

Abstract
Adenoid cystic carcinoma (AdCC) is a rare type of triple-negative breast cancer (TNBC) characterized by the presence of the MYB-NFIB fusion gene. The molecular underpinning of breast AdCCs other than the MYB-NFIB fusion gene remains largely unexplored. Here we sought to define the repertoire of somatic genetic alterations of breast AdCCs. We performed whole exome sequencing, followed by orthogonal validation, of 12 breast AdCCs to determine the landscape of somatic mutations and gene copy number alterations. Fluorescence in situ hybridization and reverse transcription PCR were used to define the presence of MYB gene rearrangements and MYB-NFIB chimeric transcripts. Unlike common forms of TNBC, we found that AdCCs have a low mutation rate (0.27 non-silent mutations/Mb), lack mutations in TP53 and PIK3CA, and display a heterogeneous constellation of known cancer genes affected by somatic mutations, including MYB, BRAF, FBXW7, SMARCA5, SF3B1 and FGFR2. MYB and TLN2 were affected by somatic mutations in two cases each. Akin to salivary gland AdCCs, breast AdCCs were found to harbor mutations targeting chromatin remodeling, cell adhesion, RNA biology, ubiquitination, and canonical signaling pathway genes. We observed that although breast AdCCs had rather simple genomes, they likely display intra-tumor genetic heterogeneity at diagnosis. Taken together, these findings demonstrate that the mutational burden and mutational repertoire of breast AdCCs are more similar to those of salivary gland AdCCs than to those of other types of TNBCs, emphasizing the importance of histologic subtyping of TNBCs. Furthermore, our data provide direct evidence that AdCCs harbor a distinctive mutational landscape and genomic structure, irrespective of disease site of origin.
Keywords: Adenoid cystic carcinoma, triple-negative breast cancer, genomics, MYB-NFIB, massively parallel sequencing
INTRODUCTION
Adenoid cystic carcinomas (AdCCs) are malignant tumors most commonly affecting the salivary glands, but can also be found in other anatomical sites including the breast, lungs and prostate[1]. AdCC of the breast is a rare (<1%) special histologic type of breast cancer that displays a triple-negative (i.e. estrogen receptor (ER)-negative, progesterone receptor (PR)-negative and HER2-negative) and basal-like phenotype[1-4]. Notably, breast AdCCs have an indolent clinical behavior, which is at variance with that of salivary gland AdCCs or common-type TNBCs (i.e. invasive ductal carcinomas of no special type, IDC-NSTs)[1,5]. Furthermore, while a subset of common-type TNBCs are sensitive to chemotherapy[5], it is thought that chemotherapy response rates in patients with breast AdCCs are low, akin to those observed in patients with salivary gland AdCCs[6,7].
AdCCs provide a clear example of genotypic-phenotypic correlation, as they display similar histologic characteristics irrespective of the site of origin and harbor the recurrent t(6;9)(q22-23;p23-24) translocation that results in the formation of the MYB-NFIB fusion gene[1,8]. The mechanistic basis for the oncogenic properties of this chimeric fusion gene, and in particular the role of the 3’-part of NFIB, has yet to be fully characterized. There is evidence, however, that this fusion gene results in activation and overexpression of MYB at mRNA and protein levels[1,8-10]. The prevalence of the MYB-NFIB fusion gene is reported to range from 28% to 100% in salivary gland AdCCs[8-13] and from 23% to 100% in breast AdCCs[8,9,14-16], however the clinical significance of this observation remains unclear. Although a substantial proportion of AdCCs lack the MYB-NFIB fusion gene, the majority of MYB-NFIB fusion gene-negative salivary gland AdCCs likely display activation of MYB due to mechanisms other than the t(6;9) chromosomal translocation[17].
Recent massively parallel sequencing studies have shown that common forms of triple-negative and basal-like breast cancers harbor recurrent TP53 (50-80%) and PIK3CA (10%) mutations, and high levels of gene copy number alterations[18,19]. The molecular underpinning of AdCCs of the breast other than the MYB-NFIB fusion gene remains largely unexplored, however. Our group has previously reported that a subset (12%) of breast AdCCs harbor mutations in BRAF[15], and that breast AdCCs display lower levels of genetic instability, as defined by gene copy number alterations, than basal-like IDC-NSTs[14,20]. The contribution of somatic mutations affecting other genes to the disease has yet to be determined.
The aims of our study were to define the landscape of somatic mutations and gene copy number alterations of breast AdCCs using whole exome sequencing, and to characterize the type and prevalence of MYB gene rearrangements and the respective MYB-NFIB chimeric transcript expression in these tumors.
MATERIALS AND METHODS
Case selection
Representative fresh/frozen and formalin-fixed paraffin-embedded (FFPE) tissues of 12 AdCCs of the breast were retrieved from the files of Institut Curie, Paris, France and Memorial Sloan Kettering Cancer Center (MSKCC), New York, USA. These cases had not been previously subjected to molecular analyses and have not been included in any previous study reported by our teams. All cases were centrally reviewed by two pathologists with an interest and expertise in breast pathology (AV-S and JSR-F). Tumors were classified according to the World Health Organization (WHO) criteria[3], and graded according to the Nottingham grading system[21]. This study was approved by the local ethics committees from the authors’ institutions. Patient consents were obtained if required by the protocols approved.
Immunohistochemical analysis
Immunohistochemical analysis of the AdCCs was performed on representative 4 μm-thick FFPE sections, using antibodies against ER, PR and HER2 as previously described[22]. Positive and negative controls were included in each slide run[22]. Immunohistochemical results were evaluated by two pathologists (AV-S and JSR-F) according to the American Society of Clinical Oncology (ASCO)/College of American Pathologists (CAP) guidelines[23,24]. MYB protein expression was assessed by immunohistochemistry as previously described[25].
DNA and RNA extraction
The AdCCs included in this study contained >70% tumor cells after manual microdissection of the frozen specimen. Tumor sections were reviewed by pathologists under a stereomicroscope (Olympus SZ61) and surrounding healthy tissue was removed using a sterile needle. Genomic DNA was extracted from the tumor samples and matched normal tissue samples, confirmed by pathology review to be devoid of any neoplastic cells, using a standard phenol/chloroform-based protocol, and RNA was extracted from the tumor tissue using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Nucleic acids were quantified using the Qubit Fluorometer assay (Life Technologies), and RNA integrity was defined using a Bioanalyzer (Agilent Technologies).
