

Abstract
Background
Pines are the most important tree species to the international forestry industry, covering 42 % of the global industrial forest plantation area. One of the most pressing threats to cultivation of some pine species is the pitch canker fungus, Fusarium circinatum, which can have devastating effects in both the field and nursery. Investigation of the Pinus-F. circinatum host-pathogen interaction is crucial for development of effective disease management strategies. As with many non-model organisms, investigation of host-pathogen interactions in pine species is hampered by limited genomic resources. This was partially alleviated through release of the 22 Gbp Pinus taeda v1.01 genome sequence (http://pinegenome.org/pinerefseq/) in 2014. Despite the fact that the fragmented state of the genome may hamper comprehensive transcriptome analysis, it is possible to leverage the inherent redundancy resulting from deep RNA sequencing with Illumina short reads to assemble transcripts in the absence of a completed reference sequence. These data can then be integrated with available genomic data to produce a comprehensive transcriptome resource. The aim of this study was to provide a foundation for gene expression analysis of disease response mechanisms in Pinus patula through transcriptome assembly.
Results
Eighteen de novo and two reference based assemblies were produced for P. patula shoot tissue. For this purpose three transcriptome assemblers, Trinity, Velvet/OASES and SOAPdenovo-Trans, were used to maximise diversity and completeness of assembled transcripts. Redundancy in the assembly was reduced using the EvidentialGene pipeline. The resulting 52 Mb P. patula v1.0 shoot transcriptome consists of 52 112 unigenes, 60 % of which could be functionally annotated.
Conclusions
The assembled transcriptome will serve as a major genomic resource for future investigation of P. patula and represents the largest gene catalogue produced to date for this species. Furthermore, this assembly can help detect gene-based genetic markers for P. patula and the comparative assembly workflow could be applied to generate similar resources for other non-model species.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-2277-7) contains supplementary material, which is available to authorized users.
Keywords: Pinus patula, De novo transcriptome assembly, Genome guided transcriptome assembly, RNA-seq
Background
Pinus species play keystone ecological roles, representing the major component of many forests across all continents [1]. These species are also the predominantly planted trees in the global commercial forestry sector [2]. One of the largest threats to global pine forestry is the pitch canker fungus Fusarium circinatum, especially where susceptible Pinus species are cultivated [3]. Consequent losses caused by this fungus have large economic impacts on commercial forestry [3, 4]. Resistance to F. circinatum varies among Pinus species [5]. Species such as Pinus patula and P. radiata, both of which are important plantation species in southern Africa, are highly susceptible, while species such as P. tecunumanii are more resistant [5]. Very little is known regarding the interaction between F. circinatum and its Pinus hosts at the molecular level. Investigation of the different responses employed by susceptible and resistant hosts, such as P. patula and P. tecunumanii [5], will improve our knowledge of responses necessary for effective defence against F. circinatum.
RNA sequencing (RNA-seq) approaches have opened the way for transcriptome-wide analysis of gene expression [6]. Accurate quantification of gene expression using RNA-seq, however, requires a high quality reference sequence for read mapping. For organisms with well characterised reference genomes, such as Arabidopsis, this requirement is easily met, while organisms lacking a well characterized reference sequence present numerous challenges. Although the P. taeda v1.01 draft genome assembly is available [7], the size and fragmented state of the assembly can limit comprehensive transcriptome analysis [8, 9]. De novo transcriptome assembly can be used to provide a reference sequence for RNA-seq analysis while circumventing potential issues arising from problems in a genome assembly [10]. De novo transcriptome assemblies are available through GenBank and the TreeGenes database [11, 12] for at least ten Pinus species (P. banksiana [13], P. contorta [13], P. lambertiana, P. massoniana, P. monticola [14], P. palustris, P. pinaster, P. radiata, P. sylvestris [15], and P. taeda), at various levels of completion. Of these, the P. taeda transcriptome is the most comprehensive, consisting of data obtained from many different tissues and developmental stages (Mockaitis et al. unpublished).
The aim of this study was to generate a resource for transcriptome profiling in P. patula by assembling the shoot transcriptome of this economically important species. We report a P. patula shoot transcriptome containing 52 112 transcripts, of which 30 844 (60 %) are annotated. This is the largest gene catalogue for P. patula to date and a major genomic resource, which will facilitate functional genomics research in this tropical pine species.
Methods
Plant material
Six month old P. patula seedlings, from a single open pollinated family, were sourced from Top Crop Nursery, South Africa. Seedlings were transferred to, and maintained for the duration of the trial in an environmentally controlled glasshouse at 25–28 °C without supplemental lighting and allowed to acclimatize for two weeks. F. circinatum isolate FCC3579 was cultured on ½ strength potato dextrose agar (½ PDA; Merck) after which spores were harvested by washing with 15 % (v/v) sterile glycerol. Spore concentration was quantified using a haemocytometer and diluted to 5×104 spores/mL by addition of 15 % (v/v) sterile glycerol. Seedlings were inoculated by clipping the apical bud and pipetting 10 μL of diluted spore solution onto the wound [16]. Seedlings inoculated with 10 μL sterile 15 % glycerol served as mock-inoculated control. Shoot tissue was harvested one day post inoculation (dpi) for three biological replicates per group (inoculated and mock-inoculated). Each biological replicate consisted of the top 4 cm of shoot tissue, measured from the wounded apical bud, harvested from 16 seedlings and pooled prior to being frozen using liquid nitrogen. Frozen tissue was stored at −80 °C until use. Disease development was monitored for six weeks post inoculation by measuring lesion and stem length from the wounded apical bud on 52 plants per group. F. circinatum infection was confirmed based on culture morphology on ½ PDA by re-isolation using tissue harvested from inoculated plants 14 dpi.
RNA isolation and sequencing
Frozen samples were homogenised using a high speed grinder (IKA-Werke, Staufen, Germany) and total RNA extracted using a modified version of Lewinsohn’s protocol [17]. Modifications were as follows: All solutions were prepared using diethylpyrolecarbonate (DEPC) treated water. Approximately 5 g homogenised shoot tissue was placed in a 50 mL conical tube containing 150 mg PVP-360 and 300 mg PVPP before adding 15 mL chilled extraction buffer. Tubes were mixed by vortexing, snap frozen in liquid nitrogen and allowed to thaw on ice. Polysaccharides were precipitated by addition of 1/9th volume 3.3 M sodium acetate and 10 % (v/v) absolute ethanol. Nucleic acids were precipitated at −20 °C for 4 h. The pellet produced from ultracentrifugation was re-suspended in 100 μL DEPC treated water and stored at −80 °C until use. Total RNA samples were treated with RNase-free DNaseI enzyme (Qiagen Inc, Valencia, CA) to digest genomic DNA and purified using the RNeasy® MinElute kit (Qiagen) according to the manufacturer’s instructions. Concentration and integrity of purified RNA samples were evaluated using a Bio-Rad Experion™ automated electrophoresis system (Bio-Rad Laboratories, Hercules, CA, USA).
High quality RNA samples (RNA Integrity Number > 7.5) for both groups were sequenced using Illumina HighSeq 2000 instruments (200 bp insert size, 101PE sequencing, 40 million reads per sample; BGI, Hong Kong). Sequence quality of raw RNA-seq data was assessed using FastQC v0.10.1 [18]. Quality trimming and filtering of data was performed using Sickle v1.210 [19] and all unpaired reads were discarded. Short reads (<40 bp) were removed from the filtered RNA-seq reads using SolexaQA LengthSort [20]. The trimmed and filtered read data for all six samples were combined, resulting in Dataset 1. FastUniq v1.1 [21] was used to reduce PCR artefacts from Dataset 1 by removing duplicate reads, resulting in Dataset 2.
