

Abstract
The stereocontrolled introduction of vicinal heteroatomic substituents into organic molecules is one of the most powerful ways of adding value and function. Whereas many methods exist for the introduction of oxygen- and nitrogen-containing substituents, the number stereocontrolled methods for the introduction of sulfur-containing substituents pales by comparison. Previous reports from these laboratories have described the sulfenofunctionalization of alkenes that construct vicinal carbon-sulfur and carbon-oxygen, carbon-nitrogen as well as carbon-carbon bonds with high levels of diastereospecificity and enantioselectivity. This process is enabled by the concept of Lewis base activation of Lewis acids that provides activation of Group 16 electrophiles. To provide a foundation for expansion of substrate scope and improved selectivities, we have undertaken a comprehensive study of the catalytically active species. Insights gleaned from kinetic, crystallographic and computational methods have led to the introduction of a new family of sulfenylating agents that provide significantly enhanced selectivities.
The importance of organosulfur compounds1, 2 manifests itself in the myriad of constructive and functional manipulations involving these as building blocks as well as in the abundance of sulfur-containing natural products3. Among a variety of methods for the introduction of sulfur groups, the vicinal sulfenofunctionalisation of alkenes using electrophilic sulfur4 reagents represents a powerful approach. Extensive studies on the mechanism of this reaction have confirmed the intermediacy of thiiranium ions5,6, which are invertively captured by a nucleophile affording 1,2-difunctionalised products with defined relative configuration. The applicability of this reaction has been demonstrated with a broad range of sulfenylating agents and nucleophiles2. However, despite the sound understanding of this transformation, asymmetric variants remain still largely underdeveloped and, for a long time, only two examples of direct enantioselective sulfenofunctionalisation have been known, both employing chiral reagents in stoichiometric amounts7,8.
Only recently have catalytic enantioselective sulfenylations of activated alkenes derived from aldehydes,9, 10 ketones,11 and amides12 been reported. In addition, the catalytic enantioselective sulfenoetherification of unactivated alkenes under chiral Brønsted acid catalysis has been described, although with moderate enantioselectivities13. The necessity for generating enantioenriched thiiranium ions has been elegantly circumvented by asymmetric desymmetrization of meso-thiiranium ions, where ring-opening by a nucleophile is the stereodetermining step14,15. Notwithstanding this progress, there is a lack of methods that control the absolute stereochemical outcome of 1,2-sulfenofunctionalisations, and this pertains especially to isolated alkenes.
During our endeavor to apply the concept of Lewis base activation of Lewis acids16 to reactions of main group elements, we have systematically investigated the stability of seleniranium17, 18 and thiiranium ions19, 20 to establish boundary conditions for catalytic, asymmetric chalcogen functionalisations. Our studies have revealed that, under certain conditions, the thiiranium ions are configurationally stable19,20 and can be captured with various heteroatom nucleophiles without erosion of enantiomeric purity20. On the basis of these findings, the first catalytic, enantioselective, sulfenofunctionalisation of unactivated alkenes was developed providing access to enantiomerically enriched tetrahydropyrans and -furans21. Under the optimized conditions, N-phenylsulfenylphthalimide serves as the electrophilic sulfur source and a BINAM-derived selenophosphoramide as the Lewis base catalyst. Moreover, a Brønsted acid is required as a co-activator for the otherwise unreactive sulfenylating agents, and its specific function has been examined in a recent study from these laboratories22.
The generality of this catalytic system has been demonstrated using various nucleophiles (Fig. 1b). In addition to the use of hydroxyl groups in sulfenoetherification reactions21, electron-rich arenes have been deployed in carbosulfenylations of olefins affording trans-tetrahydronaphthalenes with complete diastereospecificity and high enantiomeric ratios22,23,24. More recently, we have extended our protocol to tosyl-protected amines that can be engaged in sulfenoamination reactions yielding enantioenriched nitrogen heterocycles25.
Figure 1. Lewis base catalysed, enantioselective sulfenofunctionalisation of unactivated alkenes.

a, A generic reaction pathway of the 1,2-sulfenofunctionaliaztion of alkenes is presented. Initial formation of an enantiomerically enriched thiiranium ion is followed by the stereospecific (invertive) capture with a nucleophile in an intra- or intermolecular fashion. b, The substrate and nucleophile scope of the Lewis base catalysed, enantioselective sulfenofunctionalisation is shown.
Despite these positive developments, further improvement of the catalyst performance is desirable for some of the substrates. For instance, efficient discrimination between enantiotopic faces of terminal, 1,1-disubstituted and trisubstituted double bonds still remains a challenge. Beyond an empirical attempt to solve this problem, the in-depth understanding of processes occurring during the sulfenofunctionalisation as well as detailed knowledge about the structure of the active sulfenylating agent can contribute substantially to the rational design of a more selective catalytic system. Given that thiiranium ions are the common intermediate, a more enantioselective preparation thereof would be beneficial to all conceivable substrate classes of this transformation. Therefore, to advance and further generalize this method, we undertook an in depth study to elucidate the mechanism and characterise the catalytically active species in the Lewis base catalysed sulfenofunctionalisation of unactivated alkenes. Herein we report the results of kinetic, crystallographic as well as computational investigations toward this objective. The synthetic impact of our findings is illustrated by the improvement of the enantioselectivity of the process.
Results
Kinetic studies
A full investigation of the kinetic parameters was undertaken to identify the rate equation. The conversion of 1a was monitored by 19F NMR (Fig. 2). Catalyst 3a was chosen because the rates of reaction could be followed easily over a wide range of temperatures and concentrations. The order in each substrate was determined by the method of initial rate kinetics. Reactions were followed to 10% conversion and the initial rates were plotted against concentration to obtain straight lines (SI). The reaction was found to be first order in both catalyst and substrate. No order in the electrophile was observed. Interestingly, when the acid (MsOH) dependence was investigated, the rate dependence was found to be parabolic with a maximum at 0.6 equiv.
Figure 2. Kinetic study of the Lewis base catalysed sulfenofunctionalisation.

The model reaction for the kinetic analysis of the Lewis base catalysed sulfenofunctionalisation is presented.
These results suggest that transfer of the sulfenium ion to the alkene occurs from the sulfenylated selenophosphoramide [(R2N)3P=Se–SPh]+X− constituting the turnover-limiting step of the reaction. The catalytically active species [(R2N)3P=Se–SPh]+X−, in turn, is generated upon the MsOH mediated reaction of 2a with Lewis base 3 and represents the resting state of the catalyst. Its existence is supported by 31P NMR spectroscopy, in which the diagnostic signal for 3 (80–90 ppm, depending on the catalyst structure) disappears and a new resonance at ca. 60 ppm is observed. This value is in accord with previously reported, analogous compounds of the type [(R2N)3P=Se–YAr]+X−18,22.