Fluorescence in situ hybridization (FISH)
The presence of the t(6;9)(q22-23;p23-24) translocation was investigated using the ZytoLight SPEC MYB Dual Color Break Apart Probe (Zytovision) following the manufacturer’s instructions. Nuclei were counterstained using DAPI. Probes were visualized as red (Texas red) and green (FITC), and images were captured with a CDD camera, filtered and processed using a Leica Microsystems microscope. At least 50 interphase nuclei were analyzed per hybridization as previously described[14].
Reverse transcription (RT)-PCR and quantitative (q)RT-PCR
Standard end-point RT-PCR was performed in triplicate to detect the presence of the MYB-NFIB fusion transcripts, as previously described[8,9,16,22,26] (Supplementary Methods). qRT-PCR was performed to analyze the expression levels of 5’ and 3’ portions/exons of MYB using TaqMan Assay-on-Demand, as previously described[22] (Supplementary Methods).
Digital gene expression analysis
For qualitative analysis of the 5’ to 3’ expression ratio of MYB and NFIB mRNAs, 100ng of total RNA from each tumor sample was hybridized to a custom designed gene CodeSet (i.e. non-enzymatic RNA profiling using barcoded fluorescent probes; NanoString Technologies) using a validated protocol employed in the Integrated Genomics Operation (IGO) at MSKCC (Supplementary Methods, Supplementary Table S1).
Whole exome massively parallel sequencing
Matched tumor and normal DNA were subjected to whole exome capture (Agilent SureSelect Human All Exon v4) on an Illumina HiSeq 2000 using a validated protocol employed in the IGO at MSKCC, essentially as previously described[27]. For each sample, reads were aligned to the reference human genome GRCh37 using the Burrows-Wheeler Aligner (BWA, v0.6.2)[28]. Local realignment and quality score recalibration were performed using the Genome Analysis Toolkit (GATK, v3.1.1)[29]. De-duplication was performed using Picard (v1.92). Somatic single nucleotide variants (SNVs) were detected by MuTect (v1.1.4)[30], small somatic insertions and deletions (indels) by VarScan2 (v2.3.6)[31], Strelka[32] and Scalpel[33], and manually reviewed using the integrative genomic viewer (IGV)[34]. Variants found with >5% global minor allele frequency in dbSNP (Build 137) or that were supported by <5 reads were disregarded. SNVs for which the tumor variant allele fraction was <5 times than that of the normal variant allele fraction were disregarded as previously described[27]. Mutations were validated employing a targeted amplicon sequencing approach (Supplementary Methods). The potential functional effect of each SNV was assessed using previously described combination of mutation function predictors[35,36], as previously described[37] (Supplementary Methods). Validated mutations were functionally annotated into molecular pathways and networks, using the Ingenuity Pathway Analysis (IPA) software (http://www.ingenuity.com) and ConsensusPathDB-human[38] (Supplementary Methods).
To define the gene copy number alterations of breast AdCCs, whole exome sequencing data were analyzed using VarScan2[31] and GISTIC2.0[39]. In addition, ABSOLUTE was employed to infer tumor purity, tumor cell ploidy and clonal heterogeneity, as previously described[40].
Whole exome sequencing data have been deposited into the NCBI Sequence Read Archive, under accession code SRP053134.
Statistical analysis
Statistical comparisons between the mutation rates of MYB-NFIB fusion gene-positive and fusion gene-negative AdCCs, and between AdCCs and i) triple-negative and/or basal-like breast cancers and ii) salivary gland AdCCs were performed using the Mann-Whitney U test. Statistical analysis of qRT-PCR data was carried out using one-way ANOVA, Bonferroni’s multiple comparison correction, alpha: 0.05. Two-tailed p-values were employed for all comparisons. Statistical analyses were performed using GraphPad Prism (v6.0f).
RESULTS
MYB-NFIB fusion gene prevalence and expression
All breast AdCCs included in this study were of triple-negative phenotype (i.e. lacked expression of ER, PR and HER2), of histologic grade 1 (58%) or grade 2 (42%), and composed of cribriform (17%), tubular-cribriform (67%), solid-cribriform (8%) or solid-trabecular (8%) growth patterns (Supplementary Figure S1; Supplementary Table S2). Consistent with previous reports[8,9,14-16], fluorescence in situ hybridization (FISH) and reverse transcription PCR (RT-PCR) analysis revealed that 10/12 breast AdCCs (83%) harbored the MYB-NFIB fusion gene (Table 1, Figure 1A, Supplementary Figure S2). Chimeric transcripts consisting of MYB exon 14 linked to NFIB exon 8c (n=6) or exon 9 (n=3) were most prevalent in the breast AdCCs analyzed, and in one case (AdCC2T) the fusion transcript consisted of MYB exon 9 linked to NFIB exon 8c (Table 1, Figure 1A, Supplementary Figures S2 and S3). Of note, due to alternative splicing and potentially variable breakpoints in MYB and NFIB, some breast AdCCs were found to express more than one MYB-NFIB transcript or splice variant (Figure 1A, Supplementary Figure S2), as previously described[8]. Using digital gene expression analysis (NanoString Technologies), we confirmed the elevated mRNA expression levels of the 5’ portion of MYB and the 3’ portion of NFIB in all ten MYB-NFIB fusion gene-positive breast AdCCs (Figure 1B). By contrast, the two MYB-NFIB fusion gene-negative breast AdCCs did not display elevated 5’ MYB and 3’ NFIB mRNA levels, but a pattern of expression similar to those of the MYB-NFIB fusion gene-negative ER-negative breast epithelial cell lines MCF10A and MCF12A and the ER-positive MYB-expressing breast cancer cell lines T47D and MCF7. Digital gene expression profiling and quantitative RT-PCR (qRT-PCR) further revealed that the overall MYB and 5’ MYB mRNA expression levels in one of the fusion gene-negative tumors (i.e. AdCC12T) were significantly higher than in the remaining fusion gene-negative samples (i.e. case AdCC11T and breast cell lines) tested (p<0.001), but similar to those of MYB 5’ exons of chimeric transcripts in MYB-NFIB fusion gene-positive breast AdCCs (Figure 1B inset, 1C). These data suggest that MYB expression may be increased in AdCC12T due to a mechanism other than the MYB-NFIB rearrangement. Finally, the two breast AdCCs lacking the MYB-NFIB fusion gene did not show obvious histologic or clinico-pathologic differences as compared to the ten AdCCs harboring MYB rearrangements.