Transcriptome assembly
Multiple k-mer de novo transcriptome assembly
De novo transcriptome assembly was performed using three assemblers; Trinity r2013-11-10 [22], SOAPdenovo-Trans v1.04 [23] and Velvet v1.2.10/ Oases v0.2.08 [24, 25]. Assembly with Trinity was performed on both datasets using default parameters [26], except min_contig_length = 350, and repeated on Dataset 1 with the CuffFly parameter included. Trinity was applied to both Dataset 1 and 2 as Trinity allows for duplicate reads, however, SOAPdenovo-Trans and Velvet/Oases assemblers were used on Dataset 2 only, where duplicates were removed. Assembly with SOAPdenovo-Trans was performed on Dataset 2, for eight different k-mer lengths (23, 25, 31, 39, 47, 55, 63 and 71), with the parameters as follows: max_rd_len = 100, rd_len_cutoff = 100, avg_ins = 200, reverse_seq = 0, asm_flags = 3, pair_num_cutoff = 3, map_len = 35, −f and -F. Assembly with Velvet/Oases was performed on Dataset 2, for seven different k-mer lengths (23, 25, 31, 39, 47, 55, and 63), with the parameters as follows: default parameters for velveth; cov_cutoff = 10, ins_length = 200 and read_trkg = yes for velvetg; cov_cutoff = 10, min_pair_count = 5 and min_trans_lgth = 350 for Oases.
P. taeda v1.01 genome guided transcriptome assembly
Trinity genome guided transcriptome assembly was performed on Datasets 1 and 2 using the P. taeda v1.01 draft genome assembly (ca. 14.4 million scaffolds) with a minimum contig length of 350 bp. GSNAP 2014-02-28 (Genomic Short-read Nucleotide Alignment Program) [27] was used to align reads to the reference genome for transcriptome assembly using the following paramters: −-nofails, −-novelsplicing = 1, −-localsplicedist = 250000, −-npaths = 20. Transrate v0.3.1 [27] was used to calculate assembly quality metrics.
Decreasing redundancy across assemblies
The de novo and genome guided transcriptome assemblies were combined to form a redundant over-assembly. The tr2aacds pipeline, from the EvidentialGene package [28], was used to reduce redundancy in the over-assembly by selecting for the ‘optimal’ set of assembled transcripts based on coding potential. The pipeline consists of five steps: (1) prediction of coding DNA sequence (CDS) and amino acid sequences for each transcript, (2) removal of redundant sequences based on coding potential among identical sequences, (3) substring de-replication to remove sequence fragments, (4) clustering of highly similar sequences into loci and (5) classification of transcripts as ‘primary’ or ‘alternate’ and discarding of low scoring ‘drop’ transcripts. The primary assembled transcripts were used for further assessments.
Annotation
Local alignments to the National Centre for Biotechnology Information (NCBI) non-redundant (nr) and plant protein databases were generated for the primary assembled transcripts from the tr2aacds pipeline using uBLAST (Edgar RC, unpublished) [29]. Parameters used for local alignments were: −evalue 1e-10, −weak_evalue 1e-4, −id 0.9, −weak_id 0.8. Local alignment sequence descriptions were used to remove non-pine origin sequences, sequences with significant alignments to prevalent fungal, bacterial, viral and insect sequences, from the assembly to produce the P. patula v1.0 draft transcriptome assembly. Blast2GO® v2.7.2 [30] was used to predict protein domains through InterProScan 5 [31] as well as to perform Gene Ontology (GO) and Enzyme Code (EC) mapping. The P. patula transcriptome GO distribution was compared to the P. taeda v1.01 draft genome annotation using CateGOrizer [32]. Gene family memberships among species were visualized using custom scripts and Venn diagrams (http://bioinformatics.psb.ugent.be/webtools/Venn/).
Identification of orthologous protein groups
Annotated protein sequences for ten different species were retrieved from version 2.5 of the PLAZA protein database [33] and four external proteins sets, from conifer species, were also included (Table 1). The complete set of predicted P. patula v1.0 proteins from the assembled transcriptome were included. Each of the 15 protein sets were clustered to 90 % identity within species and combined. Gene families were identified and annotated for the 442 372 sequences using the approach described in [8]. Pfam domains [34] were assigned to the P. patula sequences using InterProScan 5.7 [31]. Identified gene families unique to P. patula with fewer than 5 members were discarded as these could result from assembly artefacts.
Table 1.
Protein sets used for analysis of orthologous genes
| Source | Species | Total sequences | Clustered sequencesa |
|---|---|---|---|
| Protein sets from PLAZA v2.5 | Arabidopsis thaliana | 27 403 | 26 465 |
| Glycine max | 46 324 | 36 364 | |
| Oryza sativa | 41 363 | 39 541 | |
| Physcomitrella patens | 28 090 | 26 072 | |
| Populus trichocarpa | 40 141 | 35 668 | |
| Ricins communis | 31 009 | 30 330 | |
| Selaginella moellendorffii | 18 384 | 16 876 | |
| Theobroma cacao | 28 858 | 28 294 | |
| Vitis vinifera | 26 238 | 24 635 | |
| Zea mays | 39 172 | 34 664 | |
| External protein sets | Amborella trichopoda | 25 347 | 24 643 |
| Picea abies | 22 070 | 20 869 | |
| Picea sitchensis | 10 521 | 8 770 | |
| Pinus patula | 52 112 | 41 956 | |
| Pinus taeda | 50 172 | 47 225 |
aProteins were clustered to 90 % identity and only the longest sequence was retained for each cluster
Assembly validation
The Core Eukaryotic Genes Mapping Approach (CEGMA) pipeline [35] as well as the Benchmarking Universal Single-Copy Orthologs (BUSCO) v1.1b1 tool [36] were used to identify putative core eukaryotic genes (CEGs) and universal single copy orthologs (USCOs) in the assembly as a measure of the completeness and contiguity. BUSCO analysis was performed using the early access plant dataset. In addition, conditional reciprocal best BLAST (CRBB) analysis of the P. patula draft transcriptome assembly, the P. taeda v1.01 gene models and the P. taeda draft transcriptome assembly was implemented with two different sets of reference sequences using Transrate [27]. Reference sets used were as follows: the P. taeda v1.01 predicted gene models available through the TreeGenes Database [11, 12] and the 87 P. patula protein sequences available through the NCBI and TrEMBL databases.
Sequence alignments against the P. taeda v1.01 draft genome assembly were generated to compare transcript to genome mapping of the P. patula v1.0 transcriptome assembly to that of other Pinus transcriptomes. Comparative alignments were produced using transcriptome data for seven other Pinus spp. available from the TreeGenes database [11, 12]: P. taeda (83 285 sequences), P. banksiana (21 675), P. contorta (14 375), P. pinaster (14 130), P. palustris (14 228), P. lambertiana (48 891), and P. radiata (4 742). Transcript sequences were aligned to the reference genome using GMAP 2014–02-28 (Genomic Mapping and Alignment Program) [37] with the following parameters: −-intronlength = 350000, −-no-chimeras, −-canonical-mode = 1, −-cross-species. The ‘--cross-species’ parameter was excluded for alignment of the P. taeda transcriptome. Sequence alignments were examined at two different cut-offs, the first (95 % identity, 95 % coverage) to compare mapping between species and the second (95 % identity, 50 % coverage) to account for possible effects due to genome fragmentation. The P. patula v1.0 transcriptome assembly was further validated by alignment to full-length Sanger sequenced P. taeda cDNA reference sequences present in NCBI and obtained through the TreeGenes database [11]. The 188 cDNA sequences were clustered to 90 % identity. CRBB analysis to the P. patula v1.0 transcriptome was performed using Transrate [27].
