

Abstract
Over the last several decades, the use of halogenated organic compounds has become the cause of environmental and human health concerns. Of particular notoriety has been the establishment of the neurotoxicity of polychlorinated biphenyls (PCBs) and polybrominated diphenyl ethers (PBDEs). The subsequent banning of PBDEs has led to greatly increased use of 1,2,5,6,9,10-hexabromocyclododecane (HBCDD, also known as HBCD) as a flame retardant in consumer products. The physiochemical similarities between HBCDD and PBDEs suggest that HBCDD may also be neurotoxic to the dopamine system, which is specifically damaged in Parkinson disease (PD). The purpose of this study was to assess the neurotoxicity of HBCDD on the nigrostriatal dopamine system using an in vitro and in vivo approach. We demonstrate that exposure to HBCDD (0–25 μM) for 24 hrs causes significant cell death in the SK-N-SH catecholaminergic cell line, as well as reductions in the growth and viability of TH+ primary cultured neurons at lower concentrations (0–10 μM) after 72 hrs of treatment. Assessment of the in vivo neurotoxicity of HBCDD (25 mg/kg for 30 days) resulted in significant reductions in the expression of the striatal dopamine transporter and vesicular monoamine transporter 2, both of which are integral in mediating dopamine homeostasis and neurotransmission in the dopamine circuit. However, no changes were seen in the expression of tyrosine hydroxylase in the dopamine terminal, or striatal levels of dopamine. To date, these are the first data to demonstrate that exposure to HBCDD disrupts the nigrostriatal dopamine system. Given these results and the ubiquitous nature of HBCDD in the environment, its possible role as an environmental risk factor for PD should be further investigated.
Keywords: Dopamine, Dopamine Transporter, Hexabromocyclododecane, Neurotoxicity, Parkinson disease, Vesicular Monoamine Transporter 2
1.0 Introduction
Over the last several decades, the use of halogenated organic compounds has become the cause of environmental and human health concerns. Particular attention has been given to polychlorinated biphenyls (PCBs), which were widely used for their insulating and flame retarding properties. As the toxicity of PCBs became recognized and they were phased out of use, a new class of compounds, the brominated flame retardants (BFRs), saw a precipitous influx in manufacture and use (Covaci et al., 2011). BFRs are commonly used in household and commercial products such as electronics equipment, plastics, paints, and textiles in order to reduce the flammability of these consumer products (de Wit, 2002). Until recently, the most dominant class of BFRs has been polybrominated diphenyl ethers (PBDEs). PBDEs are lipophilic, persistent, bioaccumulative, and have become distributed globally throughout the environment and population, much as has been the case for PCBs (Law et al., 2003, Covaci et al., 2011). However, investigations into the toxicity of PBDEs revealed adverse effects on learning and memory as well as neurodevelopment (Viberg et al., 2003, Viberg et al., 2006, Herbstman and Mall, 2014). They have also recently been shown by our group to damage the nigrostriatal dopamine system in mice (Bradner et al., 2013b). As a result of mounting toxicological evidence, the manufacture and use of many PBDE chemicals have been discontinued, both in Europe as well as the United States.
As manufacturers have reduced the use of PBDEs, 1,2,5,6,9,10-hexabromocyclododecane (HBCDD, also known as HBCD) has become a common flame retardant in consumer products. HBCDD is a brominated flame retardant that is used predominantly in expanded polystryrene foam for thermal insulation in buildings, as well as in upholstered furniture, automobile textiles and cushions, and electronics (Covaci et al., 2006). Similar to PBDEs, HBCDD is persistent, lipophilic, and bioaccumulative within the environment (Fonnum and Mariussen, 2009, Marvin et al., 2011). Additionally, like PBDEs, the integration of HBCDD into products is considered additive rather than being chemically bound to materials, which makes it easy for HBCDD to leach from products and deposit in the environment (de Wit, 2002). Because of this, HBCDD has been observed ubiquitously in the environment and is also commonly detected in human blood, adipose tissue, and breast milk (Covaci et al., 2006). Over the past decade, concerns regarding the toxicity of HBCDD to humans, and corresponding regulatory considerations, have increased. In May 2013, HBCDD production and use was listed to be banned under the Stockholm Convention on Persistent Organic Pollutants, and there is now a 5 year phase-out period. In light of these actions, the persistence of HBCDD in the environment and the body, similar to that seen for other halogenated chemicals including PCBs and PBDEs, presents a scenario of continual exposure and health effects that could endure for many decades (Covaci et al., 2006, Covaci et al., 2011, Marvin et al., 2011).
Studies evaluating the potential neurotoxicity of HBCDD have begun to delineate specific neurodevelopmental and behavioral effects following exposure to the compound (Lilienthal et al., 2009, Saegusa et al., 2009, Saegusa et al., 2012, Miller-Rhodes et al., 2014). Additionally, a few studies have been performed that begin to address the effects of HBCDD on the function of the dopamine system. Most notably, work by Dingemans et al. (2009) demonstrated a reduction in catecholamine release from PC12 cells following treatment with HBCDD, while Mariussen and Fonnum (2003) identified HBCDD as a potent inhibitor of dopamine uptake through the dopamine transporter (DAT) and vesicular monoamine transporter 2 (VMAT2). These studies provide valuable insight into the possible molecular targets of HBCDD neurotoxicity in the nigrostriatal dopamine system, which is specifically damaged in Parkinson disease (PD). Parkinson disease is defined by pathological damage to dopaminergic neurons and loss of dopamine in the nigrostriatal dopamine system, resulting in the hallmark clinical features of the disease, such as slowness of movement, postural instability, and resting tremor (Blandini et al., 2000). Previous work from our group as well as others has shown the nigrostriatal dopamine system to be uniquely sensitive to exposure to halogenated compounds, including PCBs and PBDEs (Caudle et al., 2006, Bradner et al., 2013b). Indeed, several studies have found exposure to PCB mixtures results in reduction in nigrostriatal dopamine content, similar to that seen in PD (Seegal et al., 1991, 1994). Related to these reductions, we have observed significant reductions in both the striatal dopamine transporter (DAT) and vesicular monoamine transporter 2 (VMAT2) following exposure to PCBs and PBDEs (Caudle et al., 2006, Bradner et al., 2013b). As evidence for the contribution of exposure to environmental toxicants and the risk for developing PD increases, it is imperative to identify and characterize emerging or current environmental hazards that could elicit damage to the dopamine system. Thus, this study sought to further address the potential neurotoxic effects of HBCDD on the nigrostriatal dopamine system by coupling in vitro and in vivo models. With these approaches we found HBCDD to be neurotoxic to dopaminergic neurons and their neuronal outgrowths in an in vitro model. These findings were further addressed in vivo, where oral exposure to HBCDD resulted in significant reductions in the expression of several synaptic proteins in the striatal dopamine nerve terminal. These findings suggest that exposure to HBCDD is neurotoxic to select dopaminergic targets in the striatum, which are involved in mediating criticial functions in dopamine handling and maintaining the integrity of the nigrostriatal dopamine system.