Table 1.
Molecular features of breast AdCCs included in this study.
| Case ID | MYB rearrangement (FISH) | MYB-NFIB fusion transcript (RT-PCR) | Non-synonymous mutations (n) | Selected gene copy number alterations |
|---|---|---|---|---|
| AdCC1T | yes | MYB Exon 14 – NFIB Exon 8c | 19 | 12q12-q14.1 loss |
| AdCC2T | yes | MYB Exon 9 – NFIB Exon 8c | 23 | 12q12-q14.1 loss |
| AdCC3T | yes | MYB Exon 14 – NFIB Exon 8c | 6 | 12q12-q14.1 loss |
| AdCC4T | yes | MYB Exon 14 – NFIB Exon 9 | 22 | 17q21-q25.1 gain; 17p13.3-p11.2 loss |
| AdCC5T | no | MYB Exon 14 – NFIB Exon 9 | 11 | 9q13-q34.2 loss |
| AdCC6T | yes | MYB Exon 14 – NFIB Exon 8c | 18 | |
| AdCC8T | yes | MYB Exon 14 – NFIB Exon 8c | 9 | 12q12-q14.1 loss |
| AdCC9T | yes | MYB Exon 14 – NFIB Exon 9 | 10 | |
| AdCC10T | yes | MYB Exon 14 – NFIB Exon 8c | 10 | 17q21-q25.1 gain; 17p13.3-p11.2 loss |
| AdCC11T | no | not detectable | 7 | 17q21-q25.1 gain |
| AdCC12T | no | not detectable | 18 | |
| AdCC32T | yes | MYB Exon 14 – NFIB Exon 8c | 14 | 12q12-q14.1 loss |
FISH, fluorescence in situ hybridization; n, number; RT-PCR, reverse transcriptase PCR.
Figure 1. Detection of the MYB-NFIB fusion gene and MYB and NFIB expression in breast AdCCs.
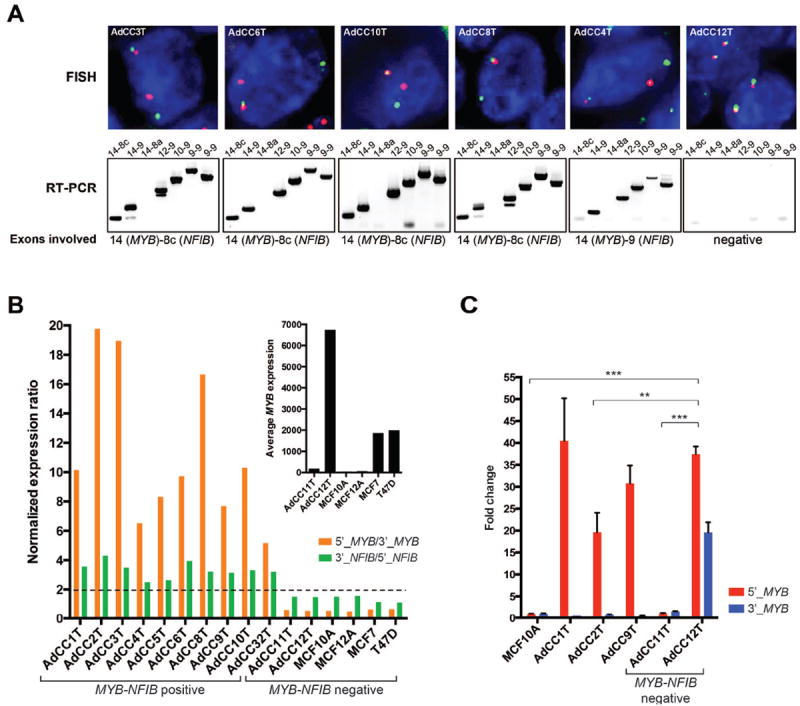
(A), Representative FISH micrographs and results of RT-PCR. A MYB dual color break-apart probe (top panel) was employed for FISH, where the green and the red probes are proximal and distal to the MYB breakpoint cluster region, respectively. The MYB-NFIB fusion transcripts (bottom panel) were detected using primers located in MYB exons 9, 10, 12 and 14, and in NFIB exons 8a, 8c and 9; lane names indicate the respective exons tested. In AdCC12T, neither a MYB split signal nor a MYB-NFIB fusion transcript could be identified. For the RT-PCR results of the remaining cases, see Supplementary Figure S2. (B) Normalized mRNA expression ratios of 5’_MYB(exons1-2)/3’_MYB(3’UTR) and 3’_NFIB(3’UTR)/5’_NFIB(exons5-6) for all breast AdCCs defined by digital gene expression. Controls included RNA derived from MCF10A and MCF12A breast epithelial cells (negative for MYB and MYB-NFIB fusion mRNA expression), and from T47D and MCF7 ER-positive breast cancer cell lines (positive for MYB and negative for MYB-NFIB fusion mRNA expression). Dotted line, 2-fold expression difference. Inset illustrating the average between 5’ and 3’ signals of MYB mRNA expression in the MYB-NFIB fusion gene-negative tumors AdCC11T and AdCC12T and in cell line controls. (C) Representative quantitative RT-PCR analysis of expression of 5’ and 3’ regions of MYB mRNA in 3 MYB-NFIB fusion gene-positive and the 2 MYB-NFIB fusion gene-negative AdCCs, and MCF10A cell line control. P-values, one-way ANOVA, Bonferroni’s multiple comparison correction, alpha: 0.05. **p<0.01, ***p<0.001. Error bars, s.d. of the mean (n=3 experimental replicates).