Differential expression analysis
RNA-seq read mapping to the P. patula v1.0 transcriptome and expression quantification was performed through RSEM v1.2.23 (RNA-Seq by Expectation-Maximum) [38] using Bowtie2 v2.2.5 [39]. Differential expression testing was performed with EBSeq v1.10.0 [40] using median normalization (FDR < 0.05).
Results and discussion
Data generation and pre-processing
Due to the expected size of the P. patula genome (ca. 22 Gb) [41], sequencing and assembly of the genome would be a costly and challenging endeavour. Therefore, transcriptome assembly was employed to generate a P. patula reference sequence. RNA-seq of shoot tissue harvested 1 dpi for inoculated and mock-inoculated samples yielded between 21 and 43 million read pairs per sample and a total of ca. 440 million reads (Table 2). Quality filtering removed ca. 13 % of reads and duplicate filtering removed a further 35 % of reads. Thus Dataset 1 consisted of ca. 36 Gb of sequence data and Dataset 2 consisted of ca. 23 Gb that passed through quality filtering and were subsequently used for transcriptome assembly.
Table 2.
Quality statistics for RNA sequencing data
| Data Set | Sample Name | Total Reads | Length (nt) | Q30a (%) | Total (Gb) |
|---|---|---|---|---|---|
| Raw Data | Mock-inoculated 1 | 78 344 666 | 100 | 87.1 | 7.83 |
| Mock-Inoculated 2 | 86 178 906 | 100 | 87.1 | 8.62 | |
| Mock-Inoculated 3 | 82 271 364 | 100 | 87.1 | 8.23 | |
| Inoculated 1 | 41 756 756 | 100 | 86.8 | 4.18 | |
| Inoculated 2 | 71 697 894 | 100 | 87.1 | 7.17 | |
| Inoculated 3 | 80 588 142 | 100 | 87.1 | 8.06 | |
| Total | 440 837 728 | 44.08 | |||
| Pre-processed Data | Mock-inoculated 1 | 68 237 135 | 41–100 | 100.0 | 6.45 |
| Mock-Inoculated 2 | 75 081 850 | 41–100 | 100.0 | 7.10 | |
| Mock-Inoculated 3 | 71 683 022 | 41–100 | 100.0 | 6.77 | |
| Inoculated 1 | 36 270 688 | 41–100 | 100.0 | 3.43 | |
| Inoculated 2 | 62 484 245 | 41–100 | 100.0 | 5.90 | |
| Inoculated 3 | 70 235 351 | 41–100 | 100.0 | 6.64 | |
| Dataset 1 | Total | 383 992 290 | 41–100 | 100.0 | 36.29 |
| Dataset 2 | Total | 248 994 870 | 41–100 | 100.0 | 23.53 |
aPercentage of reads in the library with a Phred score > 30
Comparison of assembler output
The completeness and quality of an assembled transcriptome is affected by the assembly program used as well as the assembly parameters used [42–48]. Comparative studies have also shown that the effectiveness of assembly programs can vary by input data set, with no assembler consistently outperforming any other [42, 43]. Due to this variability among assembler outputs, each variant assembly is likely to contain more accurate and complete assemblies at different loci. Therefore, in an effort to maximise diversity of assembled transcripts, we produced 18 de novo and two genome guided transcriptome assemblies using; Trinity, SOAPdenovo-Trans and Velvet/Oases. As expected from previous studies, large variation in the number, length and redundancy of contigs assembled was observed within and between assemblers (Fig. 1).
Fig. 1.
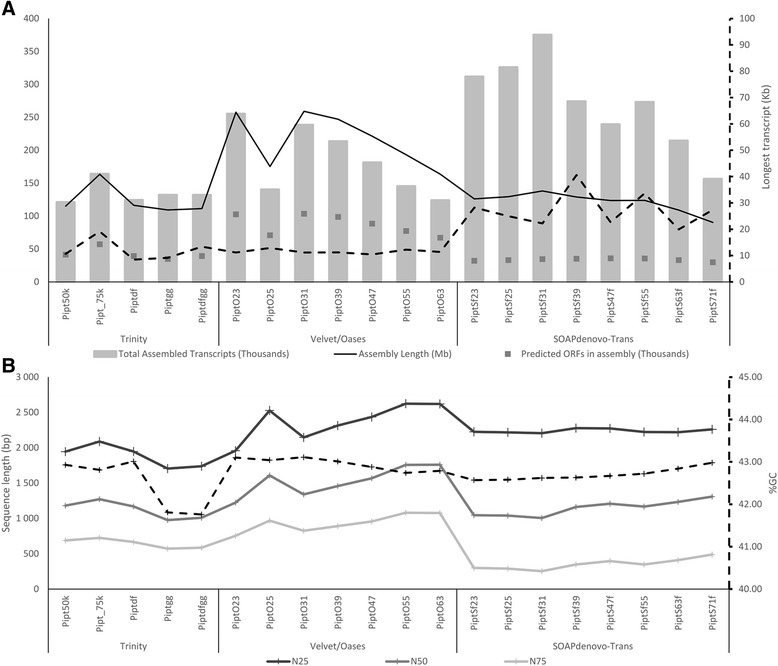
Summarised assembly statistics for all preliminary assemblies. Pipt = Pinus patula (a) – Assembly size and length statistics. (b) –Transcript N statistics and GC ratio for all assemblies. In each case the right hand y-axis only applies to the dashed line. The first three Trinity assemblies were de novo assemblies using Dataset 1 with (50 k) and without (75 k) CuffFly, and using Dataset 2 (df). The last two Trinity assemblies represent reference guided assemblies using Dataset 1 (gg) and Dataset 2 (dfgg). For Velvet/Oases and SOAPdenovo-Trans, the numbers indicated the k-mer value used. ORF = open reading frame
Trinity exhibited the most uniformity among assemblies compared to the variation among assemblies from Velvet/Oases and SOAPdenovo-Trans. An inverse relationship has been shown to exist between the number of contigs assembled and k-mer length used for assembly [43]. Therefore, the greater uniformity in assembled contig number between Trinity assemblies can likely be attributed to the program’s implementation of a fixed k-mer length for all assemblies. For each value of k used in assembly, SOAPdenovo-Trans resulted in the highest number of assembled contigs followed by Velvet/Oases and lastly, Trinity. In a comparative study, Trinity consistently assembled more contigs than Velvet/Oases and Trinity assemblies consistently had a higher N50 statistic [45]. Although the present study used newer versions of Velvet/Oases and Trinity, the difference in trends obtained illustrates the difference in performance of assemblers under different conditions, supporting the need for use of multiple assemblers during transcriptome reconstruction.
Trinity genome guided assemblies displayed lower GC ratios, as well as fewer predicted open reading frames (ORFs) compared to other assemblies (Fig. 1). This was ascribed to fragmentation of the P. taeda v1.01 genome. Nevertheless, the two genome guided Trinity assemblies were included in downstream analysis. In total, 3 447 807 assembled transcripts were used as input for the EvidentialGene tr2aacds pipeline.