The activation parameters of the reaction were obtained by carrying out an Arrhenius study in a temperature range from −20 °C to 20 °C. The enthalpy of the reaction was 8.9 +/−0.2 kcal/mol and the entropic contribution was 52.7 +/−0.6 e.u. At 298 K, the entropy term is the primary contributor to the free energy of activation of 24 kcal/mol.
Isolation of the catalytically active species
The proposed catalytically active species [(R2N)3P=Se–SPh]+X− is assumed to transfer the sulfenium ion to an alkene, forming a thiiranium ion. Since this event is the rate and, most likely, the enantiodetermining step, detailed knowledge about the stereostructure of the active species could help to understand the origin of enantioselectivity. Accordingly, the isolation and crystallographic characterisation of [(R2N)3P=Se–SPh]+X− was investigated.
Orienting experiments focused on the identification of suitable conditions for generating the active species [(R2N)3P=Se–SPh]+X− without any byproducts that would disrupt the crystallization process. The first approach relied on the reaction of a selenophosphoramide with [PhS(SMe2)]SbCl626 which had been used as an of the S-phenyl transfer agent to alkenes. This reagent was also competent in transferring the sulfenyl moiety to the catalyst forming the active species along with Me2S as the sole byproduct (Equation 1, for more details on the actual catalyst structure see Supporting Information). The formation of the active species was confirmed by the appearance of a diagnostic 31P resonance at 60.4 ppm. Unfortunately, during the crystallization attempts, a disproportionation reaction occurred reducing the sulfenyl group to PhSSPh while oxidizing the selenophosphoramide to the dicationic dimer [(R2N)3P=Se–Se=P(NR2)3]2+ 2SbCl6− (Equation 2). An X-ray crystal structure of the dimer could be obtained an initial glimpse into how the groups in the actual active species might be oriented (Supporting Information).
To avoid the disproportionation reaction caused by the lability of Se–SPh bond, the donor atom of the catalyst was replaced with a sulfur atom. Thiophosphoramides are comparably selective in the sulfenoetherification reactions21. Moreover, the disproportionation of the S–SPh bond in the active species would become thermodynamically less favorable in comparison to the selenophosphoramide analogue. However, when a thiophosphoramide Lewis base was reacted with [PhS(SMe2)]SbCl6, an equilibrium between the reactants was established (Equation 3). Besides the complication arising from reversibility, solubility was a serious problem limiting the scope of solvents that could be used for crystallization.
| (1) |
| (2) |
| (3) |
To overcome the reversible formation of the active species, thiophosphoramide (±)-3b was combined with PhSCl and NaBArF24 to furnish (±)-5b as a pale yellow, air stable solid (Fig. 3a). This approach not only enabled the irreversible generation of the active species (±)-5b, but also allowed for the introduction of the weakly coordinating BArF24− counteranion27, which greatly improved the solubility properties of complex (±)-5b. The formation of (±)-5b was initially confirmed by its 31P NMR chemical shift (65.8 ppm) as well as its kinetic competency to stoichiometrically sulfenylate substrate 1b to afford thioetherification product (±)-4b (Fig. 3a).
Figure 3. Synthesis of the catalytically active species (±)-5b and its X-ray crystal structure.

a, The catalytically active species (±)-5b was prepared from 3b, PhSCl and NaBArF24. Its sulfenylating potential is demonstrated by the reaction with alkene 1b. b, Ortep plot of (±)-5b X-ray crystal structure (35% thermal ellipsoids, BArF24− counter anion omitted for clarity). S(2)-S(1)-P(1) = 103.5°, C(1)-S(2)-S(1) = 104.1°, P(1)-S(1)-S(2)-C(1) = 96.0°, N(3)-P(1)-S(1)-S(2) = 175.1°.
Ultimately, crystals suitable for X-ray analysis were obtained by slow evaporation of a saturated solution of (±)-5b in dichloromethane. The molecular structure was confirmed by the X-ray crystal structure analysis of (±)-5b (Fig. 3b, (S)-enantiomer shown). Several features of this structure are noteworthy. First, the diisopropylamino group is rotated such that the C(32)-N(3) bond occupies a nearly antiperiplanar orientation to the S(1)-P(1) bond while the C(29)-N(3) bond is synperiplanar to this. Torsion angles of 169.1 ° for S(1)-P(1)-N(3)-C(32) and −15.0 ° for S(1)-P(1)-N(3)-C(29), respectively, define this arrangement. The sum of the angles about the N(3) atom is >359.8 ° indicating near perfect planarity. This alignment is most likely a consequence of a generalized anomeric effect in which the N(3) nonbonding electron pair is delocalized into the P(1)-N(1) and P(1)-N(3) σ* orbitals. Such anomeric effects have been previously observed in similar structures.28,29
To avoid destabilizing steric interactions with the diisopropylamino group, the S–SPh subunit is oriented back toward the binaphthyl backbone as is evidenced by a 175.1° torsion angle of the N(3)-P(1)-S(1)-S(2) subunit. With a torsion of 96.0° of the P(1)-S(1)-S(2)-C(1) moiety, the P(1)-S(1) vector is almost perpendicular to the plane defined by the S(1)-S(2)-C(1) atoms. On the basis of the X-ray crystal structures of neutral 10-S-3 compounds30,31 and by analogy to the well known 10-Se-3 T-shaped charge transfer complexes32 the active sulfenylating agent was anticipated to be a bent ion pair complex (8-S-2) which is now verified crystallographically by the S(1)-S(2)-C(1) angle of 104.1°. The S(1)-S(2) bond (2.06 Å) is in the range of a sulfur–sulfur single bond length observed in disulfides33. The P(1)-S(1) bond (2.10 Å) of the active species is significantly lengthened compared to the 1.95 Å found for the P=S double bond in (Me2N)3P=S34 and is closer to a typical P–S single bond35.
Improvement of enantioselectivity
To date, the most selective catalyst for the sulfenofunctionalisation is the BINAM-derived selenophosphoramide 3c (Table 1). Prior optimization studies identified the importance of a methyl substituent on the internal nitrogen atoms as well as the isopropyl groups on the external nitrogen.18,21 Attempts to further enhance the catalyst performance by modifying the biaryl backbone have not been successful so far. A limiting factor for the latter strategy is the lack of methods to access novel biaryl scaffolds asymmetrically and the development of new synthetic routes can be laborious.
Table 1.
Influence of the sulfenylating agent on enantioselectivity in the sulfenoetherification reaction.