Spectrum of somatic mutations
We employed massively parallel sequencing to characterize the repertoire of somatic genetic alterations affecting the exomes of the 12 breast AdCCs included in this study. Tumor and matched normal DNA were subjected to whole exome sequencing, which resulted in a median sequencing depth of 78x (range 43-129x) and a mean of 88% of the target sequence covered to at least 10x depth (Supplementary Table S3). To ensure the accuracy of our massively parallel sequencing results, we performed an independent validation of the 181 candidate non-synonymous somatic mutations identified, including all single nucleotide variations (SNVs) and small insertions and deletions (indels), using targeted amplicon resequencing (see Methods; Supplementary Tables S4 and S5). As a result, we identified 167 non-synonymous somatic mutations affecting 160 genes in the 12 AdCCs of the breast analyzed here. The majority of validated somatic mutations identified in breast AdCCs were missense mutations (n=130), but nonsense mutations (n=18), essential splice site mutations (n=6), frameshift mutations (n=10), and in-frame indels (n=3) were also found as well as a total of 73 silent mutations (Supplementary Tables S4 and S5). A median of 12.5 mutations per tumor was found (range 6-23; Table 1, Supplementary Table S5), corresponding to an average of 0.27 non-silent mutations/Mb. Reanalysis of The Cancer Genome Atlas (TCGA) breast study[19] revealed that the exonic mutation rate of breast AdCCs was significantly lower than that of common forms of basal-like breast cancers (1.41 non-silent mutations/Mb; Mann-Whitney U-test, p<0.001) or TNBCs (1.38 non-silent mutations/Mb; Mann-Whitney U-test, p<0.001). In fact, the mutation rate found in breast AdCCs was more similar to that reported for pediatric malignancies[41] and salivary gland AdCCs (0.31 non-silent mutations/Mb; Mann-Whitney U-test, p>0.1)[12].
Mutational landscape of AdCCs of the breast
Common-type triple-negative and basal-like breast cancers are characterized by the presence of mutations targeting TP53 and PIK3CA in 50%-80% and approximately 10% of tumors, respectively[18,19]. In addition, ATM mutations, BRCA1 and BRCA2 defects, and RB1 pathway deregulation have been described as being features characteristic of basal-like breast cancer[19]. In this study, we found a rather substantial heterogeneity in the landscape of somatic mutations in breast AdCCs. Importantly, unlike common-type triple-negative and basal-like breast cancers, no somatic mutations targeting TP53, PIK3CA, RB1, BRCA1 or BRCA2 were identified in breast AdCCs (Figure 2A; Supplementary Table S5).
Figure 2. Spectrum of somatic mutations in breast AdCCs.
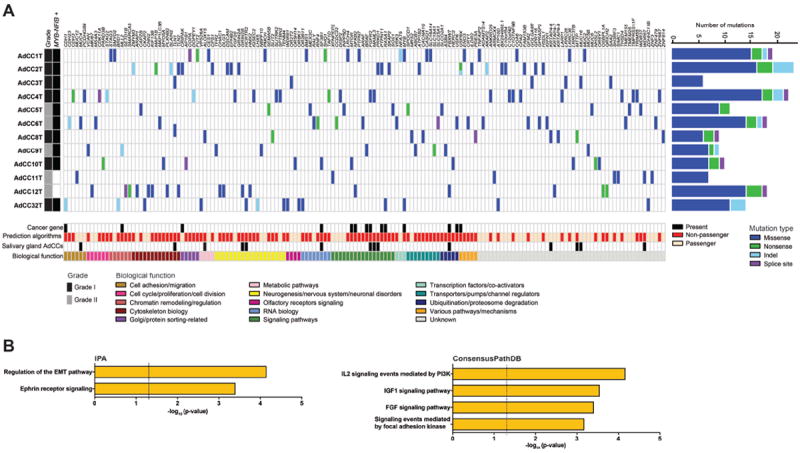
(A) Matrix of all validated and high-confidence somatic mutations identified in 12 breast AdCCs, color-coded by mutation type. The number of mutations identified per case is indicated on the right, and the histologic grade and MYB-NFIB fusion gene status on the left. Somatic mutations were classified according to their biological function, and the membership of each gene in cancer gene datasets (Kandoth et al. [43], Cancer Gene Census [42] and Lawrence et al. [44]). The results of mutation effect prediction algorithms [35-37], and the mutation status in salivary gland AdCCs [12,13] are also shown. (B) Pathways enriched for genes targeted by somatic non-passenger mutations in breast AdCCs, as defined by Ingenuity Pathway Analysis (IPA, left) and ConsensusPathDB [38] (right). Log values of the Benjamini-Hochberg corrected p-value (IPA) and of the hypergeometric test p-value (ConsensusPathDB) are shown.
We employed a combination of mutation effect prediction algorithms with a high negative predictive value[37], in combination with querying the presence of each mutated gene in the Cancer Gene Census[42], and/or in the list of cancer genes reported by Kandoth et al.[43] and Lawrence et al.[44] to discriminate passenger from potentially non-passenger mutations. This analysis resulted in the exclusion of 60 mutations, which were considered likely passengers, and in a list of 107 potentially non-passenger mutations, with at least one potentially non-passenger somatic mutation per case (Supplementary Table S5). Several of these mutations affected known cancer-related genes including BRAF, FBXW7, SF3B1, FGFR2, RASA1, PTPN11, and MTOR (Figure 2A), suggesting that AdCC of the breast likely harbor potentially pathogenic mutations in addition to the MYB-NFIB fusion gene. Only two recurrent mutations were identified in the breast AdCCs studied here, two cases (AdCC2T and AdCC12T; 17%) harbored missense mutations affecting talin 2 (TLN2), a cytoskeletal protein that plays a role in actin filament assembly and cell migration, and two other cases (AdCC1T and AdCC32T; 17%) harbored missense mutations targeting MYB itself, where the mutation in case AdCC1T occurred in the exon 13 splice site of the MYB allele that is part of the actual MYB-NFIB fusion gene (Supplementary Figure S4). Whilst only one BRAF mutation was identified in the current series analyzed (8%), our group has previously reported the presence of BRAF mutations in 12% (3/25) of an independent cohort of breast AdCCs[15], providing evidence to suggest that BRAF may also be recurrently targeted by somatic mutations in these tumors.
Somatic mutations found in breast AdCCs affected genes either not mutated (e.g. RASA1 or PTPN11) or mutated only in one of the basal-like breast cancers included in the TCGA dataset (e.g. BRAF, FBXW7, SF3B1, FGFR2, and MTOR, www.cBioPortal.org, accessed February 2015)[19,45]. On the other hand, 12 of the genes identified to harbor potentially non-passenger mutations in the breast AdCCs analyzed here were also found to be targeted by mutations in salivary gland AdCCs (12/107, 11.2% of genes affected by potentially non-passenger mutations; Figure 2A)[12,13]. The genes found to be commonly mutated between breast and salivary gland AdCCs comprised known cancer-related genes including SF3B1, FBXW7, FGFR2, MYB and PRKD1 (Figure 2A, Supplementary Table S5)[12,13].