2.0 Materials and Methods
2.1 Chemicals and Reagents
Hexabromocyclododecane (HBCDD) was purchased from Sigma-Aldrich (St. Louis, MO). SK-N-SH cells were obtained from American Type Culture Collection (ATCC; Manassas, VA). WST-1 Cytotoxicity Assay Kit was purchased from Roche (Nutley, NJ). Hibernate A and Hibernate A − Calcium were purchased from BrainBits (Springfield, IL). B27, DNaseI, and Neurobasal A were purchased from Life Technologies (Carlsbad, CA). Papain was obtained from Sigma-Aldrich (St. Louis, MO). Dispase II was purchased from Roche (Nutley, NJ). Aphidicolin was purchased from A.G Scientific (San Diego, CA). The BCA protein assay kit was obtained from Pierce (Rockford, IL). Monoclonal anti-rat dopamine transporter (DAT) and polyclonal rabbit anti-tyrosine hydroxylase (TH) antibodies were purchased from EMD Millipore (Billerica, MA). Polyclonal rabbit anti-vesicular monoamine transporter 2 (VMAT2) antibodies were generated by Covance to the C-terminal sequence in mouse (CTQNNVQPYPVGDDEESESD). Monoclonal mouse anti-β-actin antibodies were purchased from Sigma-Aldrich (St. Louis, MO). Rabbit anti-GABA transporter 1 (GAT1), vesicular GABA transporter (vGAT), and vesicular glutamate transporter (vGlut) antibodies were purchased from Synaptic Systems (Germany). Mouse anti-microtubule associated protein 2 (MAP2) antibodies were purchased from Abcam (San Francisco, CA). Secondary antibodies conjugated to horseradish peroxidase were obtained from Jackson Immunoresearch Laboratories (West Grove, PA). Secondary antibodies conjugated to fluorescent tags were obtained from Life Technologies (Carlsbad, CA). Monoamine standards for dopamine (DA), dihydroxyphenylacetic acid (DOPAC), and homovanillic acid (HVA) were purchased from Sigma-Aldrich (St. Louis, MO).
2.2 Culturing of SK-N-SH Cells
Cells were cultured in DMEM F12 media supplemented with 100 units/ml penicillin, 100 units/ml streptomycin and 10% fetal bovine serum. Cells were cultured at 37°C in a humidified atmosphere with 5% CO2 and propagated according to the protocol provided by the supplier.
2.3 WST-1 Cytotoxicity Assay
When cells were grown to confluence, they were passaged to 75,000 cells per well in 96-well plates at 100 μl for treatment with HBCDD. Cell death was assessed using the WST-1 Cell Proliferation Assay, as previously described (Wilson et al., 2014b). Following treatment for 24 hours with 0, 5, 10, 15, 20, or 25 μM of HBCDD dissolved in DMSO, 10 μl/well of Cell Proliferation Reagent was added to cells and incubated for 3 hours at 37°C and 5% CO2. Cytotoxicity was then measured by enzymatic cleavage of the tetrazolium salt WST-1 to a water-soluble formazan dye detected by spectral absorbance. Viable cells form more formazan than less viable cells. Spectral absorbance was measured at 450 nanometers on an Epoch BioTek microplate spectrophotometer and analyzed using Gen5 software (2.0) and GraphPad software.
2.4 Primary Culture of Mesencephalic Neurons
Briefly, ventral mesencephalic neuron cultures were prepared from postnatal mice (postnatal day 1–3) as previously described (Bradner et al., 2013b, Wilson et al., 2014b). Brains were dissected in ice cold Hibernate A supplemented with B27. Following isolation of the relevant region and the removal of meninges, tissue pieces were chemically treated with dissociation solution, containing Papain (1 mg/ml), Dispase II (1.2 U/ml), and DNase I (1μl/ml) dissolved in Hibernate A- Calcium for 20 minutes at 37°C and gently agitated every 5 minutes. Tissue was then rinsed in plating media, containing Neurobasal-A and 10% heat inactivated fetal bovine serum and mechanically dissociated using gentle trituration. Cells were plated on poly-d-lysine pre-coated 96-well plates at 40,000 cells per well. After 2 hours of incubation, plating media was removed and immediately switched to Neurobasal-A based culture media containing B27, 1% L-glutamine and 1% penicillin-streptomycin. The following day culture media containing aphidicolin (1μg/ml) was added to reduce the proliferation of glial cells in culture. Primary cultures were treated for 72 hours in vitro with 0, 1.75, 2.5, 5, 7.5, or 10 μM concentrations of HBCDD dissolved in DMSO. After 72 hours, cells were fixed in 4% PFA for 20 minutes and incubated overnight in rabbit anti-TH and mouse anti-MAP2 at 4°C. The following day cultures were incubated with fluorescent secondary antibodies, goat anti-rabbit 488 and goat anti-mouse 572 for 1 hour at room temperature. After staining with DAPI, cells were rinsed and stored in PBS. Images of treated cultures were taken using an Array Scan VTI HCS (Cellomics; Pittsburgh, PA). Forty-nine contiguous fields were taken per well, DAPI+ nuclei were counted, TH+ cell bodies were identified, and neurite length and branch point were recorded for all fields containing at least one TH+ cell body. Based on our previously published protocol our mesencephalic cultures were comprised of approximately 10% TH+ neurons. Objects were identified and measured using the neuronal profiling bioapplication from Thermo Scientific. Image analysis workflow was identical to that determined by Harrill et al., (2011b). In brief, cell bodies were selected or rejected based upon predefined pixel intensity and morphological parameters. Neurites emerging from selected viable cell bodies were individually identified and traced. Statistical significance between the control and treatment groups for neuron count, neurite length, and neurite branch point of TH+ neurons was determined using GraphPad analysis software.
2.5 Animals and Treatment
Eight-week-old male C57BL/6J mice were purchased from Charles River Laboratories (Wilmington, MA). Three month old mice were orally gavaged with 25 μl of corn oil control (n=4) or 25 mg/kg of HBCDD (n=6) dissolved in corn oil vehicle daily for 30 days, as previously described (Caudle et al., 2006, Bradner et al., 2013b). This dosing paradigm was intended to represent the primary route of human exposure, via oral ingestion, to HBCDD. Mice were sacrificed one day following the last exposure, and bilateral striatum was collected for subsequent analysis. Standard rodent chow and tap water were available ad libitum. All procedures were conducted in accordance with the Guide for Care and Use of Laboratory Animals (National Institutes of Health) and have been approved by the Institutional Animal Care and Use Committee at Emory University.