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| entry | product | conv.a (time) |
e.r.b | entry | product | conv.a (time) |
e.r.b | entry | product | conv.a (time) |
e.r.b |
| 1 | 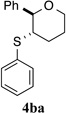 |
93% (24 h) | 95.3:4.7 | 6 | 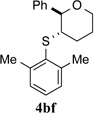 |
100% (21 h) | 97.1:2.9 | 13 | 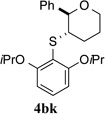 |
80% (24 h) | 98.0:2.0 |
| 2 | 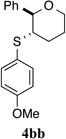 |
100% (24 h) | 95.1:4.9 | 7 |  |
>95% (42 h) | 97.5:2.5 | 14 |  |
80% (48 h) | 94.0:6.0 |
| 3 |  |
>95% (24 h) | 95.6:4.4 | 8 |  |
>95% (24 h) | 98.0:2.0 | 15 |  |
95% (21 h) | 92.6:7.4 |
| 4 |  |
88% (51 h) | 95.7:4.3 | 9 |  |
67% (48 h) | 96.3:3.7 | 16 |  |
90% (48 h) | 91.8:8.2 |
| 5 |  |
>95% (21 h) | 97.2:2.8 | 10 |  |
70% (65 h) | 99.1:0.9 | ||||
| 11 | 80%c (24 h) | 99.0:1.0 | |||||||||
| 12 | 100%d (12 h) | 98.0:2.0 | |||||||||
General reaction conditions: 5 mm NMR tube, 1b (70.0 µmol), 2 (74.0 µmol), MsOH (70.0 µmol), (R)-3c (10 mol%), CDCl3 (0.11 M), −20 °C. Conversion was determined by assuming that, besides small quantities of the respective tetrahydrofuran (<5%), 1b was converted only to 4, as no other significant product was detedcted by 1H NMR spectroscopy.
Determined by CSP-SFC analysis.
Reaction conducted at 0 °C.
Reaction conducted at room temperature.
An alternative approach to improve the enantioselectivity of the process is to change the sulfenylating agent. Insights gained from the X-ray crystal structure of 5b implicate a role for arylsulfenyl group in interaction with the approaching alkene during the stereodetermining thiiranium ion ring formation. Thus, it appeared plausible that electronic and particularly steric properties of the arylsulfenyl group could impact the stereochemical outcome of the reaction.
To test this hypothesis, a variety of electronically and sterically disparate N-arylsulfenylphthalimides 2a–n was synthesized and evaluated in the sulfenoetherification of hydroxyl substituted alkene 1b using selenophosphoramide (R)-3c as the catalyst (Table 1). The enantiomeric ratios of the resulting tetrahydropyrans 4bb–4bn were compared against the enantiomeric ratio obtained for the phenylsulfenyl substituted product 4ba (95.3:4.7 e.r., entry 1). Interestingly, almost no electronic effect on enantioselectivity was observed regardless of changes in the electron density of the phenyl ring (entries 2–4). More striking still was the lack of a significant electronic effect on reaction rate varying by only a factor of 2–3 between 2b (4-OMe) and 2d (4-NO2).
To evaluate the influence of steric effects, different ortho-substituents were installed into the arylsulfenyl moiety, 2e–j. The presence of an ortho-methyl group increased the enantioselectivity of the reaction to 97.2:2.8 e.r. for tetrahydropyran 4be (entry 5). Essentially the same ratio of enantiomers was obtained for product 4bf containing two ortho-methyl substituents (entry 6). Surprisingly, no decrease in reactivity was encountered with these sulfenylating agents. On the contrary, the rate of formation of product 4bf was faster as compared to that for 4ba and 4be, likely owing the electron donating effect of the methyl groups. Further increasing the steric bulk around the sulfur atom by incorporating ethyl substituents again led to a slight enhancement in enantioselectivity (entries 7, 8) Logically, the impact of ortho-isopropyl groups was next investigated (2i, 2j). Whereas monoisopropyl substituted product 4bi was obtained with somewhat diminished enantiomeric purity (96.3:3.7 e.r., entry 9), the sulfenoetherification with 2j bearing two isopropyl groups produced tetrahydropyran 4bj with a remarkably high enantiomeric ratio of >99:1 at −20 °C (entry 10). In this scenario, the reactivity gain from electron donation is obviously outweighed by the steric encumbrance leading to a slow conversion at −20 °C. Gratifyingly, at 0 °C the reaction proceeded at a reasonable rate (80% conv. after 24 h) accompanied by only a marginal erosion of the enantiomeric ratio (entry 11). Even at room temperature a high level of enantiocontrol was achieved (entry 12).
For the sake of completeness, other N-arylsulfenylphthalimides 2k–n were inspected (entries 13–16), all being inferior to sulfenylating agent 2j. Finally, N-alkylsulfenylphthalimides have been examined in the course of this study, however, these sulfenylating agents afforded slow conversion or no reaction at all.
The general applicability of sterically demanding N-arylsulfenylimides as efficient reagents for the highly enantioselective sulfenoetherification of other substrate classes was next evaluated (Table 2). At first, reaction of 1b was conducted on a preparative scale with 2a, 2h, 2f, and 2j to confirm the results from the initial survey (entries 1–4). Subsequently, representative alkenes 1c–1e were subjected to the sulfenoetherification using sulfenylating agent 2j. Because a different selenophosphoramide had been used under the originally reported conditions,21 reactions with the parent sulfenylating agent 2a and catalyst (R)-3c were run in parallel to provide a valid comparison with 2j. A significant improvement of enantioselectivity was seen in the reaction of terminal alkene 1c and 2j as compared to 2a (entry 5 vs. 6). Despite the already high enantiopurity of product 4da obtained from an intermolecular sulfenoetherification of 4-octene (1d), 2a and MeOH, the enantiopurity of the corresponding product 4dj was further increased by using 2j (entry 7 vs. 8). The suitability of a terminal double bond was also showcased in an intermolecular functionalisation with 1-octene (1e) and MeOH as the nucleophile (entry 9 vs. 10). Product 4ej was accessed with an excellent enantiomeric ratio. of 98.6:1.4, whereas 4ea gave only a moderate selectivity which is in line with stereochemical outcome previously observed with terminal alkene 1c. Apart from the sulfenoetherification, we were able to demonstrate the benefit of 2j in a sulfenoamination reaction25 using alkenyl sulfonamide 1f. Again an enhanced enantioselectivity was noticed with 2j forming product 4fj in 94.3:5.7 e.r. (entry 11 vs. 12). Although enantiotopic face differentiation was improved for terminal and E-1,2-disubstituted alkenes, 1,1-disubstituted and trisubstituted double bonds did not respond to this modification affording comparable results with 2a and 2j (not shown).
Table 2.