Despite the lack of recurrent mutations, annotation of the mutated genes identified in breast AdCCs revealed their convergence into several functional categories including chromatin remodeling and cell adhesion genes, akin to the genes mutated in salivary gland AdCCs[12,13], RNA biology, cell cycle/proliferation, ubiquitination/proteasome degradation, and neurogenesis/neuronal disorders (Figure 2A). Furthermore, pathway analysis of the genes affected by potentially non-passenger mutations in breast AdCCs using Ingenuity Pathway Analysis (IPA) and ConsensusPathDB revealed a significant enrichment for genes involved in the IGF1 and FGF signaling pathways (p-value<0.001), as reported for salivary gland AdCCs[12], and for genes related to the epithelial-mesenchymal transition (EMT) pathway (p-value<0.001) and Ephrin receptor signaling (p-value<0.001, Figure 2B). Taken together, our results suggest that the repertoire of somatic mutations of breast AdCCs is distinct from that of common forms of triple-negative and basal-like breast cancers but bears resemblance to that of salivary gland AdCCs.
Landscape of somatic gene copy number alterations
The exome sequencing data were also used to characterize the somatic gene copy number alterations of breast AdCCs. Consistent with previous observations by our group[14,20], breast AdCCs displayed low levels of genetic instability (Figure 3A), and no amplifications or homozygous deletions were identified. The most frequent copy number alterations were losses of 12q12-q14.1 in 5/12 cases (all MYB-NFIB fusion gene-positive) and gains of 17q21-q25.1 in 3/12 cases (two MYB-NFIB fusion gene-positive and one MYB-NFIB fusion gene-negative case, Figure 3A, Table 1, Supplementary Table S6). AdCCs of the breast were found to harbor a lower complexity in the pattern of gains and losses than that reported for common forms of triple-negative and basal-like breast cancers, and regions often altered in common-type TNBCs, such as 8q gain and 5q loss[19,46], were found not to be altered in breast AdCCs. In contrast, and consistent with the results of the mutational profiling, the landscape of gene copy number alterations found in breast AdCCs, including recurrent losses of 12q, was similar to that reported for salivary gland AdCCs[12,13,17,47].
Figure 3. Copy number alterations and clonal mutation frequencies in breast AdCCs.
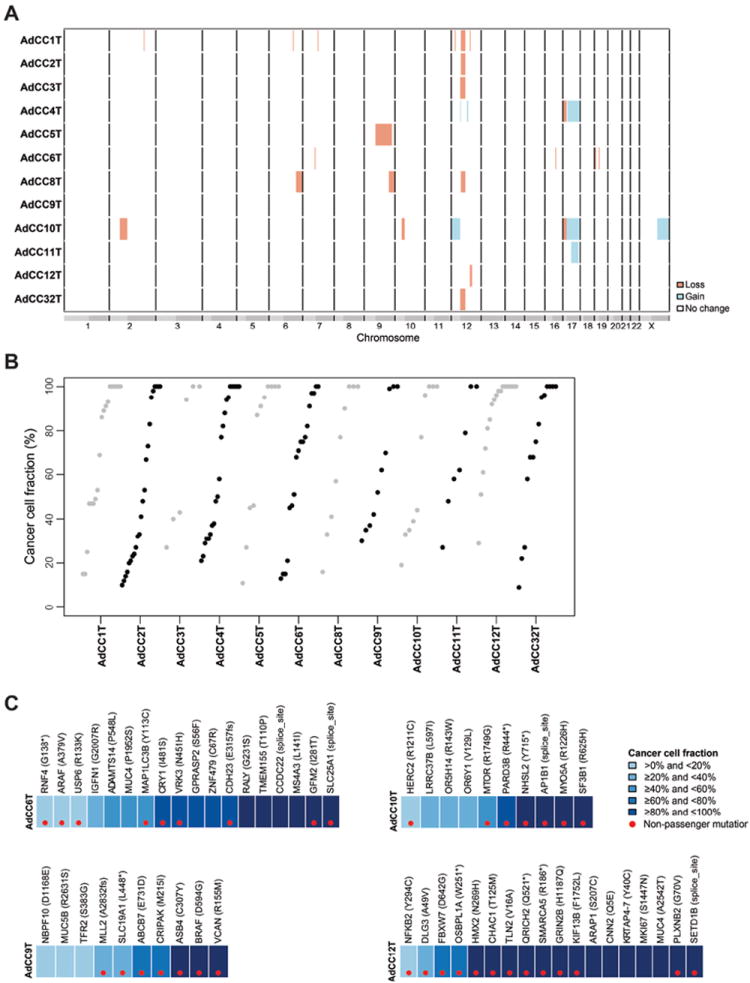
(A) Copy number profiles of breast AdCCs. The genomic position is plotted along the x-axis and the samples on the y-axis. No amplifications and homozygous deletions were found. AdCCs harbored recurrent losses of 12q12-q14.1 and gains of 17q21-q25.1. Orange, copy number loss, blue, copy number gain, white, no copy number change. (B) Clonal frequencies of mutations in breast AdCCs as defined by ABSOLUTE [40] through integration of tumor cellularity, ploidy, gene copy number and mutation data. While all cases harbored clonal mutations (cancer cell fraction ≥80%), validated subclonal mutations were also identified. (C) Representative clonal frequency plots of breast AdCCs as defined by ABSOLUTE [40] through integration of tumor cellularity, ploidy, gene copy number and mutation data. Cancer cell fractions according to the color-coding in the legend. Red dots represent likely non-passenger mutations.