2.6 High Performance Liquid Chromatography (HPLC) of Striatal Neurochemistry
HPLC analysis of neurochemistry was performed as previously described (Caudle et al., 2007a). Briefly, dissected striata were sonicated in 0.1 M perchloric acid. Homogenates were centrifuged at 15,000 ×g and the supernatant filtered through a 0.22 μm filter by centrifugation at 15,000 ×g. The supernatants were analyzed for levels of DA, DOPAC, and HVA. Quantification was made by reference to calibration curves made with individual standards.
2.7 Western Blot Analysis
Western blots were used to quantify the amount of DAT, TH, VMAT2, GAT1, vGAT, vGlut, and β-actin present in samples of striatal tissue from treated and control mice. Analysis was performed as previously described (Caudle et al., 2006). Briefly, striatum samples were homogenized and samples subjected to polyacrylamide gel electrophoresis and electrophoretically transferred to polyvinylidene difluoride membranes. Nonspecific sites were blocked in 7.5% nonfat dry milk in Tris-buffered saline and then membranes incubated overnight in a monoclonal antibody to the N-terminus of DAT. DAT antibody binding was detected using a goat anti-rat horseradish peroxidase secondary antibody (1:10,000) and enhanced chemiluminescence. The luminescence signal was captured on an Alpha Innotech Fluorochem imaging system and stored as a digital image. Densitometric analysis was performed and calibrated to coblotted dilutional standards of pooled striata from all control samples. Membranes were stripped for 15 minutes at room temperature with Pierce Stripping Buffer and sequentially reprobed with β-actin (1:1000), TH (1:1000), VMAT2 (1:10,000), GAT1 (1:1,000), vGAT (1:1,000), and vGlut (1:50,000) antibodies. β-Actin blots were used to ensure equal protein loading across samples.
2.8 Statistical analysis
Statistical analysis of the effects of HBCDD on SK-N-SH cells and primary cultured neurons was performed on raw data for each treatment group by one-way ANOVA. Analysis of the effects of HBCDD on dopaminergic and nondopaminergic endpoints by immunoblotting and HPLC was performed on raw data from each treatment group by Student’s t-test. Post hoc analysis was performed using Tukey’s post hoc test. Significance is reported at the p < 0.05 level.
3.0 Results
In order to initially establish the neurotoxic potential of HBCDD we performed our first assessement using the catecholaminergic SK-N-SH neuroblastoma cell line (Richards and Sadee, 1986, Kidd and Schneider, 2010). Cytotoxicity was assessed using the WST-1 cell viability assay following exposure to DMSO or increasing concentrations of HBCDD (0–25 μM) for 24 hours. Cell viability was reduced in a concentration dependent manner beginning at 10 μM and continuing to 25 μM, which resulted in approximately 70% reduction in cell viability (Figure 1). These findings demonstrate the general neurotoxic potential of HBCDD on a neuronally derived catecholaminergic cell line.
Figure 1.
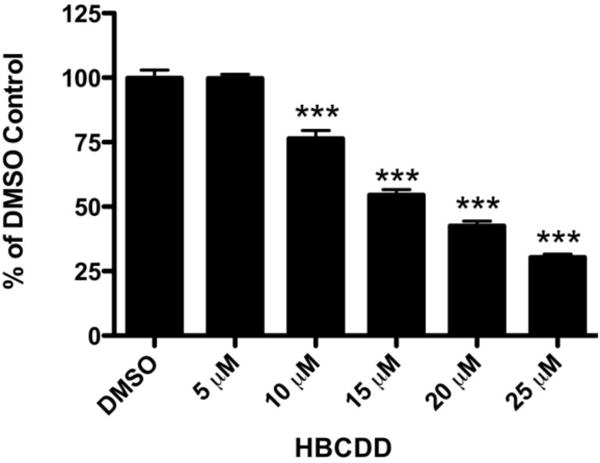
Exposure of SK-N-SH cells to HBCDD causes a reduction in cell viability. Exposure of the catecholaminergic neuroblastoma cell line, SK-N-SH to HBCDD caused a concentration-dependent decrease in cell viability beginning at 10 μM HBCDD. Columns represent percent change from DMSO control. Data represent the mean ± SEM of 8 experimental replicates performed over 3 separate experiments. ***Values that are significantly different from control (p<0.001).
We next sought to extend our investigation of the neurotoxic effects of HBCDD on dopaminergic neurons in a more biologically complex in vitro model system by exposing dopamine-rich primary cultured neurons isolated from the ventral mesencephalon and assessing these effects using high content analysis. This experimental platform allows for the efficient and sensitive evaluation of alterations to neuronal morphology that extends beyond simple quantification of cell loss. Using this approach, HBCDD exposure resulted in a significant decrease in the total number of TH+ neurons in culture at concentrations ranging from 5–10 μM HBCDD (Figure 2). In addition to loss of TH+ neurons, we also observed a reduction in neurite branching and neurite length of the TH+ neurons treated with HBCDD. These data demonstrate an elaborated neurotoxic profile of HBCDD on dopaminergic neurons in vitro.
Figure 2.
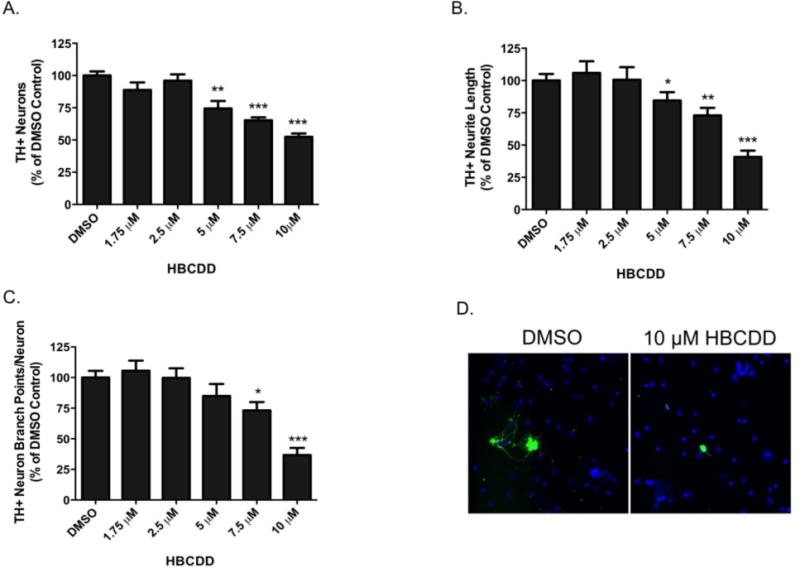
Exposure of TH+ ventral mesencephalic primary cultured neurons to HBCDD caused concentration-dependent alterations of neuron characteristics. (A) Treatment of primary cultures caused significant decreases in the number of TH+ neurons at 5, 7.5, and 10 μM HBCDD. (B) Treatment also caused significant reduction in TH+ neurite length at 5, 7.5, and 10 μM HBCDD and (C) a reduction in the number of TH+ neuron branch points per TH+ neuron at 7.5, and 10 μM HBCDD. (D) Representative micrographs of TH+ neurons treated with DMSO control or 10 μM HBCDD for 72 hrs demonstrate a reduction in the number of TH+ cell bodies in addition to a decrease in the length and complexity of neurite outgrowth. Columns represent percent change from DMSO control. Data represent the mean ± SEM of 4 experimental replicates across 3 separate experiments. *Values that are significantly different from control (p<0.05). **Values that are significantly different from control (p<0.01). ***Values that are significantly different from control (p<0.001).
