

Abstract

Herein, the synthesis and biological evaluation of dual opioid agonists–neurokinin 1 receptor (NK1R) antagonists is described. In these multitarget ligands, the two pharmacophores do not overlap, and this allowed maintaining high NK1R affinity and antagonist potency in compounds 12 and 13. Although the fusion of the two ligands resulted in slightly diminished opioid agonism at the μ- and δ-opioid receptors (MOR and DOR, respectively), as compared to the opioid parent peptide, balanced MOR/DOR activities were obtained. Compared to morphine, compounds 12 and 13 produced more potent antinociceptive effects in both acute (tail-flick) and neuropathic pain models (von Frey and cold plate). Similarly to morphine, analgesic tolerance developed after repetitive administration of these compounds. To our delight, compound 12 did not produce cross-tolerance with morphine and high antihyperalgesic and antiallodynic effects could be reinstated after chronic administration of each of the two compounds.
Keywords: Designed multiple ligands, opioid agonism, NK1 antagonism, acute pain, neuropathic pain
The adequate treatment of different pain states remains the major medical challenge in pain research. Morphine and other opioids are the most widely used analgesic drugs for the treatment of moderate to severe and chronic pain, despite the serious side effects that emerge upon chronic administration (e.g., nausea, constipation, physical dependence, and tolerance).1 These drawbacks become increasingly important during prolonged opioid administration, limiting their beneficial effect and rendering these drugs suboptimal for treatment of chronic pain.2 Moreover, of all chronic pain states, neuropathic pain (resulting from nerve tissue damage) is especially difficult to treat, as opioids lose their efficacy in attenuating common symptoms of neuropathy, such as allodynia (painful perception of neutral stimuli) or hypersensitivity to painful stimuli. When opioids are administered for a long period of time, analgesic tolerance develops; higher doses of painkillers are required to maintain the analgesic effect, and simultaneously other deleterious effects become more apparent as well. In addition, the sustained opioid administration results in an increased sensitivity to pain that occurs in undamaged tissues, also termed hyperalgesia.
The mechanisms behind opioid tolerance and opioid-induced hyperalgesia are not well understood, but neuroplastic changes that occur in the central nervous system (CNS) are being held responsible.2,3 Under neuropathic pain conditions, changes in the activity of endogenous opioid systems, reduced number of receptors or glial interactions, have been reported,4−6 and sustained exposure to opioids provokes further changes. These can, for example, lead to an increased release of neurotransmitters like substance P (SP) and the up-regulated expression of the corresponding neurokinin 1 (NK1) receptor.3,7 SP, an endogeneous neuropeptide, is a known pronociceptive neurotransmitter involved in chronic pain states, and it has been shown to contribute to the development of hyperalgesia.8 It activates the NK1 receptors that are expressed both in the CNS and in peripheral tissues. The analgesics currently in use do not counteract these changes in the CNS, rendering them ineffective for chronic pain treatment. The strategy to regulate pain transmission through a simultaneous interaction with the opioid and NK1 receptors, has hence been proposed as an approach for improved/prolonged therapy.9−11
One alternative to the coadministration of different drugs consists of using designed multiple ligands (DMLs), or single molecules exerting multiple pharmacological effects.12−14 DMLs specifically targeting both opioid and NK1 receptors were designed in the past.3,10,15−18 In these DMLs the opioid agonist pharmacophore activates the μ and δ opioid receptors (MOR and DOR, respectively) and is responsible of the analgesic effect, while the NK1 antagonist ligand blocks pain signaling induced by SP, thus potentially increasing and extending the antinociceptive effect of the drug.
A potent hybrid opioid-NK1 peptidomimetic (Dmt-d-Arg-Aba-Gly-NMe-3′,5′-(CF3)2-Bn 1), containing an opioid pharmacophore at the N-terminus and a NK1 pharmacophore at the C-terminus, was designed by our group,19 and its structure was recently optimized via the preparation of Dmt-d-Arg-Aba-βAla-NMe-Bn 2, which proved to be more active in the neuropathic pain models.20 Both compounds belong to the subclass of “merged” DMLs, in which the two pharmacophores, interacting with two different targets, overlap (Figure 1). The opioid agonist part is derived from the μ opioid receptor agonist Dmt-d-Arg-Phe-Lys-NH2 ([Dmt]1-DALDA 3)21 and contains the 4-amino-2-benzazepin-3-one (Aba) scaffold, a conformationally constrained amino acid used in the design of very potent opioid agonists.22−25 The Aba scaffold is also part of the NK1 pharmacophore and enabled the design of very compact chimeric compounds.
Figure 1.
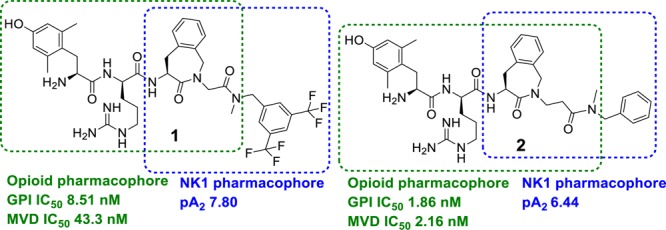
Previously reported opioid agonist-NK1 antagonist hybrid ligands with merged frameworks.
The in vitro evaluation of peptidomimetics 1 and 2 showed that these bifunctional ligands have high (1) to subnanomolar (2) binding affinity for both the μ and δ opioid receptors and good binding affinity for the NK1 receptor.19,20,26 Moreover, the in vivo studies showed activity in acute pain26 as well as in neuropathic pain models.20 The potent analgesic responses after intravenous (i.v.) administration indicated that these hybrid compounds were capable of crossing the blood–brain barrier (BBB), but unfortunately they still produced analgesic tolerance and cross-tolerance with morphine.20,26
The activity profile of DMLs, such as the opioid agonist-NK1 antagonist hybrids, is often dependent on the relative potency at the two target receptors, the modulation of which is a very challenging aspect of DML design.
In this letter we report the synthesis and the biological evaluation of new hybrid opioid-NK1 ligands, in which potent opioid agonist pharmacophores based on the [Dmt]1-DALDA peptide 3 are conjugated to three different NK1 antagonist pharmacophores with the aim to obtain hybrids producing a more potent NK1 antagonism than 1 or 2. The 3-arylpiperidine pharmacophores 4, 5,27,28 and 6(29,30) are known potent NK1 antagonists or fragments thereof (Figure 2).
Figure 2.