Clonal heterogeneity
A subset of common forms of TNBC has been reported to display intra-tumor genetic heterogeneity at diagnosis[18]. Analysis of the clonal frequencies by integration of the gene copy number alterations and validated mutations identified in the breast AdCCs studied here using ABSOLUTE[40] revealed that the ploidy of all cases was ~2n (data not shown), and that many of the mutations identified were likely clonal with a cancer cell fraction >80% (Figure 3B). It should be noted, however, that a subset of mutations were likely subclonal with cancer cell fractions ranging from 9-79% (Figure 3B, Supplementary Table S5). These subclonal mutations also affected known cancer genes, such as FBXW7, MTOR, MLL2 (KMT2D), ARAF or CDH1 (Figure 3C, Supplementary Table S5). These data provide evidence to suggest that although breast AdCCs have rather simple genomes, they may be composed of mosaics of cancer cells harboring subclonal mutations at diagnosis
DISCUSSION
Here we show that AdCCs of the breast, a rare type of TNBC, have a low exonic mutation rate, low levels of genetic instability, show a heterogeneous repertoire of somatic genetic alterations, and, in addition to the MYB-NFIB fusion gene, harbor mutations targeting chromatin remodeling, cell adhesion, and canonical signaling pathway genes including known cancer genes such as BRAF, FBXW7, FGFR2 and MTOR. Furthermore, we observed that mutations and copy number alterations characteristic of common-type basal-like and TNBCs, such as mutations affecting TP53 and PIK3CA, and 5q losses and 8q gains[18,19,46], are not found in breast AdCCs. On the other hand, AdCCs of the breast were found to harbor mutations in genes rarely mutated in basal-like breast cancers, including RASA1, PTPN11 or BRAF, and recurrent 12q losses. Importantly, recurrent losses of 12q and somatic mutations affecting SF3B1, MYB, PRKD1 and FGFR2 have been documented in salivary gland AdCCs[12,13,17,47]. These results provide evidence to support the contention that breast AdCCs are more similar to salivary gland AdCCs than to common forms of triple-negative and basal-like breast cancers, and that TNBC is a mere operational term, encompassing a spectrum of lesions with distinct histologic features, clinical behaviors, and genomic landscapes[4,48]. In fact, contrary to common forms of TNBCs, patients with breast AdCCs have a favorable outcome[1,3,4,47].
Our genomic analysis of breast AdCCs has uncovered multiple alterations affecting known cancer genes and the involvement of several canonical signaling pathways that may play a role in their development. In addition to MYB overexpression, not driven exclusively by the MYB-NFIB fusion gene, we observed that chromatin remodeling, cell adhesion/migration, RNA biology and neurogenesis-related genes were targeted by potentially pathogenic somatic mutations in breast AdCCs. Although only two genes (i.e. MYB and TLN2) were recurrently mutated in the 12 breast AdCCs studied here, annotation of the mutated genes in functional pathways and networks provided evidence to suggest that the mutations in AdCCs converged and preferentially affected genes involved in EMT, Ephrin receptor signaling, and IGF1 and FGF signaling pathways.
Given that AdCCs irrespective of the anatomical site are characterized by similar histologic features and the presence of the MYB-NFIB fusion gene[1,8], which was present in 83% of cases studied here, it is not entirely unexpected that some of the genes and signaling pathways reported to be targeted by mutations in salivary AdCCs are also affected in breast AdCCs. Breast and salivary gland AdCCs displayed many similarities in regards to their constellation of somatic genetic alterations, including similar mutation rates (0.27 non-silent mutations/Mb in breast vs. 0.31 non-silent mutations/Mb in salivary gland AdCCs[12]), recurrent losses of 12q, mutations affecting SF3B1, FBXW7, FGFR2, MYB and PRKD1, and enrichment for mutations affecting genes playing a role in chromatin remodeling, cell adhesion, and the FGF signaling pathway[12,13]. It should be noted, however, that at the genomic level, differences between the AdCCs of the breast and the salivary gland were also observed; whilst salivary gland AdCCs were reported to harbor mutations in NOTCH1 and/or NOTCH2, and in SPEN[12,13], a downstream effector of NOTCH signaling, these genes were not found to be altered in breast AdCCs. Another important distinction between breast and salivary AdCCs relates to the enrichment for mutations affecting DNA damage response signaling genes in the latter but not in the former[12]. Using a binomial distribution, based on a reported 30% mutation rate in DNA damage response and NOTCH signaling pathway related genes in salivary gland AdCCs[12,13], the probability of observing at least one mutation in these genes in the 12 breast AdCCs analyzed here was 98.6% (Supplementary Methods), rendering a type II or β error unlikely. Taken together, although breast AdCCs appear to be more similar to AdCCs of the salivary glands than to triple-negative and basal-like breast cancers, important differences are also observed between breast and salivary gland AdCCs. Further studies are warranted to confirm these findings in larger series and to define whether these genetic differences may account for the reported distinct behaviors of AdCCs of the breast and salivary glands[1].
Despite the low level of genetic instability observed in breast AdCCs, our results provide evidence to suggest that in a way akin to other forms of cancer[18,49,50], at diagnosis, AdCCs may be constituted by a mosaic of cancer cell clones, with some potentially non-passenger mutations affecting known cancer genes being restricted to minor subclones within the tumor bulk. Although the biological and clinical significance of this observation remains to be fully elucidated, our findings demonstrate that even tumors with low levels of genetic instability may display intra-tumor genetic heterogeneity.
This study has several limitations. First, given the rarity of breast AdCCs (approximately 0.1% of all invasive breast cancers)[3,4], the number of cases analyzed here is relatively small. It is noteworthy, however, that the current study represents the largest cohort of breast AdCCs subjected to massively parallel sequencing to date. Second, the use of normal breast tissue as the source of germline DNA for massively parallel sequencing analyses may lead to false-negative results. To ensure the absence of contaminating tumor cells in normal breast tissue, however, only normal tissue distant from the lesion was used, and the normal breast tissue was reviewed by pathologists and microdissected if required to be entirely devoid of any neoplastic cells. Third, the mutational signatures[51] in the MYB-NFIB fusion gene-driven cancers could not be studied given the limited number of somatic mutations present in a given breast AdCC. The development of breast AdCC cell lines or ER-negative non-malignant breast epithelial cell lines harboring the MYB-NFIB fusion gene may be required to elucidate the mechanistic basis of the oncogenic properties of the MYB-NFIB fusion gene, and the contribution of somatic genetic alterations other than the MYB-NFIB fusion gene for the tumorigenesis of these rare forms of TNBC. Furthermore, whole genome sequencing analysis of MYB-NFIB fusion gene-negative AdCCs is warranted to define alternative genetic mechanisms resulting in MYB overexpression and activation.