Scope of the sulfenofunctionalisation using sterically demanding sulfenylating reagents.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | alkene | sulf. agent |
temp, °C | time, h | product | yield,d % | e.r.h | |
| 1 | 1b | 2a | −20 | 36 | 4baa | 78 | 95.6:4.4j | |
| 2 | 1b | 2f | −20 | 36 | 4bf | 84 | 97.6:2.4j | |
| 3 | 1b | 2h | −20 | 36 | 4bh | 87e | 98.4:1.6i,j | |
| 4 | 1b | 2j | 0 | 48 | 4bj | 87f | 99.3:0.7i,j | |
| 5 | 2a | 0 | 36 |  |
4ca (R = H)a | 83 | 88.9:11.1j | |
| 6 | 2j | 0 | 36 | 4cj (R = iPr) | 94 | 98.6:1.4k | ||
| 7 | 2a | −20 | 48 |  |
4da (R = H)a,b | 77 | 97.4:2.6k | |
| 8 | 2j | 0 | 36 | 4dj (R = iPr)b | 85 | 99.2:0.8k | ||
| 9 | 2a | 23 | 24 |  |
4ea (R = H)a,b | 75g | 87.8:12.2k | |
| 10 | 2j | 23 | 24 | 4ej (R = iPr)b | 84g | 98.6:1.4k | ||
| 11 |  |
2a | 0 | 48 |  |
4fa (R = H)c | 85 | 83.7:16.3j |
| 12 | 2j | 23 | 24 | 4fj (R = iPr)c | 72 | 94.3:5.7k | ||
General reaction conditions: 1 (1.0 mmol), 2 (1.0 mmol), MsOH (1.0 mmol), (R)-3c (10 mol%), CH2Cl2 (0.2 M).
Reaction of literature described compounds21 conducted on 0.2 mmol scale.
MeOH used as external nucleophile.
0.5 equiv MsOH used.
Yield of isolated products.
Combined yield of a 97:3 mixture of tetrahydropyran/-furan.
Combined yield of a 93:7 mixture of tetrahydropyran/-furan.
Combined yield of a 4:1 mixture of 4e and its constitutional isomer (Supporting Information).
Absolute configuration assigned by comparison and in analogy to literature described compounds.
e.r. of the major isomer.
Determined by CSP-SFC analysis.
Determined by HPLC analysis.
Discussion
The kinetic studies undertaken here have allowed for a better understanding of the reaction mechanism (Fig. 4). First-order dependence was observed for both the catalyst 3a and the substrate 1a, implying a simple bimolecular turnover-limiting step. In addition, the zeroth order dependence of the rate on the concentration of electrophile 2a (even at only 3 equiv of 2a with respect to catalyst) implies that the catalytically active sulfenylating agent (5, Fig. 4) is at saturated equilibrium concentration22. When the reaction was tested at different acid concentrations, no overall linear relationship was found. Instead, the rate appeared to be dependent on two competing processes at low and high acid concentrations. At concentrations corresponding to the catalyst-unsaturated system (< 4 equiv of MsOH with respect to 3a), a first order dependence on the acid concentration is observed. In contrast, the rate dependence of the catalyst-saturated system (≥ 6 equiv of MsOH with respect to 3a), an inverse dependence with a slope of −0.35 is observed. This behavior could be a result of substrate protonation by the strong acid. Once a sufficient amount of acid has been added such that the catalyst is fully saturated as I, further addition increases the fraction of the protonated substrate. Therefore at high acid concentrations the available amount of unprotonated substrate is likely to control the rate.
Figure 4. Proposed catalytic cycle of the Lewis base catalysed sulfenofunctionalisation.

The catalytic cycle commences with the sulfenylation of Lewis base 3 mediated by MsOH to generate the catalytically active species 5 which is the resting state of the catalyst with a diagnostic 31P NMR chemical shift at 59.9 ppm for 5c. Subsequently, in the turnover-limiting and stereodetermining step, the arylsulfenyl group is transferred to the alkene forming the enantiomerically enriched thiiranium ion. Its stereospecific capture by an internal or external nucleophile delivers the sulfenofunctionalised product and regenerates catalyst 3.
The enantiodetermining step of the sulfenylation is most likely an irreversible step, as the high selectivity precludes a dynamic process that can interconvert the enantiomers. The capture of the thiiranium ion formed from 1b, e.g., in the presence of excess MsOH is known to be reversible even at low temperatures. Isolation of tetrahydropyranyl and -furanyl thio ethers followed by resubjection to the reaction conditions resulted in no change in enantiomeric composition, although the ratio of constitutional isomers was affected21. Therefore it is unlikely that capture can be rate-determining, leaving thiiranium ion formation as the only possibility for the enantiodetermining step. The formation of the thiiranium ion II (Fig. 4) thereby constitutes the irreversible setting of both stereocenters, which are not affected by downstream processes. The observed improvements in enantioselectivity of the overall reaction when more hindered sulfenylating agents are employed further support this conclusion, as avoidance of increased steric interactions leads to better differentiated transition states.
The activation parameters of ΔH‡ = 8.9 kcal/mol and ΔS‡= 52.7 e.u. imply a highly ordered transition state in which the reaction barrier is primarily entropy-driven. This profile results from the organizational requirement wherein the alkene must approach the catalyst in a highly restricted conformational landscape. In fact, the entropic barrier is comparable to those of other highly-conformationally-restricted, bimolecular transition states such as those of Diels-Alder reactions36, MBH reactions37 or the Lewis base promoted aldol addition of trichlorosilyl enolates38. Notably, such highly restricted transition states bode well for computational approaches as the degrees of freedom of the reaction partners are significantly reduced.
Although the existence of thiiranium ions as reactive intermediates of the sulfenofunctionalization is generally accepted5,6, computational studies on their formation are virtually non-existent. The lack of computational insights is surprising given the fact that this event constitutes the enantiodetermining step of the reaction unless a meso-thiiranium ion is generated14,15. Radom39,40,41 and Modena42,43 have calculated the transfer of a sulfenium ion from a thiiranium ion to both σ- and π-nucleophiles which represents the microscopic reverse of the process. However, the availability of the X-ray crystallographic structural information of the catalytically active species 5b enables a detailed examination of the forward reaction, namely the formation of the thiiranium ion, with the aid of computational methods to gain a more quantitative understanding of the origin of enantioselectivity. In addition, the enhanced enantioselectivity obtained with sterically more encumbered sulfenylating agents could be investigated with a higher level of precision (Table 1 and Table 2).