The effect of HBCDD on the dopamine system was next assessed using an in vivo model. For this approach we orally gavaged 3-month old male mice with 25 mg/kg HBCDD for 30 days and then evaluated alterations to select dopaminergic proteins in the striatum of control and HBCDD treated mice. Using this dosing paradigm we found mice exposed to HBCDD exhibited a significant reduction in the expression of the plasmalemmal DAT, which functions to terminate the dopaminergic signal by recycling dopamine from the synapse (Figure 3). Additionally, similar reductions were observed for VMAT2, which resides on synaptic vesicles and functions to sequester dopamine from the cytosol and into synaptic vesicles for neurotransmitter release in the striatum. In contrast, we did not observe a change in the expression of the dopamine synthesis enzyme, TH, which is a marker of terminal integrity in the striatal dopamine neurons. To further assess the effects of HBCDD on the dopamine system, levels of dopamine and its metabolites were analyzed in the striatum. As shown in Figure 4, exposure to HBCDD for 30 days did not affect the concentrations of dopamine, DOPAC, or HVA in this region.
Figure 3.
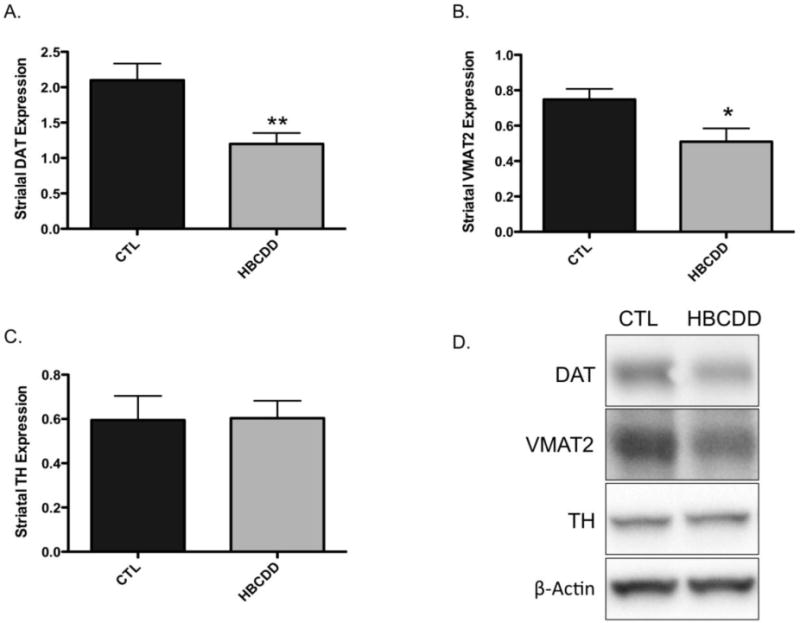
In vivo exposure of mice to HBCDD causes a reduction in proteins involved in dopamine handling. Animals received either 0 (control) or 25 mg/kg HBCDD for 30 days and were then evaluated for alterations in striatal expression of (A) DAT, (B) VMAT2, and (C) TH. (D) Representative immunoblot bands for each marker. Data represents mean ± SEM (4 control and 6 HBCDD treated animals per experimental group). *Values that are significantly different from control (p<0.05). **Values that are significantly different from control (p<0.01).
Figure 4.
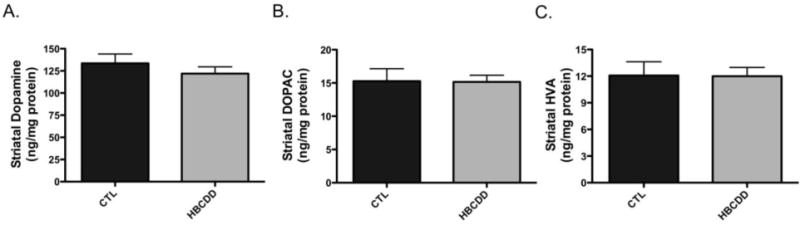
In vivo exposure to HBCDD does not alter the concentration of dopamine or its metabolites in the striatum. Animals received either 0 (control) or 25 mg/kg HBCDD for 30 days and were then evaluated for alterations to (A) dopamine, (B) DOPAC, or (C) HVA in the striatum. Columns represent the mean ± SEM of raw values (ng/mg protein) of 4 control and 6 HBCDD treated animals per experimental group.
Given our findings related to alterations in the dopamine system we were interested in determining the selectivity of these changes for dopamine neurons. The striatum is heavily innervated with GABAergic and glutamatergic projections and interneurons, which further refine the motor output of the basal ganglia (Obeso et al., 2008). Evaluation of transporter proteins involved in GABAergic and glutamatergic signaling in the striatum demonstrated no change in expression of GAT1, vGAT, or vGlut following exposure to HBCDD (Figure 5). These data suggest that the neurotoxic effects of HBCDD are preferential for the dopamine neurons in the striatum, in vivo.
Figure 5.
