. Sequist, Pasi A. Jänne, John R. Walker, Christian Fritz, Robert W. Ross, David Grayzel
Financial support: Robert W. Ross
Administrative support: Robert W. Ross
Provision of study materials or patients: Lecia V. Sequist, Scott Gettinger, Neil N. Senzer, Renato G. Martins, Pasi A. Jänne, Rogerio Lilenbaum, Jhanelle E. Gray, Robert W. Ross
Collection and assembly of data: Lecia V. Sequist, Scott Gettinger, Renato G. Martins, Pasi A. Jänne, Jhanelle E. Gray, A. John Iafrate, Ryohei Katayama, Nafeeza Hafeez, Jennifer Sweeney, John R. Walker, Robert W. Ross, Jeffrey A. Engelman, Guillermo Paez
Data analysis and interpretation: Lecia V. Sequist, Pasi A. Jänne, A. John Iafrate, Ryohei Katayama, Nafeeza Hafeez, John R. Walker, Christian Fritz, Robert W. Ross, David Grayzel, Jeffrey A. Engelman, Darrell R. Borger, Guillermo Paez
Manuscript writing: Lecia V. Sequist
Final approval of manuscript: Lecia V. Sequist
REFERENCES
- 1.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 2.Xu W, Neckers L. Targeting the molecular chaperone heat shock protein 90 provides a multifaceted effect on diverse cell signaling pathways of cancer cells. Clin Cancer Res. 2007;13:1625–1629. doi: 10.1158/1078-0432.CCR-06-2966. [DOI] [PubMed] [Google Scholar]
- 3.Workman P, Burrows F, Neckers L, et al. Drugging the cancer chaperone HSP90: Combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann N Y Acad Sci. 2007;1113:202–216. doi: 10.1196/annals.1391.012. [DOI] [PubMed] [Google Scholar]
- 4.Nathan DF, Lindquist S. Mutational analysis of Hsp90 function: Interactions with a steroid receptor and a protein kinase. Mol Cell Biol. 1995;15:3917–3925. doi: 10.1128/mcb.15.7.3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rutherford SL, Lindquist S. Hsp90 as a capacitor for morphological evolution. Nature. 1998;396:336–342. doi: 10.1038/24550. [DOI] [PubMed] [Google Scholar]
- 6.Grbovic OM, Basso AD, Sawai A, et al. V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc Natl Acad Sci U S A. 2006;103:57–62. doi: 10.1073/pnas.0609973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shimamura T, Lowell AM, Engelman JA, et al. Epidermal growth factor receptors harboring kinase domain mutations associate with the heat shock protein 90 chaperone and are destabilized following exposure to geldanamycins. Cancer Res. 2005;65:6401–6408. doi: 10.1158/0008-5472.CAN-05-0933. [DOI] [PubMed] [Google Scholar]
- 8.Suda K, Tomizawa K, Mitsudomi T. Biological and clinical significance of KRAS mutations in lung cancer: An oncogenic driver that contrasts with EGFR mutation. Cancer Metastasis Rev. 2010;29:49–60. doi: 10.1007/s10555-010-9209-4. [DOI] [PubMed] [Google Scholar]
- 9.Sharma SV, Bell DW, Settleman J, et al. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 10.Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27:4247–4253. doi: 10.1200/JCO.2009.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 12.Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non–small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 13.Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–128. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 14.Kwak EL, Bang Y-J, Camidge R, et al. Anaplastic lymphoma kinase inhibition in non–small-cell lung cancer. N Engl J Med. doi: 10.1056/NEJMoa1006448. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ge J, Normant E, Porter JR, et al. Design, synthesis, and biological evaluation of hydroquinone derivatives of 17-amino-17-demethoxygeldanamycin as potent, water-soluble inhibitors of Hsp90. J Med Chem. 2006;49:4606–4615. doi: 10.1021/jm0603116. [DOI] [PubMed] [Google Scholar]
- 16.Sydor JR, Normant E, Pien CS, et al. Development of 17-allylamino-17-demethoxygeldanamycin hydroquinone hydrochloride (IPI-504), an anti-cancer agent directed against Hsp90. Proc Natl Acad Sci U S A. 2006;103:17408–17413. doi: 10.1073/pnas.0608372103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sequist LV, Janne PA, Sweeney J, et al. Phase 1/2 trial of the novel Hsp90 Inhibitor, IPI-504, in patients with relapsed and/or refractory stage IIIb or stage IV non-small cell lung cancer (NSCLC) stratified by EGFR mutation status. AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics; October 22-26, 2007; San Francisco, CA. [Google Scholar]
- 18.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors: European Organisation for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 19.Oken MM, Creech RH, Tormey DC, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol. 1982;5:649–655. [PubMed] [Google Scholar]
- 20.Demetri GD, LeCesne A, Von Mehren M, et al. Final results from a phase III study of IPI-504 (retaspimycin hydrochloride) versus placebo in patients with gastrointestinal stromal tumors (GIST) following failure of kinase inhibitor therapies. American Society of Clinical Oncology Gastrointestinal Cancers Symposium; January 22-24, 2010; Orlando, FL. [Google Scholar]
- 21.Dias-Santagada D, Akhavanfard S, David SS, et al. Rapid targeted mutational analysis of human tumors: A clinical platform to guide personalized cancer medicine. EMB Mol Med. 2010;2:146–158. doi: 10.1002/emmm.201000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Faber AC, Li D, Song Y, et al. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc Natl Acad Sci U S A. 2009;106:19503–19508. doi: 10.1073/pnas.0905056106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 1994;263:1281–1284. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- 24.Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 25.Bonvini P, Gastaldi T, Falini B, et al. Nucleophosmin-anaplastic lymphoma kinase (NPM-ALK), a novel Hsp90-client tyrosine kinase: Down-regulation of NPM-ALK expression and tyrosine phosphorylation in ALK(+) CD30(+) lymphoma cells by the Hsp90 antagonist 17-allylamino,17-demethoxygeldanamycin. Cancer Res. 2002;62:1559–1566. [PubMed] [Google Scholar]
- 26.Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038–1042. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 27.Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 28.Shimamura T, Li D, Ji H, et al. Hsp90 inhibition suppresses mutant EGFR-T790M signaling and overcomes kinase inhibitor resistance. Cancer Res. 2008;68:5827–5838. doi: 10.1158/0008-5472.CAN-07-5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sawai A, Chandarlapaty S, Greulich H, et al. Inhibition of Hsp90 down-regulates mutant epidermal growth factor receptor (EGFR) expression and sensitizes EGFR mutant tumors to paclitaxel. Cancer Res. 2008;68:589–596. doi: 10.1158/0008-5472.CAN-07-1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brain J, Slocum K, Cole J, et al. IPI-504, a novel orally administered Hsp90 inhibitor, demonstrates anti-tumor effects in EGFR mutant, kinase inhibitor resistant NSCLC. Annual Meeting of the American Association of Cancer Research; April 14-18, 2007; Los Angeles, CA. [Google Scholar]
- 31.Horn L, Pao W. EML4-ALK: Honing in on a new target in non-small-cell lung cancer. J Clin Oncol. 2009;27:4232–4235. doi: 10.1200/JCO.2009.23.6661. [DOI] [PMC free article] [PubMed] [Google Scholar]