Initial clues to understand the origin of enantioselectivity are provided by the solid-state structure of the active species 5b. As already implied, the plane defined by the C(32)-N(3)-C(29) atoms of the diisopropylamino group is coincident with the P(1)-S(1) bond. This anomerically stabilised conformation positions the two isopropyl groups such that rotation of the P(1)-S(1)-S(2)-phenyl subunit around the P(1)-S(1) bond is restricted. As a consequence, the transferable S-phenyl group is oriented toward the binaphthyl backbone which is reflected in the N(3)-P(1)-S(1)-S(2) torsion angle of 175.1°. This orientation dictates specific steric interactions of the approaching alkene with the one of the naphthyl rings as well as with the aryl group of the arylsulfenyl moiety and contributes to the enantiotopic face discrimination. In contrast, if the steric demand of the dialkylamino substituent of the catalyst is decreased, as it is the case with a piperidinyl group, the arylsulfenyl moiety can rotate away from the binaphthyl backbone. This increased flexibility now, leads to reduced steric interactions between the approaching alkene and the active species during the thiiranium ion formation and is ultimately responsible for the moderate enantioselectivity seen e.g. with a the piperidinyl-substituted catalyst (79:21 e.r.)21.
To further refine this picture, two assumptions were made with regard to the directionality of the alkene approach. First, to maximize orbital overlap between the LUMO of the electrophile and the HOMO of the alkene, an approach of the bonding π-orbital of the alkene along the S–S σ*-orbital is expected.38–40 Second, for such group-transfer reactions two limiting transition state geometries must be considered. In the planar transition state (Fig. 5a, left) the alkene double bond is located in the plane that is defined by the Caryl–S–Se subunit whereas in the spiro transition state (Fig. 5a, right) the double bond is positioned perpendicular to this plane. By analogy to the oxygen atom transfer to alkenes with dioxiranes and peracids (epoxidation), the spiro transition state is favored due to stabilizing interaction of a sulfur lone pair with π*-orbital of the alkene44,45,46.
Figure 5. Calculations of the transition states leading to the formation of the thiiranium ion.

a, Limiting transition state geometries are shown wherein the spiro transition state is favored over the planar transition state due to stabilizing interaction of a sulfur lone pair with π*-orbital of the alkene in the former. b, Free energies of the four transition states with two different arylsulfenyl groups are given for the reaction at −20 °C. c, Three transition states are presented to illustrate the steric interactions between the alkene and the catalytically active species during thiiranium ion formation. Both of the transition states that lead to the respective minor enantiomer (H-TS-minor1 and Me-TS-minor1, respectively) suffer from destabilising repulsions between the binaphthyl rings and alkene substituents.
Combining these considerations, four limiting transition states for the formation of the thiiranium ion can be formulated (Fig. 5b). For the purposes of calculating their energies, the coordinates from the X-ray crystal structure of active species 5b were employed wherein the sulfur donor atom of the Lewis base was replaced by a selenium atom (5c) to more accurately reflect the active species generated from the most selective catalyst 3c (Note the (S)-enantiomer of the catalyst was used for the calculations). With regard to the arylsulfenyl moiety, the parent phenysulfenyl group (R = H, H-TS) was compared to the 2,6-dimethylphenylsulfenyl group (R = Me, Me-TS) to secure greater insight into the observed differences in enantioselectivity between different sulfenylating agents (Table 1 and Table 2). For the alkene, trans β-methylstyrene was chosen as surrogate for substrate 1b to embody the energetic contributions of aryl and alkyl substituents, with the active species. All of the transition states were investigated at B3LYP level using 6–31G(d) basis set and the results are summarized in Fig. 5b and Table 3 (the full structures of all transition states are provided in the Supporting Information).
Table 3.
Distortion Interaction and NBO Analysis: (B3LYP/6-31G(d) energies at 253.15K in kcal/mol).
| ΔEdist_Aa | ΔEdist_Bb | ΔEd | ΔEi | ΔEactc | ΔH | ΔG | π(C=C)- σ*(S-Se)d |
|
|---|---|---|---|---|---|---|---|---|
| H-TS-major1 | 9.6 | 26.1 | 35.7 | −24.8 | 10.9 (0.0) | 11.9 (0.0) | 23.9 (0.0) | 63.3 (6.7) |
| H-TS-major2 | 10.7 | 27.4 | 38.1 | −25.6 | 12.5 (1.6) | 13.5 (1.6) | 25.4 (1.5) | 57.5 (0.9) |
| H-TS-minor1 | 10.4 | 28.1 | 38.5 | −25.9 | 12.6 (1.7) | 13.6 (1.7) | 25.6 (1.7) | 64.2 (7.6) |
| H-TS-minor2 | 13.3 | 33.5 | 46.8 | −32.2 | 14.6 (3.7) | 15.2 (3.3) | 26.9 (3.0) | 56.6 (0.0) |
| Me-TS-major1 | 7.8 | 22.8 | 30.7 | −19.9 | 10.8 (0.0) | 11.2 (0.0) | 25.0 (0.0) | 61.7 (7.3) |
| Me-TS-major2 | 9.0 | 27.2 | 36.1 | −22.4 | 13.7 (2.9) | 14.7 (3.5) | 26.8 (1.8) | 55.9 (1.5) |
| Me-TS-minor1 | 9.4 | 28.6 | 38.0 | −23.0 | 15.0 (4.2) | 15.3 (4.1) | 28.5 (3.5) | 62.1 (7.7) |
| Me-TS-minor2 | 9.1 | 29.9 | 39.0 | −23.6 | 15.4 (4.6) | 15.5 (4.3) | 30.4 (5.4) | 54.4 (0.0) |
A = β-methylstyrene.
B = 5c (H = C6H5; Me = 2,6-Me2C6H3)
ΔEact = ΔEd + ΔEi; ΔEd = ΔEdist_A + ΔEdist_B.
π(C=C)-σ*(S-Se) orbital interaction energy is calculated by NBO analysis.
For R = H, the lowest calculated transition state H-TS-major1 accounts for the formation of the (2S,3R)-enantiomer of 4ba which is in agreement with the experimental findings using catalyst (S)-3c. A depiction of the full transition state structure of H-TS-major1 is presented in Fig. 5c (left). Destabilising steric repulsion with the upper naphthyl ring is most effectively avoided by the approach of the alkene with the given enantiotopic face and the methyl group being placed on the side of the binaphthyl backbone. Transition state H-TS-major2 is 1.5 kcal/mol less stable than H-TS-major1 (see SI) and leads to the same enantiomer of 4ba. Interestingly, significantly different lengths of the developing bonds of the thiiranium ion between sulfur and the methyl substituted carbon on the one hand, and sulfur and the phenyl substituted carbon on the other hand are observed (2.11 Å vs. 2.53 Å, resp. for H-TS-major1). Obviously, the nascent positive charge is more effectively stabilised at the benzylic position and, assuming a similar charge distribution in the thiiranium ion, also biases the nucleophilic opening to occur at this carbon.