The EvidentialGene pipeline selects the ‘best’ transcripts based on coding potential, thus selecting for the best ORFs assembled. Open reading frames were successfully predicted for ca. 2.7 million (77 %) of the input transcripts. Of these, 49 % were classified as redundant and 51 % were classified as differing in CDS (non-redundant). A further 55 % of non-redundant sequences were classified as perfect fragments of other longer CDS, leaving 23 % of the predicted 2.7 million CDS as informative. Of the informative CDS, 60 % were assigned to the ‘drop’ category and discarded. Overall, this brought about a 14-fold reduction in assembled transcript number, with only 7 % of the original input sequences maintained. The resulting merged assembly contained 247 035 transcripts grouped into 66 377 predicted loci (Additional file 1: Table S1). This assembly was compared to the average assembly statistics across assemblies for each assembly program respectively (Fig. 2; Additional file 2: Table S2). Despite the decrease in transcript number, the proportion of transcripts containing a predicted ORF in the merged assembly was 10–40 % higher compared to the average ORFs per assembly for all three assemblers. This indicates that a higher proportion of transcripts in the merged set have been accurately assembled to near-full or full length. The average length among the 1 000 longest predicted proteins in the merged assembly was 1 425 amino acids. Due to the high resource expenditure required to produce these long proteins they are often well-conserved and a biological maximum has been observed for their average length [28]. For plants this maximum observed average is ca. 1 500 amino acids (based on the average between Arabidopsis, banana, cacao and poplar) [28]. This indicates that these proteins are well assembled in the merged assembly, although there is room for improvement.
Fig. 2.
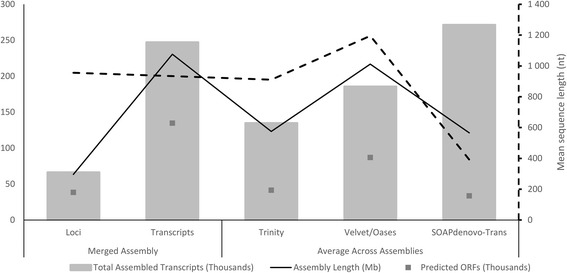
Assembly statistics for tr2aacds pipeline merged assembly compared to average assembly statistics for each assembler. Assembly size and length statistics. The dashed y-axis only applies to the dashed line. Unfiltered output assemblies from Trinity, Velvet/Oases and SOAPdenovo-Trans were used
Annotation
Sequence homology searches against the NR and plant protein databases successfully obtained significant local alignments for 32 416 loci. Of these, 14 255 were classified as non-pine origin transcripts and filtered from the transcriptome assembly. The majority of non-pine origin transcripts (71.62 %) aligned to transcripts from Fusarium species (Additional file 3: Table S3), with most aligning to either F. fujikuroi (4 698 transcripts) or F. oxysporum (3 290 transcripts). A further 22.81 % of non-pine origin transcripts aligned to Bipolaris maydis (1 309 transcripts), Pyrenophora tritici-repentis (992 transcripts) and Leptosphaeria maculans (949 transcripts), while the remaining 5.57 % (798 transcripts) aligned to 146 different species.
Removal of non-pine origin transcripts resulted in 52 112 putative loci, classified as the P. patula v1.0 shoot transcriptome. The current estimates for conifer gene numbers lie between ca. 32 000 for Picea glauca [49] and ca. 50 000 for P. taeda and P. pinaster [8, 50]. Roughly 60 % of assembled P. patula transcript sequences were successfully annotated, representing a wide array of molecular functions, biological processes and cellular compartment GO terms. The remaining 40 % of the assembled transcript sequences could not be annotated through similarity searches, however, each sequence contained an ORF predicted by the EvidentialGene pipeline and could potentially be expressed. Thus, these sequences were not removed from the assembly as they could represent uncharacterised or conifer specific genes. The top molecular function terms for P. patula v1.0 transcriptome were protein binding, transferase activity and nucleic acid binding (Additional file 4: Figure S1), similar to what has been observed for P. taeda, P. glauca and Picea mariana [8, 51].
Identification of putative NB-ARC defence related gene families
Orthologous protein groups were identified by comparing 41 956 clustered P. patula protein sequences from the assembled transcriptome to the 400 416 clustered protein sequences from 14 other plant species [52] (Table 1). Tribe-MCL analysis [53] resulted in 21,492 unique gene families, with an average of 18 members per family (Additional file 5: Table S4). Gene families were identified for 396 684 (89.6 %) sequences and ranged in size from 6 258 members from 15 species to 2 members from one species. Genes from the P. patula v1.0 transcriptome assembly initially clustered to 9 677 gene families (35 433 genes). This was reduced to 8 743 families (33 367 genes) by removing P. patula specific gene families with less than 5 members. While there are likely valid families in the removed set, these families were removed as most are likely to have arisen due to the remaining heterozygosity in the assembly. Of the total gene families, 2 165 were unique to conifers (Fig. 3). Although this is higher than the 1 554 reported by the P. taeda genome project [7, 8], it is a similar increase from the 1 021 reported by the P. abies genome project [54]. Included in the conifer-specific gene families are 130 that were unique to P. patula. The largest family identified in P. patula (1 794 members) contained leucine rich repeat (LRR), toll/interleukin-1 receptor (TIR), nucleotide binding domain with an ARC motif (NB-ARC), golgi transport complex 5 (COG5) and poxvirus A32 protein motifs. This gene family was also one of the largest observed for P. taeda and had low representation among the angiosperms while representation in the moss species differed. In total, 35 NB-ARC families were identified, of which 13 were present in conifers. NB-ARC gene families with higher representation of angiosperm genes had little to no representation from the conifers and vice versa (Fig. 4). The NB-ARC family of genes are associated with disease resistance as the majority of resistance proteins (R proteins) characterized are members of the NB-ARC and NB-LRR families [55]. Thus this difference could result from divergent R gene evolution between the plant lineages.
Fig. 3.
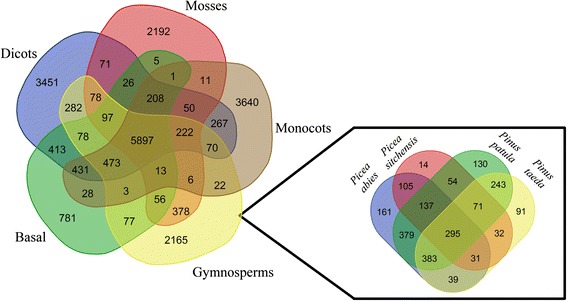
Unique orthologous protein groups identified through Tribe-MCL analysis. Left Comparison of protein family counts for all identified orthologous protein groups between five different plant classifications. Right Comparison of conifer specific protein counts between four conifer species. Dicots = Arabidopsis thaliana, Glycine max, Populus trichocarpa, Ricinus communis, Theobroma cacao, Vitis vinifera. Mosses = Selaginella moellendorffii, Physcomitrella patens. Monocots = Oryza sativa, Zea mays. Gymnosperms = Picea abies, Picea sitchensis, Pinus patula, Pinus taeda. Basal = Amborella trichopoda
Fig. 4.