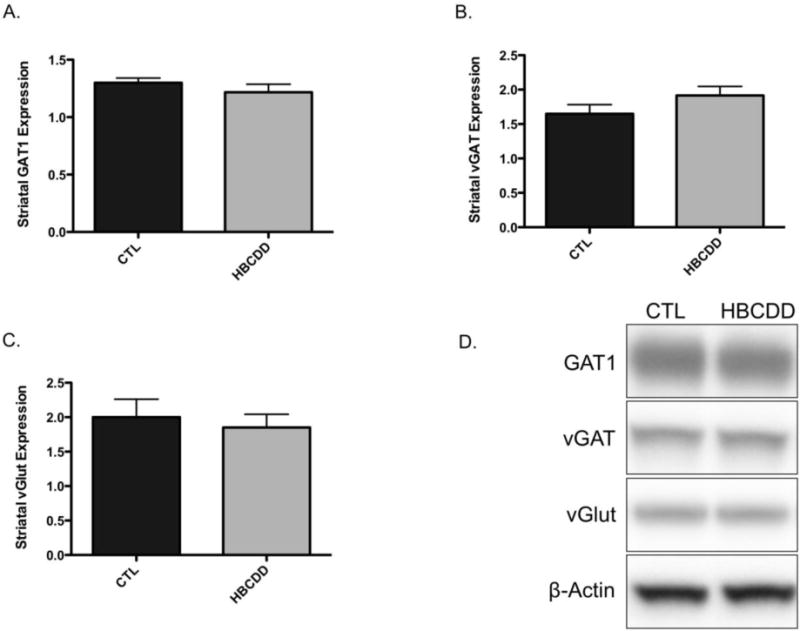
In vivo exposure to HBCDD does not affect the expression of GABAergic or glutamatergic markers in the striatum. Animals received either 0 (control) or 25 mg/kg HBCDD for 30 days and were then evaluated for alterations to (A) GAT1, (B) vGAT, and (C) vGlut in the striatum. (D) Representative immunoblot bands for each marker. Data represents mean ± SEM (4 control and 6 HBCDD treated animals per experimental group).
4.0 Discussion
Exposure to a variety of environmental chemicals has been shown to damage the nigrostriatal dopamine system and be a significant contributor to the etiopathogenesis of Parkinson disease (PD) (Hatcher et al., 2008, Wirdefeldt et al., 2011, Caudle et al., 2012). Work from our group has specifically demonstrated alterations to this circuit following exposure to PCBs, as well as PBDEs (Caudle et al., 2006, Bradner et al., 2013b). As the manufacture and use of these compounds has been or is currently being reduced, the manufacture of chemically similar compounds, such as HBCDD, has been increased. While some studies have addressed the potential neurotoxicity of HBCDD on the dopamine system, many questions still remain regarding the explicit targets and mechanisms involved in neurotoxicity. Thus, our current study sought to investigate the impact of HBCDD exposure on dopamine neurons using an in vitro and in vivo approach.
Initial assessments to better understand the general neurotoxicity of HBCDD focused on acute exposures of catecholaminergic neuroblastoma SK-N-SH cells, resulting in significant decreases in cell viability even at low micromolar concentrations. These reductions in cell viability align very well with those previously observed by our group with another brominated flame retardant compound, DE-71, a commercially available mixture of PBDEs (Bradner et al., 2013b). Similar HBCDD-mediated reductions in cell viability have also been observed in other catecholaminergic cell lines, such as SH-SY5Y, which is a derivative of the SK-N-SH line used in our study (Al-Mousa and Michelangeli, 2012). In contrast, exposure of a liver-derived clonal cell line, HepG2, did not demonstrate overt cytotoxicity following exposure to HBCDD under similar conditions (An et al., 2014). These results suggest the relative sensitivity of catecholaminergic cells to HBCDD neurotoxicity.
We expanded upon the SK-N-SH data with in vitro HBCDD exposure of dopaminergic primary cultured ventral mesencephalic neurons. In our assay, TH+ dopamine neurons also showed sensitivity to HBCDD, with significant reductions in a number of different neuronal metrics, including the number of TH+ neurons as well as the length and arborization of the neurite outgrowths. These results are interesting as they begin to address specific aspects of the dopaminergic morphology, beyond cell number, that are sensitive to damage by HBCDD. While HBCDD treatment resulted in a significant loss of TH+ neurons, HBCDD also caused a reduction in the length and complexity of the TH+ neurite outgrowths that arise from the cell body and are involved in mediating synapse formation and communication with adjacent neurons. Like our SK-N-SH data, these findings are congruent with our previously published observations of PBDE-mediated neurotoxicity in TH+ neurons and follow with additional studies in our lab that have paired primary cultured neurons with high content analysis to delineate specific alterations in neuronal morphology (Bradner et al., 2013a, Bradner et al., 2013b, Wilson et al., 2014a, Wilson et al., 2014b). It should be appreciated that our findings do not suggest that HBCDD-mediated neurotoxicity is selective for dopaminergic neurons and endpoints. Although we did not assess other neuronal populations that might be altered in our cultures, other studies have found HBCDD to cause significant loss of cell viability in cerebellar granule cells, in vitro (Reistad et al., 2006, Ibhazehiebo et al., 2011a, Ibhazehiebo et al., 2011b). Future work will seek to delineate the relative neurotoxicity of HBCDD in other brain regions, including striatum, cortex, and hippocampus, using in vitro and in vivo approaches.
It is critical to note that analysis of neurite morphology in our HBCDD study was not dependent upon loss of TH+ neurons. Our evaluative parameters for these endpoints are performed on intact and viable TH+ neurons, which means a reduction in TH+ neuron number does not underlie a reduction in other morphological endpoints, (Harrill et al., 2011a, Harrill et al., 2011b). These findings are interesting as they suggest that HBCDD-mediated neurotoxicity may be affecting multiple targets on the dopamine neurons. Alterations in neurite length and branching are especially relevant to PD pathogenesis as these dopaminergic processes that innervate the striatum are severely damaged prior to loss of dopaminergic cell bodies in the substantia nigra pars compacta. This suggests that dysfunction of the presynaptic dopaminergic terminal may be a significant component of PD pathogenesis.