In conclusion, our findings demonstrate that at the genomic level, breast AdCCs are similar to salivary gland AdCCs and are likely distinct from common types of triple-negative or basal-like breast cancers. Our results provide further evidence of the heterogeneity of triple-negative and basal-like breast cancers at the molecular level, and that histologic subtyping of triple-negative/basal-like breast cancers provides useful information. In fact, our findings emphasize the importance of histologic subtyping in the context of triple-negative disease, given that breast AdCCs have outcomes and response to chemotherapy distinct from those of common forms of TNBC, and that agents targeting the driver genes most frequently mutated in common forms of TNBC may be of limited value for patients with breast AdCCs, given their distinctive repertoire of somatic mutations and gene copy number alterations. Finally, owing to the heterogeneity of TNBCs, their sub-stratification according to histologic and molecular subtypes may be required for the identification of disease drivers and therapeutic targets[48].
Supplementary Material
Supplementary Figure S1: Adenoid cystic carcinomas of the breast.
Supplementary Figure S2: MYB-NFIB transcript detection by end-point (gel) RT-PCR in breast AdCCs.
Supplementary Figure S3: Illustration of the MYB-NFIB fusion gene and detection of the MYB-NFIB fusion transcript variants using RT-PCR.
Supplementary Figure S4: Presence of a somatic MYB mutation in the MYB-NFIB fusion gene transcript of case AdCC1T.
Supplementary Table S1: List of primers used for RT-PCR MYB-NFIB transcript detection and for the validation of mutations using amplicon resequencing (MiSeq), and Nanostring Codesets.
Supplementary Table S2: Clinico-pathologic characteristics of breast AdCCs included in this study.
Supplementary Table S3: Whole exome massively parallel sequencing statistics.
Supplementary Table S4: Non-silent and silent mutations identified in breast AdCC by whole exome massively parallel sequencing.
Supplementary Table S5: Targeted amplicon resequencing validation of somatic mutations identified by massively parallel sequencing in breast AdCCs, clonal frequencies, and mutation function prediction.
Supplementary Table S6: Recurrent copy number gains and losses identified in AdCCs of the breast.
Acknowledgments
FINANCIAL SUPPORT
RN is funded by a Breast Cancer Campaign Career Development Fellowship (2011MaySF01), SP by a Susan G Komen Postdoctoral Fellowship Grant (PDF14298348), and AMS by a stipend from the German Cancer Aid (Dr. Mildred Scheel Stiftung). AV-S is supported by an Interface INSERM grant, and GS by the Swedish Cancer Society and BioCARE (University of Gothenburg). This study was funded in part by the Adenoid Cystic Carcinoma Research Foundation. Research reported in this publication was supported in part by the Cancer Center Support Grant of the National Institutes of Health/National Cancer Institute under award number P30CA008748. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
AUTHORS’ CONTRIBUTIONS
BW, AV-S and JSR-F conceived and supervised the study. LF, JC, OM, P-EC and AV-S provided and curated samples. AV-S and JSR-F performed the histopathologic review of cases. LGM, RN, LF, JC, JSR-F and AV-S carried out experiments and acquired data. LGM, MRDF, CKYN, RN, SP, H-CW, RSL, RS, AMS, YHW, ME, GS, TAC, LN, AV-S, JSR-F and BW analyzed and interpreted data. The first draft of the manuscript was written by LGM and BW, which was subsequently reviewed, edited and approved by all authors.
Conflict of interest: The authors have no conflicts of interest to declare.
References
- 1.Marchio C, Weigelt B, Reis-Filho JS. Adenoid cystic carcinomas of the breast and salivary glands (or ‘The strange case of Dr Jekyll and Mr Hyde’ of exocrine gland carcinomas) J Clin Pathol. 2010;63:220–228. doi: 10.1136/jcp.2009.073908. [DOI] [PubMed] [Google Scholar]
- 2.Weigelt B, Horlings HM, Kreike B, et al. Refinement of breast cancer classification by molecular characterization of histological special types. J Pathol. 2008;216:141–150. doi: 10.1002/path.2407. [DOI] [PubMed] [Google Scholar]
- 3.Lakhani SR, Ellis IO, Schnitt SJ, Tan PH, van de Vijver MJ. WHO Classification of Tumours of the Breast. IARC; Lyon: 2012. [Google Scholar]
- 4.Weigelt B, Reis-Filho JS. Histological and molecular types of breast cancer: is there a unifying taxonomy? Nat Rev Clin Oncol. 2009;6:718–730. doi: 10.1038/nrclinonc.2009.166. [DOI] [PubMed] [Google Scholar]
- 5.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363:1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 6.Yerushalmi R, Hayes MM, Gelmon KA. Breast carcinoma--rare types: review of the literature. Ann Oncol. 2009;20:1763–1770. doi: 10.1093/annonc/mdp245. [DOI] [PubMed] [Google Scholar]
- 7.Papaspyrou G, Hoch S, Rinaldo A, et al. Chemotherapy and targeted therapy in adenoid cystic carcinoma of the head and neck: a review. Head Neck. 2011;33:905–911. doi: 10.1002/hed.21458. [DOI] [PubMed] [Google Scholar]
- 8.Persson M, Andren Y, Mark J, et al. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc Natl Acad Sci U S A. 2009;106:18740–18744. doi: 10.1073/pnas.0909114106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brill LB, 2nd, Kanner WA, Fehr A, et al. Analysis of MYB expression and MYB-NFIB gene fusions in adenoid cystic carcinoma and other salivary neoplasms. Mod Pathol. 2011;24:1169–1176. doi: 10.1038/modpathol.2011.86. [DOI] [PubMed] [Google Scholar]
- 10.West RB, Kong C, Clarke N, et al. MYB expression and translocation in adenoid cystic carcinomas and other salivary gland tumors with clinicopathologic correlation. Am J Surg Pathol. 2011;35:92–99. doi: 10.1097/PAS.0b013e3182002777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mitani Y, Li J, Rao PH, et al. Comprehensive analysis of the MYB-NFIB gene fusion in salivary adenoid cystic carcinoma: Incidence, variability, and clinicopathologic significance. Clin Cancer Res. 2010;16:4722–4731. doi: 10.1158/1078-0432.CCR-10-0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ho AS, Kannan K, Roy DM, et al. The mutational landscape of adenoid cystic carcinoma. Nat Genet. 2013;45:791–798. doi: 10.1038/ng.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stephens PJ, Davies HR, Mitani Y, et al. Whole exome sequencing of adenoid cystic carcinoma. J Clin Invest. 2013;123:2965–2968. doi: 10.1172/JCI67201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wetterskog D, Lopez-Garcia MA, Lambros MB, et al. Adenoid cystic carcinomas constitute a genomically distinct subgroup of triple-negative and basal-like breast cancers. J Pathol. 2012;226:84–96. doi: 10.1002/path.2974. [DOI] [PubMed] [Google Scholar]