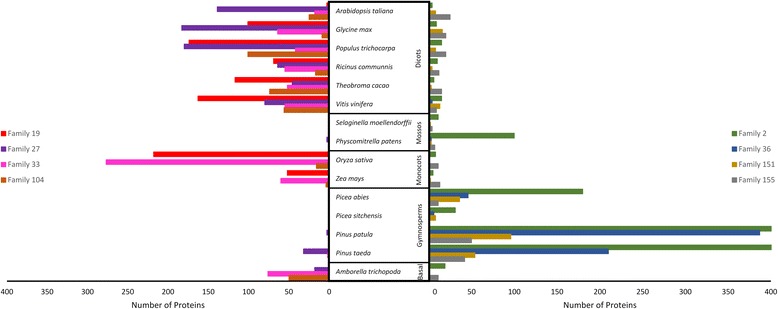
Number of proteins per species for the eight most populated NB-ARC motif containing gene families identified. Gene families were identified using Tribe-MCL. Left – NB-ARC families most prominent in conifers. Right – NB-ARC families most prominent in angiosperms. Each color represents a different gene family (Additional file 2: Table S2). Family 2 (green bars) for P. taeda and P. patula had 852 and 1 794 members respectively
Assembly validation
Many well established metrics exist for assessment of genome assembly quality, the majority of which are based on size, such as contig and N50 size. Size based metrics such as N50 have been used in the past as a measure of transcriptome completeness [56], yet these metrics have no real biological relevance and are ineffective without prior knowledge of the actual size distribution in the sequenced data set. These metrics are also highly sensitive to assembly parameters and assembled isoforms (Fig. 1), which can bias quality assessment. For this reason, three reference based metrics were used to assess the transcriptome assembly; completeness, contiguity and accuracy [10]. Completeness and contiguity are closely linked. Completeness is the percentage of a reference set that has been assembled. Contiguity is the percentage of assembled reference sequences covered to X%, where X is an arbitrary minimum threshold [56]. In this study, contiguity and completeness of the P. patula transcriptome assembly was measured by comparison against four data sets.
Comparison to the CEGMA core eukaryotic proteins identified 217 (88 % completeness) of the 248 core genes, whereas 203 (82 % completeness) were identified in the P. taeda v1.01 genome [7]. At the same time, of the identified core genes, 91 % showed full length alignments to the P. taeda v1.01 genome, while 93 % (contiguity) of those from the P. patula v1.0 shoot transcriptome were full length. The higher completeness and contiguity obtained for CEGs in the P. patula transcriptome assembly compared to the P. taeda v1.01 genome can most likely be attributed to genome fragmentation. This illustrates the value of de novo transcriptome assembly for analysing genes missing from an incomplete genome. CEGMA analysis also identified multiple orthologs for 90 % of the identified CEGs. This is likely due to the presence of high allelic variation in the data used for assembly arising from the pooled nature of the samples (pooled RNA from seedlings) used for sequencing.
BUSCO analysis against the early access plant data set identified 850 (88 %) complete BUSCOs, out of 956 groups searched, of which 307 (32 %) were duplicated. A further 26 fragmented BUSCOs were identified. The high amount of duplicated complete BUSCOs further indicate the presence of assembled haplotypes still present in the transcriptome [36].
CRBB analysis to the P. patula reference proteins showed a similar pattern as above when comparing completeness (49 %) and contiguity (92 %) of the P. patula transcriptome to that of the P. taeda transcriptome (43; 92 %). This indicates higher completeness of the P. patula transcriptome for P. patula origin proteins as would be expected. The completeness (46 %) of the P. taeda gene models was intermediate between the transcriptomes, however, its contiguity (13 %) was notably lower. This low contiguity most likely arose due to the presence of partial genes in the high confidence gene models. The completeness and contiguity of the P. patula transcriptome assembly was also investigated through CRBB analysis to the P. taeda v1.0 transcriptome assembly using the gene models extracted from the P. taeda v1.01 genome assembly and the 87 available P. patula protein sequences as the reference sets. Compared to the P. taeda transcriptome, the P. patula transcriptome covered a higher proportion of the P. taeda gene models (at 95 % coverage), had a higher proportion of reference sequences with a CRBB result and had the lowest reciprocal best hit (RBH) ratio (Table 3). Overall CRBB statistics for comparison to the P. patula proteins were higher for the P. patula transcriptome compared to both the P. taeda transcriptome and gene models (Table 3).
Table 3.
Conditional reciprocal best BLAST (CRBB) comparisonsa of assembled Pinus patula transcripts to available Pinus taeda gene models and transcripts
| Query | P. taeda gene models | P. taeda v1.0 | P. patula v1.0 |
|---|---|---|---|
| Reference | P. taeda gene models (n = 48 391) | ||
| Hits at 85 % coverage | 99.7 % | 6.6 % | 8.3 % |
| Hits at 95 % coverage | 99.7 % | 3.7 % | 4.2 % |
| Contigs with CRBB | 48 363 | 29 052 | 28 491 |
| % Contigs with CRBB | 99.9 % | 34.9 % | 54.7 % |
| References with CRBB | 48 269 | 12 339 | 15 958 |
| % Reference CRBB | 99.7 % | 25.5 % | 33.0 % |
| Reciprocal Best Hit Ratio | 1.00 | 2.35 | 1.79 |
| Reference | P. patula proteins (n = 87) | ||
| Hits at 85 % coverage | 5.8 % | 39.1 % | 43.7 % |
| Hits at 95 % coverage | 2.3 % | 34.5 % | 40.2 % |
| Contigs with CRBB | 73 | 80 | 71 |
| % Contigs with CRBB | 0.2 % | 0.1 % | 0.1 % |
| References with CRBB | 40 | 37 | 43 |
| % Reference CRBB | 46.0 % | 42.5 % | 49.4 % |
| Reciprocal Best Hit Ratio | 1.83 | 2.16 | 1.65 |
aCRBB alignments for query sequences were generated against the available high confidence P. taeda gene models and the available P. patula protein sequences
The third metric assessed was accuracy, defined as the percentage of correctly assembled bases in an assembly compared to a reference [10]. This was estimated through high-identity mapping of the assembled P. patula transcriptome, along with seven other pine transcriptomes, to the P. taeda v1.01 genome (Table 4). Mapping to the genome precluded calculation of completeness and contiguity, due to genome fragmentation and lack of exact gene number and location. At 95 % sequence identity and query coverage thresholds a total of 64 % of P. patula sequences mapped. The highest total mapping rates were observed for P. banksiana and P. contorta, while the lowest mapping rate was obtained for P. lambertiana, as expected from their phylogenetic relationship and previous studies [8]. Mapping rates obtained for the P. patula transcriptome were similar to the mapping rates obtained for the P. radiata and P. taeda transcriptome assemblies. These alignment metrics serve as a measure of transcriptome accuracy. Lowering the minimum coverage threshold to 50 % increased mapping by between 2 % and 15 %. The P. banksiana (3.8 %), P. contorta (2.5 %) and P. patula (2.8 %) transcriptomes were the least affected, while the transcriptomes for P. radiata (15.1 %), P. sylvestris (10.3 %) and P. taeda (13.1 %) showed the largest increase in mapping rates, suggesting that these transcriptomes have a higher content of genes that were fragmented in the genome assembly. Comparison of accuracy metrics between assemblies should be done with care, however, as even though the P. taeda transcriptome showed a lower accuracy (57 %) than P. patula, the size of the transcriptome means that it still contains ca. 10 000 more accurately assembled sequences. This illustrates the importance of considering assembly size when comparing between datasets, such as the high mapping rates to the P. taeda v1.01 genome obtained for P. contorta, P. pinaster and P. radiata (Table 4). Still, more than 33 000 (64 %) of the assembled P. patula sequences were shown to be accurately assembled and this number is expected to increase as fragmentation in the genome decreases.