Our findings in TH+ primary cultured neurons served to guide our in vivo experiments, which focused on interrogating the effects of HBCDD on select presynaptic dopaminergic proteins in the striatum. While prenatal and neonatal HBCDD exposures have been used to analyze dopamine-related behavioral effects of HBCDD in rodents, there have been no adult exposures or ensuing laboratory measurements of the dopamine circuit in vivo (Lilienthal et al., 2009, Miller-Rhodes et al., 2014). In our study, exposure of 3-month old mice to HBCDD resulted in a precipitous reduction in the expression of DAT and VMAT2, both of which are integral in mediating dopamine homeostasis and neurotransmission in the dopamine circuit. In contrast to these reductions, we did not observe a change in TH expression or levels of dopamine and its major metabolites in the striatum. Similar alterations to presynaptic proteins were also seen by our group in mice that were exposed to the PCB mixtures Aroclor 1254 and 1260, as well as in mice exposed to the PBDE mixture DE-71 (Caudle et al., 2006, Bradner et al., 2013b). Based on our previous work these data suggest that HBCDD selectively targets the expression of DAT and VMAT2. Further, these alterations occur prior to explicit damage or loss of the dopamine terminal, as assessed by expression of the TH protein and dopamine concentrations. Of these alterations, the reductions in VMAT2 raise specific questions regarding the potential effects HBCDD exposure may have on dopamine handling in the cytosolic compartment of the dopamine terminal. Reductions in the expression and function of VMAT2 have demonstrated the importance of dopamine sequestration by VMAT2 (Caudle et al., 2008). Animals that have a 95% reduction in VMAT2 show an age dependent neurodegeneration in the nigrostriatal dopamine circuit that is defined by accumulation of reactive oxygen species as well as a differential loss of DAT and TH expression in the striatum (Caudle et al., 2007a). Related effects of HBCDD on VMAT2 have also been identified by Mariussen and Fonnum (2003), who found HBCDD to be a potent inhibitor of VMAT2 function in an acute synaptic vesicle preparation. Based on these findings, it can be speculated that HBCDD-mediated alteration in dopamine handling could lead to an accumulation of dopamine in the cytosol of dopaminergic terminals, over time. Through normal metabolic actions dopamine can form neurotoxic dopamine-quinones as well as other reactive oxygen and nitrogen species, facilitating further damage to the dopamine terminal and cell bodies, as previously shown by our group (Caudle et al., 2007a, Caudle et al., 2008). The impact of HBCDD-mediated alterations to DAT and VMAT2 on dopaminergic neurotransmission in our model remains to be evaluated. However, findings from Dingemans et al., (2009) has demonstrated treatment of PC12 cells with HBCDD, as well as its single isomers, causes a reduction in depolarization-induced catecholamine neurotransmission. While their findings point to alterations in calcium handling related to transmitter release, dysfunction of other cellular aspects of neurotransmission may be involved. Although not explicitly evaluated in our current study, the behavioral manifestations of these alterations in the nigrostriatal dopamine system will be important to deduce in future experiments. Most notably, it will be critical to assess potential alterations in locomotor activity or other measures of locomotor performance that we have previously coupled with neurotoxic models of the dopamine system and PD, as alterations in movement are premier clinical signs in this disorder (Tillerson et al., 2002, 2003, Caudle et al., 2007a, Caudle et al., 2007b).
Finally, the alterations observed in the striatum appear to be specific for the dopamine circuit as no change in GABAergic or glutamatergic transporters was recorded. The preferential damage to the striatal dopamine system was also observed by our group following treatment with PCBs and PBDEs, and is further supported by a lack of effect of HBCDD on GABAergic and glutamatergic transport in isolated synaptosomes and synaptic vesicles (Mariussen and Fonnum, 2003, Caudle et al., 2006, Bradner et al., 2013b). The reason for the selective vulnerability of the dopamine terminals in the striatum to HBCDD-mediated neurotoxicity is unclear. However, the inherent functional properties of dopamine neurons that predispose them to toxicological insult, including an elevated potential for oxidative stress from metabolism of cytosolic dopamine and a concomitant reduction in antioxidant capacity to attenuate oxidative stress, could contribute (Caudle et al., 2007a, Caudle et al., 2008, Dias et al., 2013).
With these findings in mind, it is important to consider how our exposure paradigm relates to levels of HBCDD found in the human population compared to tissue levels of HBCDD in animal models of exposure. To date, several studies have determined relative levels of HBCDD in various biofluids, including breast milk, plasma, and adipose tissue. In general, these data suggest that HBCDD levels in the human population range from approximately 1 ng/g–20 ng/g (Aylward and Hays, 2011, Kicinski et al., 2012). Although we did not evaluate levels of HBCDD in the plasma or other tissues of our mice in this current study, previous work using a similar paradigm as ours has delineated internal HBCDD concentrations following exposure. Indeed, oral exposure of rats to HBCDD for 28 days found that exposure to 30 mg/kg/day HBCDD resulted in approximately 100 mg/g concentration of HBCDD isomers in the liver of male rats, which would most likely align with internal concentrations in our animals (van der Ven et al., 2006). The discrepancy between HBCDD concentrations observed in the human population compared with animal models addresses the limitations associated with mimicking and recapitulating a lifetime human exposure to a specific chemical in an animal model. Future work in our group will be focused on determining HBCDD concentrations in biofluids of our animal model and better equating them with concentrations found in the human population. More importantly, especially in the context of neurological disease, we will assess HBCDD concentrations in postmortem brain tissue and compare these to levels found in our animal model. With this information in hand we will be better able to refine our exposure paradigm to align with human exposure.
The in vitro and in vivo studies of the nigrostriatal dopamine system presented here demonstrate the neurotoxic effects of HBCDD on dopamine homeostasis. In particular, proteins involved with dopamine handling are significantly reduced in mice exposed orally with HBCDD. This is concerning given the chemical similarities between HBCDD and PBDEs, which have also been demonstrated as dopaminergic neurotoxicants, and the ubiquitous nature of HBCDD in the environment (Fonnum and Mariussen, 2009, Marvin et al., 2011). Future implications of HBCDD exposure to humans should continue to be investigated, starting with its possible role as an environmental risk factor for PD.
Highlights.