Inspection of the two transition states for reaction on the opposite face of the alkene, reveals that more stable H-TS-minor1 (Fig. 5, middle) is 1.7 kcal/mol (≅ 96.7:3.3 e.r.) higher in energy than H-TS-major1 which correlates well with the enantioselectivity observed (95.6:4.4 e. r.; ΔΔG† ≅1.55 kcal/mol) for 4ba at −20 °C (Table 2, entry 1). The least stable of the four transition states is H-TS-minor2 (2.9 kcal/mol) in which the phenyl ring is positioned in the proximity of the binaphthyl backbone.
Computational analysis also provided insights into the origin of improved enantioselectivity with bulkier arylsulfenyl groups (Table 1). Here, the enhanced differentiation between the enantiotopic alkene faces obviously benefits from more pronounced steric interactions between the alkene and the S-aryl moiety as compared to the parent S-phenyl subunit. To probe this feature, methyl substituents were attached to the ortho-positions of the S-phenyl group in the active species and the transition state energies were calculated as before. This modification increases the energy gap difference between the two most stable transition states that lead to enantiomeric products to 3.5 kcal/mol (Fig. 5b, Me-TS-major1 vs. Me-TS-minor1). The experimentally observed enantiomeric composition of 4bf (97.6:2.4 e.r.; ΔΔG† = 1.86 kcal/mol, Table 2, entry 2) is, however, below the expected value of >99.9:0.1 e.r. that is predicted by the magnitude of the energy difference between Me-TS-major1 and Me-TS-minor1. This disparity could arise from several reasons primarily associated with assumptions implicit in these calculations. Nevertheless, the fact that the calculations reflect the trend of higher enantioselectivity with increased steric demand of the arylsulfenyl group is satisfying.
In both of these systems, careful inspection of the competing transition structures failed to identify any obvious interactions that would disfavor H-TS-major1 or Me-TS-major1 with respect to vs. H-TS-minor1 or Me-TS-minor1. The highly unsymmetrical transition states allow for a significant distance between the aryl residue and the catalytically active species obviating any severe non-bonding interactions.
To provide additional insight into the origin of enantioselectivity, distortion-interaction47 and NBO48 analyses were carried out (Table 3). These results mirror those from the DFT analysis and suggest that a more subtle effect may be operative. As highlighted in red, H-TS-major1 possesses the lowest activation energy, 1.7 kcal/mol lower than H-TS-minor1. Interestingly, event though H-TS-minor1 benefits from a greater interaction energy (ΔΔEi = −1.1 kcal/mol), this advantage is offset by a greater distortion energy (ΔΔEd = 2.8 kcal/mol). The greater interaction energy associated with H-TS-major1 was substantiated by the NBO analysis which provided the stabilization energies arising from orbital overlap from the π-bond of the alkene to the antibonding (σ*) orbital of the sulfur-selenium bond. The stabilization energy for H-TS-minor1 is slightly greater (0.9 kcal/mol) than H-TS-major1 indicating similar levels of orbital overlap. However, to achieve those levels of overlap requires greater distortion of the orbitals in H-TS-minor1 (likely resulting from non-ideal approach of the alkene). Thus, whereas unfavorable steric interactions are not the apparent cause of the enantioselectivity, it is likely that the avoidance of unfavorable steric interactions leads to a non-ideal approach of the alkene that manifests in the greater distortion energy contribution to that transition state.
The exact same trends are seen for the corresponding transition states calculated for the 2,6-dimethyl substituted sulfenylating agent, but with a much greater energy differences throughout. Interestingly, Me-TS-minor1 enjoys significantly greater interaction energy (ΔΔEi = −3.1 kcal/mol) and marginally larger orbital overlap energy (0.4 kcal/mol) but at a much greater cost in distortion energy (ΔΔEd = 7.3 kcal/mol) consistent with a more drastic change in the approach vector for the alkene to avoid nonbonding interactions with the methyl substituents.
In conclusion, kinetic, spectroscopic, crystallographic and computational investigations have shed light on the mechanistic pathway of the Lewis base catalysed sulfenofunctionalisation. Initially, the catalytically active species was identified by studying the kinetic parameters of the reaction. Its isolation and crystallographic characterization, provided crucial insights into the factors that govern the stereochemical course of the reaction. Using the information gained from the solid state structure of the catalytically active species, the enantioselectivity of the process was improved by optimizing the sulfenylating agent. A computational analysis of the enantiodetermining thiiranium ion formation substantiated the experimental findings. The major contributor to the enantiotopic face discrimination is the avoidance of steric repulsion between the binaphthyl backbone and one of the substituents of the approaching alkene. These insights are guiding the design of more selective Lewis base catalysts.
Methods
Following the literature procedure,21 an oven-dried Schlenk flask was charged with sulfenylating agent 2j (351 mg, 1.03 mmol, 1.03 equiv), catalyst (R)-3c (51.0 mg, 0.098 mmol, 0.098 equiv), substrate 1b and CH2Cl2 (5.0 mL). The flask was capped with a septum and placed into an i-PrOH bath, which was cooled to 0 °C using a cryocool unit. The temperature of the mixture was monitored via a thermocouple digital temperature probe. After the temperature stabilized, MsOH (65 µL, 1.0 mmol, 1.0 equiv) was added via syringe and the mixture was allowed to stir for 48 h at 0 °C. Upon complete reaction (TLC monitoring), the mixture was quenched while cold by the addition of Et3N (0.20 mL). The mixture was poured into aqueous HCl (1.0 M, 20 mL) in a separatory funnel, CH2Cl2 (30 mL) was added and the layers were thoroughly mixed. The organic layer was poured into aqueous NaOH (1.0 M, 20 mL), and the layers were thoroughly mixed and then separated. The acidic layer was back-extracted with CH2Cl2 (30 mL) which was poured into the basic layer and used to extract that layer as well. Both organic portions were combined, dried over MgSO4, filtered through glass wool and concentrated in vacuo (20–23 °C, 20 mmHg). Purification by flash column chromatography (SiO2, 65 g, 35 mm ∅, hexane/MTBE, 60:1) afforded 310 mg (87%) of a 93:7 mixture of 4bj and the corresponding tetrahydrofurane (4bj') as a pale yellow oil. Partial separation of isomers was accomplished by flash column chromatography using high porosity silica gel (SiO2, 65 g, 35 mm ∅, hexane/MTBE, 80:1→5:1) yielding 271 mg of a 97:3 mixture of 4bj and 4bj' and 30 mg of a 60:40 mixture of 4bj and 4bj'.
X-ray crystallographic data
CCDC 1006824 contains the crystallographic data for the dimer in equation 2 and CCDC 1006831 contains the crystallographic data for compound 5b. These data can be obtained free of charge from the Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk.