Table 4.
Mapping statistics to the P. taeda v1.01 genome
| Assembly | Total Sequences | Identity | Coverage | Unique Hits | Non-unique hits | Total % mapped |
|---|---|---|---|---|---|---|
| Pinus patula (EviGene Loci) | 66 377 | 95 | 95 | 33.53 % | 16.78 % | 50.31 % |
| 66 377 | 95 | 50 | 34.44 % | 18.10 % | 52.55 % | |
| Pinus patula v1.0 | 52 112 | 95 | 95 | 42.68 % | 21.35 % | 64.03 % |
| 52 112 | 95 | 50 | 43.84 % | 23.04 % | 66.87 % | |
| Pinus banksiana | 21 675 | 95 | 95 | 73.25 % | 15.26 % | 88.51 % |
| 21 675 | 95 | 50 | 74.72 % | 17.62 % | 92.34 % | |
| Pinus contorta | 14 375 | 95 | 95 | 70.23 % | 14.91 % | 85.15 % |
| 14 375 | 95 | 50 | 70.37 % | 17.27 % | 87.64 % | |
| Pinus lambertiana | 48 891 | 95 | 95 | 25.16 % | 1.07 % | 26.23 % |
| 48 891 | 95 | 50 | 31.04 % | 2.07 % | 33.11 % | |
| Pinus pinaster | 14 130 | 95 | 95 | 56.24 % | 12.76 % | 69.00 % |
| 14 130 | 95 | 50 | 61.27 % | 16.28 % | 77.54 % | |
| Pinus radiata | 4 742 | 95 | 95 | 46.06 % | 11.66 % | 57.72 % |
| 4 742 | 95 | 50 | 57.11 % | 15.67 % | 72.78 % | |
| Pinus sylvestris | 11 248 | 95 | 95 | 47.75 % | 16.79 % | 64.54 % |
| 11 248 | 95 | 50 | 53.90 % | 20.96 % | 74.87 % | |
| Pinus taeda | 83 285 | 95 | 95 | 48.72 % | 7.82 % | 56.54 % |
| 83 285 | 95 | 50 | 57.64 % | 11.99 % | 69.63 % |
The assembled P. patula transcripts were further compared to corresponding P. taeda complete CDS sequences to ascertain the quality of the assembly against experimentally validated data (Additional file 6: Table S5). Of the 121 cDNA sequences, 89 (73.5 %) mapped to the P. patula transcriptome with greater than 89 % identity and 80 % subject coverage (Additional file 6: Table S5). Of the mapped sequences, 47 had a query coverage of more than 80 % with an average sequence identity of 98.4 ± 1.9 % and an average coverage of 97.9 ± 3.5 % and 91.5 ± 6.8 % for the subject and query sequences respectively (Additional file 7: Figure S2). Thus, of the P. taeda cDNA sequences present in the assembled P. patula transcriptome, 52.8 % have been assembled to near full-length.
Differential expression analysis
Comparison of inoculated and mock-inoculated data sets using EBSeq identified 166 transcripts as differentially expressed between conditions (Additional file 8: Table S6). The small number of detectable differentially expressed transcripts is likely a reflection of the very early time-point investigated, where small amounts of pathogen would have been in contact with the host tissue.
Ten transcripts were up-regulated (log2(fold change) > 1) in the inoculated set, relative to mock-inoculated, while 156 transcripts were down-regulated (log2(fold change) < −0.25; 77 had log2(fold change) < −1). Among the up-regulated genes four had putative annotations (Additional file 9: Table S7). Two of these genes are involved in folate metabolism (methylenetetrahydrofolate dehydrogenase) and stomatal closure (PF03595), while the other two are linked to sugar metabolism (PREDICTED: alpha-galactosidase-like; sucrose synthase-like protein).
In the down-regulated set 83 transcripts had putative annotations (Additional file 9: Table S7). Some of these are related to plant defence such as a putative WRKY76 encoding transcript, implicated in susceptibility against Magnoporthe oryzae but increased tolerance to cold in rice [57] and a putative phenylalanine ammonia-lyase (PAL) encoding transcript. PAL is an important enzyme for salicylic acid production and is a key enzyme in the phenylpropanoid pathway, shown to be induced in response to wounding and fungal infection in Pinus sylvestris [58]. A putative map kinase 4 is also down-regulated. In Arabidopsis, map kinase 4 is known to regulate the salicylic acid and jasmonic acid/ ethylene defence signaling [59]. Although it is tempting to speculate that the down-regulation of such important transcripts in defence may, in part, contribute to susceptibility against F. circinatum, a detailed time-course of infection in P. patula is necessary to determine the full suite of host responses during this susceptible interaction.
Conclusions
This study presents the first transcriptome sequencing and assembly analysis for Pinus patula. The P. patula v1.0 transcriptome assembly constitutes the largest gene catalogue for this economically important species to date. More than 23 Gb of data was used to assemble 52 112 sequences with a total length of 52 Mb and an average coverage of more than 200×. Of these sequences, 30 844 could be assigned annotations. This transcriptome represents a major genomic resource for future studies on this tropical Pinus species, and will be used as the basis for further investigation of the host pathogen interaction between P. patula and F. circinatum. The workflow used for transcriptome assembly can in future be reapplied and altered as new sequencing data becomes available for P. patula to produce a more comprehensive and complete assembly. Furthermore, the workflow implemented during this study could be applied to other species where a high quality genome sequence is not available. One species to which the workflow could be applied in future is P. tecunumanii, a species that is closely related to P. patula [60] but which displays resistance to F. circinatum. Assembly of the P. tecunumanii transcriptome would thus allow for further investigation of the mechanisms differentiating resistance and susceptibility through comparison of defence responses in these closely related species.
Availability of supporting data
The data sets supporting the results of this article are available through the NCBI BioProject repository, [PRJNA301922; http://www.ncbi.nlm.nih.gov/bioproject/301922].
Acknowledgements
The author would like to thank the following people for their contributions to the study: Dr Kitt Payn and Dr Nicky Jones for help in obtaining plant material and Ms Thandekile Mamni for assistance with infection trials. The author would also like to thank Forestry South Africa (for seed funding), the Genomics Research Institute (GRI) and the National Research Foundation’s (NRF) Bioinformatics and Functional Genomics Programme (NBFG, UID:71255) as well as Innovation, Thuthuka and THRIP grants (Grant numbers: 84951, 86936, 87912) for financing various aspects of this study. Opinions expressed and conclusions arrived at, are those of the author(s) and are not necessarily to be attributed to the NRF.