Exposure to environmental toxicants is a risk factor for Parkinson disease (PD)
Flame retardant chemicals, like PCBs and PBDEs alter the dopamine system, as in PD
Manufacture and use of HBCDD has increased as PCBs and PBDEs have been phased out
Our study found HBCDD damages dopamine neurons using in vitro and in vivo models
Future work will further identify HBCDD-mediated alterations to dopamine synapses
Acknowledgments
This work was supported by National Institutes of Health grants R00 ES017477 (WMC), P30 ES019776 (WMC), RO1 ES021800 (JRR), and P30 ES005022 (JRR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al-Mousa F, Michelangeli F. Some commonly used brominated flame retardants cause Ca2+-ATPase inhibition, beta-amyloid peptide release and apoptosis in SH-SY5Y neuronal cells. PloS one. 2012;7:e33059. doi: 10.1371/journal.pone.0033059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J, Chen C, Wang X, Zhong Y, Zhang X, Yu Y, Yu Z. Oligomeric proanthocyanidins alleviate hexabromocyclododecane-induced cytotoxicity in HepG2 cells through regulation on ROS formation and mitochondrial pathway. Toxicology in vitro: an international journal published in association with BIBRA. 2014;28:319–326. doi: 10.1016/j.tiv.2013.11.009. [DOI] [PubMed] [Google Scholar]
- Aylward LL, Hays SM. Biomonitoring-based risk assessment for hexabromocyclododecane (HBCD) International journal of hygiene and environmental health. 2011;214:179–187. doi: 10.1016/j.ijheh.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Blandini F, Nappi G, Tassorelli C, Martignoni E. Functional changes of the basal ganglia circuitry in Parkinson’s disease. Progress in neurobiology. 2000;62:63–88. doi: 10.1016/s0301-0082(99)00067-2. [DOI] [PubMed] [Google Scholar]
- Bradner JM, Suragh TA, Caudle WM. Alterations to the circuitry of the frontal cortex following exposure to the polybrominated diphenyl ether mixture, DE-71. Toxicology. 2013a;312:48–55. doi: 10.1016/j.tox.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner JM, Suragh TA, Wilson WW, Lazo CR, Stout KA, Kim HM, Wang MZ, Walker DI, Pennell KD, Richardson JR, Miller GW, Caudle WM. Exposure to the polybrominated diphenyl ether mixture DE-71 damages the nigrostriatal dopamine system: role of dopamine handling in neurotoxicity. Experimental neurology. 2013b;241:138–147. doi: 10.1016/j.expneurol.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudle WM, Colebrooke RE, Emson PC, Miller GW. Altered vesicular dopamine storage in Parkinson’s disease: a premature demise. Trends in neurosciences. 2008;31:303–308. doi: 10.1016/j.tins.2008.02.010. [DOI] [PubMed] [Google Scholar]
- Caudle WM, Guillot TS, Lazo CR, Miller GW. Industrial toxicants and Parkinson’s disease. Neurotoxicology. 2012;33:178–188. doi: 10.1016/j.neuro.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudle WM, Richardson JR, Delea KC, Guillot TS, Wang M, Pennell KD, Miller GW. Polychlorinated biphenyl-induced reduction of dopamine transporter expression as a precursor to Parkinson’s disease-associated dopamine toxicity. Toxicological sciences: an official journal of the Society of Toxicology. 2006;92:490–499. doi: 10.1093/toxsci/kfl018. [DOI] [PubMed] [Google Scholar]
- Caudle WM, Richardson JR, Wang MZ, Taylor TN, Guillot TS, McCormack AL, Colebrooke RE, Di Monte DA, Emson PC, Miller GW. Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007a;27:8138–8148. doi: 10.1523/JNEUROSCI.0319-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudle WM, Tillerson JL, Reveron ME, Miller GW. Use-dependent behavioral and neurochemical asymmetry in MPTP mice. Neuroscience letters. 2007b;418:213–216. doi: 10.1016/j.neulet.2006.03.045. [DOI] [PubMed] [Google Scholar]
- Covaci A, Gerecke AC, Law RJ, Voorspoels S, Kohler M, Heeb NV, Leslie H, Allchin CR, De Boer J. Hexabromocyclododecanes (HBCDs) in the environment and humans: a review. Environmental science & technology. 2006;40:3679–3688. doi: 10.1021/es0602492. [DOI] [PubMed] [Google Scholar]
- Covaci A, Harrad S, Abdallah MA, Ali N, Law RJ, Herzke D, de Wit CA. Novel brominated flame retardants: a review of their analysis, environmental fate and behaviour. Environment international. 2011;37:532–556. doi: 10.1016/j.envint.2010.11.007. [DOI] [PubMed] [Google Scholar]
- de Wit CA. An overview of brominated flame retardants in the environment. Chemosphere. 2002;46:583–624. doi: 10.1016/s0045-6535(01)00225-9. [DOI] [PubMed] [Google Scholar]
- Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson’s disease. Journal of Parkinson’s disease. 2013;3:461–491. doi: 10.3233/JPD-130230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingemans MM, Heusinkveld HJ, de Groot A, Bergman A, van den Berg M, Westerink RH. Hexabromocyclododecane inhibits depolarization-induced increase in intracellular calcium levels and neurotransmitter release in PC12 cells. Toxicological sciences: an official journal of the Society of Toxicology. 2009;107:490–497. doi: 10.1093/toxsci/kfn249. [DOI] [PubMed] [Google Scholar]
- Fonnum F, Mariussen E. Mechanisms involved in the neurotoxic effects of environmental toxicants such as polychlorinated biphenyls and brominated flame retardants. Journal of neurochemistry. 2009;111:1327–1347. doi: 10.1111/j.1471-4159.2009.06427.x. [DOI] [PubMed] [Google Scholar]
- Harrill JA, Freudenrich TM, Robinette BL, Mundy WR. Comparative sensitivity of human and rat neural cultures to chemical-induced inhibition of neurite outgrowth. Toxicology and applied pharmacology. 2011a;256:268–280. doi: 10.1016/j.taap.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Harrill JA, Robinette BL, Mundy WR. Use of high content image analysis to detect chemical-induced changes in synaptogenesis in vitro. Toxicology in vitro: an international journal published in association with BIBRA. 2011b;25:368–387. doi: 10.1016/j.tiv.2010.10.011. [DOI] [PubMed] [Google Scholar]
- Hatcher JM, Pennell KD, Miller GW. Parkinson’s disease and pesticides: a toxicological perspective. Trends in pharmacological sciences. 2008;29:322–329. doi: 10.1016/j.tips.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbstman JB, Mall JK. Developmental Exposure to Polybrominated Diphenyl Ethers and Neurodevelopment. Current environmental health reports. 2014;1:101–112. doi: 10.1007/s40572-014-0010-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibhazehiebo K, Iwasaki T, Shimokawa N, Koibuchi N. 1,2,5,6,9,10-alphaHexabromocyclododecane (HBCD) impairs thyroid hormone-induced dendrite arborization of Purkinje cells and suppresses thyroid hormone receptor-mediated transcription. Cerebellum. 2011a;10:22–31. doi: 10.1007/s12311-010-0218-1. [DOI] [PubMed] [Google Scholar]
- Ibhazehiebo K, Iwasaki T, Xu M, Shimokawa N, Koibuchi N. Brain-derived neurotrophic factor (BDNF) ameliorates the suppression of thyroid hormone-induced granule cell neurite extension by hexabromocyclododecane (HBCD) Neuroscience letters. 2011b;493:1–7. doi: 10.1016/j.neulet.2011.01.062. [DOI] [PubMed] [Google Scholar]