- 15.Wetterskog D, Wilkerson PM, Rodrigues DN, et al. Mutation profiling of adenoid cystic carcinomas from multiple anatomical sites identifies mutations in the RAS pathway, but no KIT mutations. Histopathology. 2013;62:543–550. doi: 10.1111/his.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.D’Alfonso TM, Mosquera JM, MacDonald TY, et al. MYB-NFIB gene fusion in adenoid cystic carcinoma of the breast with special focus paid to the solid variant with basaloid features. Hum Pathol. 2014;45:2270–2280. doi: 10.1016/j.humpath.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 17.Persson M, Andren Y, Moskaluk CA, et al. Clinically significant copy number alterations and complex rearrangements of MYB and NFIB in head and neck adenoid cystic carcinoma. Genes Chromosomes Cancer. 2012;51:805–817. doi: 10.1002/gcc.21965. [DOI] [PubMed] [Google Scholar]
- 18.Shah SP, Roth A, Goya R, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–399. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horlings HM, Weigelt B, Anderson EM, et al. Genomic profiling of histological special types of breast cancer. Breast Cancer Res Treat. 2013;142:257–269. doi: 10.1007/s10549-013-2740-6. [DOI] [PubMed] [Google Scholar]
- 21.Elston CW, Ellis IO. Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience from a large study with long-term follow-up. Histopathology. 1991;19:403–410. doi: 10.1111/j.1365-2559.1991.tb00229.x. [DOI] [PubMed] [Google Scholar]
- 22.Piscuoglio S, Ng CK, Martelotto LG, et al. Integrative genomic and transcriptomic characterization of papillary carcinomas of the breast. Mol Oncol. 2014;8:1588–1602. doi: 10.1016/j.molonc.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hammond ME, Hayes DF, Dowsett M, et al. American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. 2010;28:2784–2795. doi: 10.1200/JCO.2009.25.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolff AC, Hammond ME, Hicks DG, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol. 2013;31:3997–4013. doi: 10.1200/JCO.2013.50.9984. [DOI] [PubMed] [Google Scholar]
- 25.Fusco N, Colombo P-E, Martelotto LG, et al. Resolving quandaries: basaloid adenoid cystic carcinoma or breast cylindroma? The role of massively parallel sequencing. Histopathology. 2015 doi: 10.1111/his.12735. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fehr A, Kovacs A, Loning T, et al. The MYB-NFIB gene fusion-a novel genetic link between adenoid cystic carcinoma and dermal cylindroma. J Pathol. 2011;224:322–327. doi: 10.1002/path.2909. [DOI] [PubMed] [Google Scholar]
- 27.Weinreb I, Piscuoglio S, Martelotto LG, et al. Hotspot activating PRKD1 somatic mutations in polymorphous low-grade adenocarcinomas of the salivary glands. Nat Genet. 2014;46:1166–1169. doi: 10.1038/ng.3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saunders CT, Wong WS, Swamy S, et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 33.Narzisi G, O’Rawe JA, Iossifov I, et al. Accurate de novo and transmitted indel detection in exome-capture data using microassembly. Nat Methods. 2014;11:1033–1036. doi: 10.1038/nmeth.3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwarz JM, Rodelsperger C, Schuelke M, et al. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 36.Carter H, Chen S, Isik L, et al. Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Cancer Res. 2009;69:6660–6667. doi: 10.1158/0008-5472.CAN-09-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martelotto LG, Ng C, De Filippo MR, et al. Benchmarking mutation effect prediction algorithms using functionally validated cancer-related missense mutations. Genome Biol. 2014;15:484. doi: 10.1186/s13059-014-0484-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kamburov A, Stelzl U, Lehrach H, et al. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 2013;41:D793–800. doi: 10.1093/nar/gks1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mermel CH, Schumacher SE, Hill B, et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carter SL, Cibulskis K, Helman E, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Futreal PA, Coin L, Marshall M, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lawrence MS, Stojanov P, Mermel CH, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Turner N, Lambros MB, Horlings HM, et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene. 2010;29:2013–2023. doi: 10.1038/onc.2009.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.El-Rifai W, Rutherford S, Knuutila S, et al. Novel DNA copy number losses in chromosome 12q12--q13 in adenoid cystic carcinoma. Neoplasia. 2001;3:173–178. doi: 10.1038/sj.neo.7900158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Turner NC, Reis-Filho JS. Tackling the diversity of triple-negative breast cancer. Clin Cancer Res. 2013;19:6380–6388. doi: 10.1158/1078-0432.CCR-13-0915. [DOI] [PubMed] [Google Scholar]
- 49.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Bruin EC, McGranahan N, Mitter R, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014;346:251–256. doi: 10.1126/science.1253462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1: Adenoid cystic carcinomas of the breast.
Supplementary Figure S2: MYB-NFIB transcript detection by end-point (gel) RT-PCR in breast AdCCs.
Supplementary Figure S3: Illustration of the MYB-NFIB fusion gene and detection of the MYB-NFIB fusion transcript variants using RT-PCR.
Supplementary Figure S4: Presence of a somatic MYB mutation in the MYB-NFIB fusion gene transcript of case AdCC1T.
Supplementary Table S1: List of primers used for RT-PCR MYB-NFIB transcript detection and for the validation of mutations using amplicon resequencing (MiSeq), and Nanostring Codesets.
Supplementary Table S2: Clinico-pathologic characteristics of breast AdCCs included in this study.
Supplementary Table S3: Whole exome massively parallel sequencing statistics.
Supplementary Table S4: Non-silent and silent mutations identified in breast AdCC by whole exome massively parallel sequencing.
Supplementary Table S5: Targeted amplicon resequencing validation of somatic mutations identified by massively parallel sequencing in breast AdCCs, clonal frequencies, and mutation function prediction.
Supplementary Table S6: Recurrent copy number gains and losses identified in AdCCs of the breast.