Abbreviations
- BUSCO
Benchmarking Universal Single-Copy Orthologs
- ca
circa
- CDS
DNA Coding Sequence
- CEGMA
core eukaryotic genes mapping approach
- CEGs
core eukaryotic genes
- CRBB
conditional reciprocal best BLAST
- DEPC
diethylpyrolecarbonate
- dpi
days post inoculation
- EC
enzyme code
- GMAP
genomic mapping and alignment program
- GO
gene ontology
- GSNAP
genomic short-read nucleotide alignment program
- NCBI
National Centre for Biotechnology Information
- nr
non-redundant
- ORF
Open Reading Frame
- PCR
polymerase chain reaction
- PDA
potato dextrose agar
- RBH
reciprocal best hit
- RNA-seq
RNA sequencing
- USCOs
Universal Single-Copy Orthologs
Additional files
EvidentialGene tr2aacds pipeline output summary. (PDF 138 kb)
Assembly statistics for EvidentialGene tr2aacds pipeline merged assembly compared to average statistics for each assembler. (PDF 150 kb)
Predicted species distribution for non-pine origin sequences removed from the Pinus patula v1.0 transcriptome. (TSV 7 kb)
Molecular function gene ontology distribution for the Pinus patula v1.0 transcriptome. (TIF 713 kb)
Tribe-MCL gene families and annotations for all 15 species used. (CSV 1210 kb)
Conditional reciprocal best BLAST alignment results between full-length Sanger sequenced Pinus taeda cDNA and representative Pinus patula transcripts for each cDNA. (CSV 20 kb)
Summary statistics for alignment of Pinus taeda complete CDS sequences to assembled Pinus patula transcripts. Pita = P. taeda. The x-axis represents the query P. taeda cDNA sequence. The solid y-axis (left) illustrates: cDNA query sequence length (pink circle), P. patula subject sequence length (blue square), conditional reciprocal best BLAST alignment length (gold triangle). The dashed y-axis (right) depicts the: percentage identity between sequences (black line), percentage coverage of the P. taeda cDNA by the corresponding P. patula transcript (green cross) and vice versa (purple plus). (TIF 2325 kb)
EBSeq differential expression analysis results comparing expression between inoculated and mock-inoculated data. (TSV 6517 kb)
Summarized list of differentially expressed genes between inoculated and mock-inoculated data with annotations. (TSV 68 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EAV performed all data generation, analysis, and prepared this manuscript. JLW provided technical support, supervision and guidance for data analysis and interpretation. ETS and AAM participated in the design of the study and edited the manuscript. SN conceived the study, provided valuable advice, direction and supervision in the planning and execution of the project and edited the final manuscript. All other technical assistance is listed in the acknowledgements.
Contributor Information
Erik A. Visser, Email: erik.visser@up.ac.za
Jill L. Wegrzyn, Email: jill.wegrzyn@uconn.edu
Emma T. Steenkmap, Email: emma.steenkamp@up.ac.za
Alexander A. Myburg, Email: zander.myburg@up.ac.za
Sanushka Naidoo, Email: sanushka.naidoo@up.ac.za.
References
- 1.Critchfield W, Little E. Geographic distribution of pines of the world. USDA For Serv. 1966;991:1–97. [Google Scholar]
- 2.Indufor: Forest Stewardship Council (FSC) Strategic Review on the Future of Forest Plantations. 2012:121.
- 3.Wingfield MJ, Coutinho TA, Roux J, Wingfield BD. The future of exotic plantation forestry in the tropics and southern Hemisphere: Lessons from pitch canker. South Afr Forestry J. 2002;195:79–82. doi: 10.1080/20702620.2002.10434607. [DOI] [Google Scholar]
- 4.Wingfield MJ, Hammerbacher A, Ganley RJ, Steenkamp ET, Gordon TR, Wingfield BD, et al. Pitch canker caused by Fusarium circinatum - A growing threat to pine plantations and forests worldwide. Australas Plant Pathol. 2008;37:319–334. doi: 10.1071/AP08036. [DOI] [Google Scholar]
- 5.Hodge GR, Dvorak WS. Differential responses of Central American and Mexican pine species and Pinus radiata to infection by the pitch canker fungus. New For. 2000;19:241–258. doi: 10.1023/A:1006613021996. [DOI] [Google Scholar]
- 6.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zimin A, Stevens KA, Crepeau MW, Holtz-Morris A, Koriabine M, Marçais G, et al. Sequencing and assembly of the 22-Gb loblolly pine genome. Genetics. 2014;196:875–890. doi: 10.1534/genetics.113.159715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wegrzyn JL, Liechty JD, Stevens KA, Wu LS, Loopstra CA, Vasquez-Gross HA, et al. Unique features of the loblolly pine (Pinus taeda L.) megagenome revealed through sequence annotation. Genetics. 2014;196:891–909. doi: 10.1534/genetics.113.159996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salzberg SL, Yorke JA. Beware of mis-assembled genomes. Bioinformatics. 2005;21:4320–4321. doi: 10.1093/bioinformatics/bti769. [DOI] [PubMed] [Google Scholar]
- 10.Martin JA, Wang Z. Next-generation transcriptome assembly. Nat Rev Genet. 2011;12:671–682. doi: 10.1038/nrg3068. [DOI] [PubMed] [Google Scholar]
- 11.Wegrzyn JL, Lee JM, Tearse BR, Neale DB. TreeGenes: A forest tree genome database. Int J Plant Genomics. 2008;2008:412875. doi: 10.1155/2008/412875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wegrzyn JL, Main D, Figueroa B, Choi M, Yu J, Neale DB, et al. Uniform standards for genome databases in forest and fruit trees. Tree Genet Genomes. 2012;8:549–557. doi: 10.1007/s11295-012-0494-7. [DOI] [Google Scholar]
- 13.Hall DE, Yuen MMS, Jancsik S, Quesada AL, Dullat HK, Li M, et al. Transcriptome resources and functional characterization of monoterpene synthases for two host species of the mountain pine beetle, lodgepole pine (Pinus contorta) and jack pine (Pinus banksiana) BMC Plant Biol. 2013;13:80. doi: 10.1186/1471-2229-13-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu J, Sturrock RN, Benton R. Transcriptome analysis of Pinus monticola primary needles by RNA-seq provides novel insight into host resistantce to Cronartium ribicola. BMC Genomics. 2013;14:884. doi: 10.1186/1471-2164-14-884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Canales J, Bautista R, Label P, Gómez-Maldonado J, Lesur I, Fernández-Pozo N, et al. De novo assembly of maritime pine transcriptome: implications for forest breeding and biotechnology. Plant Biotechnol J. 2014;12:286–299. doi: 10.1111/pbi.12136. [DOI] [PubMed] [Google Scholar]
- 16.Porter B. Pathogenicity and competition studies on Fusarium circinatum, a pathogen of pine trees. South Africa: University of Pretoria; 2010.
- 17.Lewinsohn E, Steele CL, Croteau R. Simple isolation of functional RNA from woody stems of gymnosperms. Plant Mol Biol Report. 1994;12:20–25. doi: 10.1007/BF02668660. [DOI] [Google Scholar]
- 18.Andrews S. FastQC a quality control tool for high throughput sequence data. 2012. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- 19.Joshi NA, Fass JN: Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ. (Version 1.33) [Software] 2011, Available at https://github.com/najoshi/sickle.