- Kicinski M, Viaene MK, Den Hond E, Schoeters G, Covaci A, Dirtu AC, Nelen V, Bruckers L, Croes K, Sioen I, Baeyens W, Van Larebeke N, Nawrot TS. Neurobehavioral function and low-level exposure to brominated flame retardants in adolescents: a cross-sectional study. Environmental health: a global access science source. 2012;11:86. doi: 10.1186/1476-069X-11-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd SK, Schneider JS. Protection of dopaminergic cells from MPP+-mediated toxicity by histone deacetylase inhibition. Brain research. 2010;1354:172–178. doi: 10.1016/j.brainres.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law RJ, Alaee M, Allchin CR, Boon JP, Lebeuf M, Lepom P, Stern GA. Levels and trends of polybrominated diphenylethers and other brominated flame retardants in wildlife. Environment international. 2003;29:757–770. doi: 10.1016/S0160-4120(03)00110-7. [DOI] [PubMed] [Google Scholar]
- Lilienthal H, van der Ven LT, Piersma AH, Vos JG. Effects of the brominated flame retardant hexabromocyclododecane (HBCD) on dopamine-dependent behavior and brainstem auditory evoked potentials in a one-generation reproduction study in Wistar rats. Toxicology letters. 2009;185:63–72. doi: 10.1016/j.toxlet.2008.12.002. [DOI] [PubMed] [Google Scholar]
- Mariussen E, Fonnum F. The effect of brominated flame retardants on neurotransmitter uptake into rat brain synaptosomes and vesicles. Neurochemistry international. 2003;43:533–542. doi: 10.1016/s0197-0186(03)00044-5. [DOI] [PubMed] [Google Scholar]
- Marvin CH, Tomy GT, Armitage JM, Arnot JA, McCarty L, Covaci A, Palace V. Hexabromocyclododecane: current understanding of chemistry, environmental fate and toxicology and implications for global management. Environmental science & technology. 2011;45:8613–8623. doi: 10.1021/es201548c. [DOI] [PubMed] [Google Scholar]
- Miller-Rhodes P, Popescu M, Goeke C, Tirabassi T, Johnson L, Markowski VP. Prenatal exposure to the brominated flame retardant hexabromocyclododecane (HBCD) impairs measures of sustained attention and increases age-related morbidity in the Long-Evans rat. Neurotoxicology and teratology. 2014;45c:34–43. doi: 10.1016/j.ntt.2014.06.009. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Marin C, Rodriguez-Oroz C, Blesa J, Benitez-Temino B, Mena-Segovia J, Rodriguez M, Olanow CW. The basal ganglia in Parkinson’s disease: current concepts and unexplained observations. Annals of neurology. 2008;64(Suppl 2):S30–46. doi: 10.1002/ana.21481. [DOI] [PubMed] [Google Scholar]
- Reistad T, Fonnum F, Mariussen E. Neurotoxicity of the pentabrominated diphenyl ether mixture, DE-71, and hexabromocyclododecane (HBCD) in rat cerebellar granule cells in vitro. Archives of toxicology. 2006;80:785–796. doi: 10.1007/s00204-006-0099-8. [DOI] [PubMed] [Google Scholar]
- Richards ML, Sadee W. Human neuroblastoma cell lines as models of catechol uptake. Brain research. 1986;384:132–137. doi: 10.1016/0006-8993(86)91228-x. [DOI] [PubMed] [Google Scholar]
- Saegusa Y, Fujimoto H, Woo GH, Inoue K, Takahashi M, Mitsumori K, Hirose M, Nishikawa A, Shibutani M. Developmental toxicity of brominated flame retardants, tetrabromobisphenol A and 1,2,5,6,9,10-hexabromocyclododecane, in rat offspring after maternal exposure from mid-gestation through lactation. Reproductive toxicology. 2009;28:456–467. doi: 10.1016/j.reprotox.2009.06.011. [DOI] [PubMed] [Google Scholar]
- Saegusa Y, Fujimoto H, Woo GH, Ohishi T, Wang L, Mitsumori K, Nishikawa A, Shibutani M. Transient aberration of neuronal development in the hippocampal dentate gyrus after developmental exposure to brominated flame retardants in rats. Archives of toxicology. 2012;86:1431–1442. doi: 10.1007/s00204-012-0824-4. [DOI] [PubMed] [Google Scholar]
- Seegal RF, Bush B, Brosch KO. Sub-chronic exposure of the adult rat to Aroclor 1254 yields regionally-specific changes in central dopaminergic function. Neurotoxicology. 1991;12:55–65. [PubMed] [Google Scholar]
- Seegal RF, Bush B, Brosch KO. Decreases in dopamine concentrations in adult, non-human primate brain persist following removal from polychlorinated biphenyls. Toxicology. 1994;86:71–87. doi: 10.1016/0300-483x(94)90054-x. [DOI] [PubMed] [Google Scholar]
- Tillerson JL, Caudle WM, Reveron ME, Miller GW. Detection of behavioral impairments correlated to neurochemical deficits in mice treated with moderate doses of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Experimental neurology. 2002;178:80–90. doi: 10.1006/exnr.2002.8021. [DOI] [PubMed] [Google Scholar]
- Tillerson JL, Caudle WM, Reveron ME, Miller GW. Exercise induces behavioral recovery and attenuates neurochemical deficits in rodent models of Parkinson’s disease. Neuroscience. 2003;119:899–911. doi: 10.1016/s0306-4522(03)00096-4. [DOI] [PubMed] [Google Scholar]
- van der Ven LT, Verhoef A, van de Kuil T, Slob W, Leonards PE, Visser TJ, Hamers T, Herlin M, Hakansson H, Olausson H, Piersma AH, Vos JG. A 28-day oral dose toxicity study enhanced to detect endocrine effects of hexabromocyclododecane in Wistar rats. Toxicological sciences: an official journal of the Society of Toxicology. 2006;94:281–292. doi: 10.1093/toxsci/kfl113. [DOI] [PubMed] [Google Scholar]
- Viberg H, Fredriksson A, Eriksson P. Neonatal exposure to polybrominated diphenyl ether (PBDE 153) disrupts spontaneous behaviour, impairs learning and memory, and decreases hippocampal cholinergic receptors in adult mice. Toxicology and applied pharmacology. 2003;192:95–106. doi: 10.1016/s0041-008x(03)00217-5. [DOI] [PubMed] [Google Scholar]
- Viberg H, Johansson N, Fredriksson A, Eriksson J, Marsh G, Eriksson P. Neonatal exposure to higher brominated diphenyl ethers, hepta-, octa-, or nonabromodiphenyl ether, impairs spontaneous behavior and learning and memory functions of adult mice. Toxicological sciences: an official journal of the Society of Toxicology. 2006;92:211–218. doi: 10.1093/toxsci/kfj196. [DOI] [PubMed] [Google Scholar]
- Wilson WW, Onyenwe W, Bradner JM, Nennig SE, Caudle WM. Developmental exposure to the organochlorine insecticide endosulfan alters expression of proteins associated with neurotransmission in the frontal cortex. Synapse. 2014a;68:485–497. doi: 10.1002/syn.21764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WW, Shapiro LP, Bradner JM, Caudle WM. Developmental exposure to the organochlorine insecticide endosulfan damages the nigrostriatal dopamine system in male offspring. Neurotoxicology. 2014b;44:279–287. doi: 10.1016/j.neuro.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirdefeldt K, Adami HO, Cole P, Trichopoulos D, Mandel J. Epidemiology and etiology of Parkinson’s disease: a review of the evidence. European journal of epidemiology. 2011;26(Suppl 1):S1–58. doi: 10.1007/s10654-011-9581-6. [DOI] [PubMed] [Google Scholar]