Supplementary Material
Acknowledgements
We are grateful to the National Institutes of Health (GM R01-085235) for generous financial support. E. H. thanks the Deutscher Akademischer Austausch Dienst for a postdoctoral fellowship. D. J.-P. K. thanks the University of Illinois for a Seemon H. Pines Graduate Fellowship in Synthetic Organic Chemistry. We are grateful to Dr. Larry M. Wolf (MPI-Mühlheim) for assistance with the computational analysis.
Footnotes
Author Contributions
E. H. planned and carried out the experimental work and obtained the X-ray structure of 5b. D. J.-P. K. carried out the kinetic analysis and H. W. performed the transition state calculations. S.E.D. initiated and directed the project. E. H. wrote the manuscript with the assistance of the other authors.
The authors declare no competing financial interests.
Supplementary information accompanies this paper at www.nature.com/naturechemistry.
References
- 1.Metzner P, Thuillier A. Sulfur Reagents in Organic Synthesis. San Diego, CA: Academic Press; 1994. [Google Scholar]
- 2.Page P, editor. Organosulfur Chemistry: Synthetic Aspects. London: Academic Press; 1995. [Google Scholar]
- 3.Prinsep MR. Sulfur-containing natural products from marine invertebrates. Studies in Natural Products Chemistry. 2003;28:617–751. [Google Scholar]
- 4.de la Mare PBD, Bolton R. In: Electrophilic Additions to Unsaturated Systems. 2nd ed. de la Mare P, B D, Bolton R, editors. Vol. 9. Amsterdam: Elsevier; 1982. pp. 198–246. [Google Scholar]
- 5.Smit WA, Zefirov NS, Bodrikov IV, Krimer MZ. Episulfonium Ions: Myth and Reality. Acc. Chem. Res. 1979;12:282–288. [Google Scholar]
- 6.Rayner CM. In: Organosulfur Chemistry: Synthetic Aspects. Page P, editor. Chapter 3. London: Academic Press; 1995. [Google Scholar]
- 7.Archer NJ, Rayner CM, Bell D, Miller D. Synthetic routes to novel homochiral sulfenyl sulfonium salts and their use as potential enantioselective sulfenylating agents. Asymmetric synthesis via homochiral thiiranium ions. Synlett. 1994:617–619. [Google Scholar]
- 8.Lucchini V, Modena G, Pasquato L. Enantiopure thiosutfonium salts in asymmetric synthesis. Face selectivity in electrophilic additions to unfunctionalised olefins. J. Chem. Soc. Chem. Commun. 1994:1565–1566. [Google Scholar]
- 9.Marigo M, Wabnitz TC, Fielenbach D, Jørgensen KA. Enantioselective organocatalyzed α-sulfenylation of aldehydes. Angew. Chem. Int. Ed. 2005;44:794–797. doi: 10.1002/anie.200462101. [DOI] [PubMed] [Google Scholar]
- 10.Zhao GL, Rios R, Vesely J, Eriksson L, Córdova A. Organocatalytic enantioselective aminosulfenylation of α,β-unsaturated aldehydes. Angew. Chem. Int. Ed. 2008;47:8468–8472. doi: 10.1002/anie.200802335. [DOI] [PubMed] [Google Scholar]
- 11.Sobhani S, Fielenbach D, Marigo M, Wabnitz TC, Jørgensen KJ. Direct organocatalytic asymmetric α-sulfenylation of activated C–H Bonds in lactones, lactams, and β-dicarbonyl compounds. Chem.–Eur. J. 2005;11:5689–5694. doi: 10.1002/chem.200500512. [DOI] [PubMed] [Google Scholar]
- 12.Han Z, Chen W, Dong S, Yang C, Liu H, Pan Y, Yan L, Jiang Z. Highly enantioselective organocatalytic sulfenylation of 3-aryloxindoles. Org. Lett. 2012;14:4670–4673. doi: 10.1021/ol3021176. [DOI] [PubMed] [Google Scholar]
- 13.Guan H, Wang H, Huang D, Shi Y. Enantioselective oxysulfenalytion and oxyselenylation of olefins catalyzed by chiral Brønsted acids. Tetrahedron. 2012;68:2728–2735. [Google Scholar]
- 14.Hamilton GL, Kanai T, Toste FD. Chiral anion-mediated asymmetric ring opening of meso-aziridinium and episulfonium ions. J. Am. Chem. Soc. 2008;130:14984–14986. doi: 10.1021/ja806431d. [DOI] [PubMed] [Google Scholar]
- 15.Lin S, Jacobsen EN. Thiourea-catalysed ring opening of episulfonium ions with indole derivatives by means of stabilizing non-covalent interactions. Nature Chem. 2012;4:817–824. doi: 10.1038/nchem.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Denmark SE, Beutner GL. Lewis base catalysis in organic synthesis. Angew. Chem. Int. Ed. 2008;47:1560–1638. doi: 10.1002/anie.200604943. [DOI] [PubMed] [Google Scholar]
- 17.Denmark SE, Collins WR. Lewis base activation of Lewis acids: development of a Lewis base catalyzed selenolactonization. Org. Lett. 2007;9:3801–3804. doi: 10.1021/ol701617d. [DOI] [PubMed] [Google Scholar]
- 18.Denmark SE, Kalyani D, Collins WR. Preparative and mechanistic studies toward the rational development of catalytic, enantioselective selenoetherification Reactions. J. Am. Chem. Soc. 2010;132:15752–15765. doi: 10.1021/ja106837b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Denmark SE, Collins WR, Cullen MD. Observation of direct sulfenium and selenenium group transfer from thiiranium and seleniranium ions to alkenes. J. Am. Chem. Soc. 2009;131:3490–3492. doi: 10.1021/ja900187y. [DOI] [PubMed] [Google Scholar]
- 20.Denmark SE, Vogler T. Synthesis and reactivity of enantiomerically enriched thiiranium ions. Chem. Eur. J. 2009;15:11737–11745. doi: 10.1002/chem.200901377. [DOI] [PubMed] [Google Scholar]
- 21.Denmark SE, Kornfilt DJP, Vogler T. Catalytic asymmetric thiofunctionalization of unactivated alkenes. J. Am. Chem. Soc. 2011;133:15308–15311. doi: 10.1021/ja2064395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Denmark SE, Chi HM. Catalytic, enantioselective, intramolecular carbosulfenylation of olefins. Mechanistic aspects: a remarkable case of negative catalysis. J. Am. Chem. Soc. 2014;136:3655–3663. doi: 10.1021/ja413270h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Denmark SE, Jaunet A. Catalytic, enantioselective, intramolecular carbosulfenylation of olefins. J. Am. Chem. Soc. 2013;135:6419–6422. doi: 10.1021/ja401867b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Denmark SE, Jaunet A. Catalytic, enantioselective, intramolecular carbosulfenylation of olefins. Preparative and stereochemical aspects. J. Org. Chem. 2014;79:140–171. doi: 10.1021/jo4023765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Denmark SE, Chi HM. J. Am. Chem. Soc. 2014 in press, doi: [Google Scholar]