- 20.Cox MP, Peterson DA, Biggs PJ. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics. 2010;11:485. doi: 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu H, Luo X, Qian J, Pang X, Song J, Qian G, et al. FastUniq: A Fast De Novo Duplicates Removal Tool for Paired Short Reads. PLoS One. 2012;7:1–6. doi: 10.1371/annotation/7a1b01e4-b894-40ef-9c31-584e9c24ee0a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat Biotechnol. 2013;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie Y, Wu G, Tang J, Luo R, Patterson J, Liu S, et al. SOAPdenovo-Trans: De novo transcriptome assembly with short RNA-Seq reads. Bioinformatics. 2014;30:1660–1666. doi: 10.1093/bioinformatics/btu077. [DOI] [PubMed] [Google Scholar]
- 24.Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schulz MH, Zerbino DR, Vingron M, Birney E. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics. 2012;28:1086–1092. doi: 10.1093/bioinformatics/bts094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Philip D, Bowden J, Couger MB, Eccles D, Li B, Macmanes MD, Ott M, Orvis J, Pochet N: Reference Generation and Analysis with Trinity. Volume 8; 2014. [DOI] [PMC free article] [PubMed]
- 27.Smith-Unna RD, Boursnell C, Patro R, Hibberd JM, Kelly S. TransRate: reference free quality assessment of de-novo transcriptome assemblies. bioRxiv. 2015. http://dx.doi.org/10.1101/021626. [DOI] [PMC free article] [PubMed]
- 28.Gilbert D. EvidentialGene: tr2aacds, mRNA transcript assembly software. 2013. http://arthropods.eugenes.org/EvidentialGene/.
- 29.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 30.Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 31.Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu Z, Bao J, Reecy JM. CateGOrizer: a web-based program to batch analyse gene ontology classification categories. Online J Bioinforma. 2008;9:108–112. [Google Scholar]
- 33.Van Bel M, Proost S, Wischnitzki E, Movahedi S, Scheerlinck C, Van de Peer Y, et al. Dissecting Plant Genomes with the PLAZA Comparative Genomics Platform. Plant Physiol. 2012;158:590–600. doi: 10.1104/pp.111.189514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: The protein families database. Nucleic Acids Res. 2014;42:222–230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parra G, Bradnam K, Korf I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–1067. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- 36.Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM: BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, doi:10.1093/bioinformatics/btv351 [DOI] [PubMed]
- 37.Wu TD, Watanabe CK. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- 38.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:232. doi: 10.1093/bioinformatics/btq629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie2. Nat Protoc. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leng N, Dawson JA, Thomson JA, Ruotti V, Rissman AI, Smits BMG, et al. EBSeq: An empirical bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics. 2013;29:1035–43. doi: 10.1093/bioinformatics/btt087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hall SE, Dvorak WS, Johnston JS, Price HJ, Williams CG. Flow cytometric analysis of DNA content for tropical and temperate New World pines. Ann Bot. 2000;86:1081–1086. doi: 10.1006/anbo.2000.1272. [DOI] [Google Scholar]
- 42.Duan J, Xia C, Zhao G, Jia J, Kong X. Optimizing de novo common wheat transcriptome assembly using short-read RNA-Seq data. BMC Genomics. 2012;13:392. doi: 10.1186/1471-2164-13-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang Y, Smith SA. Optimizing de novo assembly of short-read RNA-seq data for phylogenomics. BMC Genomics. 2013;14:328. doi: 10.1186/1471-2164-14-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gruenheit N, Deusch O, Esser C, Becker M, Voelckel C, Lockhart PJ. Cutoffs and k-mers: Implications from a transcriptome study in allopolyploid plants. BMC Genomics. 2012;13:92. doi: 10.1186/1471-2164-13-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clarke K, Yang Y, Marsh R, Xie LL, Zhang KK. Comparative analysis of de novo transcriptome assembly. Sci China Life Sci. 2013;56:156–162. doi: 10.1007/s11427-013-4444-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oono Y, Kobayashi F, Kawahara Y, Yazawa T, Handa H, Itoh T, et al. Characterisation of the wheat (Triticum aestivum L.) transcriptome by de novo assembly for the discovery of phosphate starvation-responsive genes: gene expression in Pi-stressed wheat. BMC Genomics. 2013;14:77. doi: 10.1186/1471-2164-14-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steijger T, Abril JF, Engström PG, Kokocinski F, The RGASP Consortium. Hubbard TJ, et al. Assessment of transcript reconstruction methods for RNA-seq. Nat Methods. 2013;10:1177–1184. doi: 10.1038/nmeth.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vijay N, Poelstra JW, Künstner A, Wolf JBW. Challenges and strategies in transcriptome assembly and differential gene expression quantification. A comprehensive in silico assessment of RNA-seq experiments. Mol Ecol. 2013;22:620–634. doi: 10.1111/mec.12014. [DOI] [PubMed] [Google Scholar]
- 49.Rigault P, Boyle B, Lepage P, Cooke JEK, Bousquet J, Mackay JJ. A white spruce gene catalog for conifer genome analyses. Plant Physiol. 2011;157:14–28. doi: 10.1104/pp.111.179663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fernández-Pozo N, Canales J, Guerrero-Fernández D, Villalobos DP, Díaz-Moreno SM, Bautista R, et al. EuroPineDB: a high-coverage web database for maritime pine transcriptome. BMC Genomics. 2011;12:366. doi: 10.1186/1471-2164-12-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pavy N, Pelgas B, Laroche J, Rigault P, Isabel N, Bousquet J. A spruce gene map infers ancient plant genome reshuffling and subsequent slow evolution in the gymnosperm lineage leading to extant conifers. BMC Biol. 2012;10:84. doi: 10.1186/1741-7007-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30(7):1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Van Dongen S, Abreu-Goodger C. Using MCL to Extract Clusters from Networks. In: Van Helden J, Toussaint A, Thieffry D, editors. Bacterial Molecular Networks SE - 15. New York: Springer; 2012. pp. 281–295. [DOI] [PubMed] [Google Scholar]
- 54.Nystedt B, Street NR, Wetterbom A, Zuccolo A, Lin Y-C, Scofield DG, et al. The Norway spruce genome sequence and conifer genome evolution. Nature. 2013;497:579–584. doi: 10.1038/nature12211. [DOI] [PubMed] [Google Scholar]
- 55.Głowacki S, Macioszek V, Kononowicz A. R proteins as fundamentals of plant innate immunity. Cell Mol Biol Lett. 2011;16:1–24. doi: 10.2478/s11658-010-0024-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martin J, Bruno VM, Fang Z, Meng X, Blow M, Zhang T, et al. Rnnotator: an automated de novo transcriptome assembly pipeline from stranded RNA-Seq reads. BMC Genomics. 2010;11:663. doi: 10.1186/1471-2164-11-663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yokotani N, Sato Y, Tnabe S, Chujo T, Shimizu T, Okada K, et al. WRKY76 is a rice transcriptional repressor playing opposite roles in blast disease resistance and cold stress response. J Exp Bot. 2013;64:5085–5097. doi: 10.1093/jxb/ert298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Adomas A, Heller G, Guosheng LI, Olson A, Tzu-Ming C, Osborne J, et al. Transcript profiling of a conifer pathosystem: response of Pinus sylvestris root tissues to pathogen (Heterobasidion annosum) invasion. Tree Physiol. 2007;27:1441–1458. doi: 10.1093/treephys/27.10.1441. [DOI] [PubMed] [Google Scholar]
- 59.Broderson P, Peterson M, Bjorn Nielsen H, Zhu S, Newman MA, Shokat KM, et al. Arabidopsis MAP kinase 4 regulates salicylic acid- and jasmonic acid/ethylene-dependent responses via EDS1 and PAD4. Plant J. 2006;47:532–546. doi: 10.1111/j.1365-313X.2006.02806.x. [DOI] [PubMed] [Google Scholar]
- 60.Eckert AJ, Hall BD. Phylogeny, historical biogeography, and patterns of diversification for Pinus (Pinaceae): Phylogenetic tests of fossil-based hypotheses. Mol Phylogenet Evol. 2006;40:166–182. doi: 10.1016/j.ympev.2006.03.009. [DOI] [PubMed] [Google Scholar]