- 26.Kal'yan YB, Krimer MZ, Cherepanova EG, Bogdanov VS, Smit VA. Arylthiosulfonium salts as transfer agents of the S-aryl group to double bonds. Russ. Chem. Bull. 1982;31:342–349. [Google Scholar]
- 27.Krossing I, Raabe I. Noncoordinating Anions – Fact or Fiction? A Survey of Likely Candidates. Angew. Chem. Int. Ed. 2004;43:2066–2090. doi: 10.1002/anie.200300620. [DOI] [PubMed] [Google Scholar]
- 28.Hartmann F, Dahlems T, Mootz D. Crystal Structure of hexamethylphosphoric triamide (C2H3N)3PO. Z. Kristallogr.-New Cryst. Struct. 1998;213:639–640. [Google Scholar]
- 29.Denmark SE, Eklov BM. Neutral and Cationic Phosphoramide Adducts of Silicon Tetrachloride: Synthesis and Characterization of Their Solution and Solid-State Structures. Chem. Eur. J. 2008;14:234–239. doi: 10.1002/chem.200701466. [DOI] [PubMed] [Google Scholar]
- 30.Arduengo AJ, Burgess EM. Tricoordinate Hypervalent Sulfur-Compounds. J. Am. Chem. Soc. 1977;99:2376–2378. [Google Scholar]
- 31.Kuhn N, Bohnen H, Fahl J, Bläser D, Boese R. Koordination oder Reduktion? Zur Reaktion von 1,3-Diisopropy1-4,5-dimethylimidazol-Zyliden mit Schwefelhalogeniden und Schwefeloxidhalogeniden. Chem. Ber. 1996;129:1579–1586. [Google Scholar]
- 32.Godfrey SM, Ollerenshaw RTA, Pritchard RG, Richards CL. Structural isomerism in R3PSe(Ph)I compounds: the ionic structure of [(Me2N)3PSePh]I. J. Chem. Soc., Dalton Trans. 2001:508–509. [Google Scholar]
- 33.Sacerdoti M, Gilli G, Domiano P. Comparison of two independent structure determinations of diphenyl disulphide. Acta Cryst. 1975;B31:327–329. [Google Scholar]
- 34.Rudd MD, Lindeman SV, Husebye S. Structural characteristics of three-coordinate arylhalide tellurium(II) complexes with chalcogen ligands. Synthesis, spectroscopic characterization and X-ray structural studies of bromo[N-methylbenzothiazole-2(3H)selone]phenyltellurium(II), bromophenyl[tris-(dimethylamino)phosphaneselenide]tellurium(II) and tris(dimethylamino)phosphanesulfide. Acta Chem. Scand. 1996;50:759–774. [Google Scholar]
- 35.Acheson RM, Lines CT, Bryce MR, Dauter Z, Reynolds CD, Schmidpeter A. Synthesis and X-Ray crystal structures of 2,3-dihydro-2-mercapto-2,1,3-benzophosphadiazine-4(1H)-thione 2-sulphide derivatives. J. Chem. Soc. Perkin Trans. 1985;II:1913–1917. [Google Scholar]
- 36.Goldstein E, Beno B, Houk KN. Density functional theory prediction of the relative energies and isotope effects for the concerted and stepwise mechanisms of the Diels-Alder reaction of butadiene and ethylene. J. Am. Chem. Soc. 1996;118:6036–6034. [Google Scholar]
- 37.Cantillo D, Kappe CO. A unified mechanistic view on the Morita-Baylis-Hillman reaction: computational and experimental investigations. J. Org. Chem. 2010;75:8615–8625. doi: 10.1021/jo102094h. [DOI] [PubMed] [Google Scholar]
- 38.Denmark SE, Pham SM. Kinetic analysis of the divergence of reaction pathways in the chiral Lewis base promoted aldol addition of trichlorosilyl enolates: a rapid injection NMR study. Helv. Chim. Acta. 2000;83:1846–1853. [Google Scholar]
- 39.Sølling TI, Radom L. A G2 study of SH+ exchange reactions involving lone-pair donors and unsaturated hydrocarbons. Chem. Eur. J. 2001;7:1516–1524. doi: 10.1002/1521-3765(20010401)7:7<1516::aid-chem1516>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 40.Sølling TI, Wild SB, Radom L. Are pi-ligand exchange reactions of thiirenium and thiiranium ions feasible? An ab initio investigation. Chem. Eur. J. 1999;5:509–514. [Google Scholar]
- 41.Sølling TI, Wild SB, Radom L. Are the approach directions of s and p nucleophiles to the sulfur atom of Thiiranium and Thiirenium Ions Different? Chem. Eur. J. 2000;6:590–591. [Google Scholar]
- 42.Modena G, Pasquato L, Lucchini V. Different approaching directions of s and p nucleophiles to the sulfur atom of thiiranium and thiirenium Ions. Chem. Eur. J. 2000;6:589–590. doi: 10.1002/(sici)1521-3765(20000218)6:4<589::aid-chem589>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 43.Fachini M, Lucchini V, Modena G, Pasi M, Pasquato L. Nucleophilic reactions at the sulfur of thiiranium and thiirenium ions. New insight in the electrophilic additions to alkenes and alkynes. Evidence for an episulfurane intermediate. J. Am. Chem. Soc. 1999;121:3944–3950. [Google Scholar]
- 44.Bach RD, Andres JL, Owensby AL, Schlegel HB, McDouall RD. Electronic structure and reactivity of dioxirane and carbonyl oxide. J. Am. Chem. Soc. 1992;114:7207–7217. [Google Scholar]
- 45.Houk KN, Liu J, Demello NC, Condroski KD. Transition states of epoxidations: diradical character, spiro geometries, transition state flexibility, and the origins of stereoselectivity. J. Am. Chem. Soc. 1997;119:10147–10152. [Google Scholar]
- 46.Dmitrenko O, Bach RD. Reassessment of the level of theory required for the epoxidation of ethylene with dioxiranes. J. Phys. Chem. A. 2004;108:6886–6892. [Google Scholar]
- 47.Ess DH, Houk KN. J. Am. Chem. Soc. 2008;130:10187–10198. doi: 10.1021/ja800009z. [DOI] [PubMed] [Google Scholar]
- 48.Glendening ED, Reed AE, Carpenter JE, Weinhold F. NBO Version 3.1. Madison, WI: Theoretical Chemistry Institute, University of Wisconsin; 2001. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.