NK1 antagonist pharmacophores used in the hybrids.
The hybrid peptidomimetics 7–9 and 11–14 (Scheme 1) were prepared in two steps: the fully protected tetrapeptide precursors were synthesized via solid phase peptide synthesis (SPPS) using the 2-chlorotrityl chloride resin as solid support, and subsequently coupled to the secondary amines 4–6 in solution. Boc-Dmt-d-Arg(Pbf)-Phe-Lys(Boc)-OH and Boc-Dmt-d-Arg(Pbf)-Phe-β3homoLys(Boc)-OH, precursors of compounds 7–9 and 11, respectively, were prepared via standard SPPS using Nα-Fmoc (or Nα-Boc for terminal residue Dmt) protected amino acids. The Boc-Dmt-d-Arg(Pbf)-Aba-βAla-OH peptide, precursor of compounds 12–14, was prepared by direct assembly of the Aba scaffold on the solid support following a protocol recently described.20 The coupling of Boc-Dmt-OH was performed using 3 equiv of DIC/3 equiv of HOBt as the coupling mixture to avoid side reactions that occur when uronium reagents (e.g., TBTU/DIPEA) are used. The fully protected tetrapeptides were cleaved from the resin using a mixture of 1% TFA in DCM for 1 h. The protected peptide acids were coupled to the secondary amines (1.5 equiv of amines 4–6) using 1.5 equiv of DIC/1.5 equiv of HOBt (for 4 and 6) or HOAt (for 5) in DCM at room temperature. After overnight reaction, the reaction mixture was evaporated and the residue was purified by flash chromatography, using a mixture of DCM/MeOH as mobile phase. The removal of the Boc and Pbf protecting groups was achieved using a mixture of TFA/TES/H2O 95:2.5:2.5 v/v for 3 h (Scheme 1). After evaporation of the cleavage mixture and purification by preparative RP-HPLC, all compounds were obtained in high purity (>95%). The intermediate purification of the fully protected peptidomimetics was mandatory because of the overlap between the peaks of the desired unprotected hybrids and their corresponding parent amines 4–6, in the HPLC chromatogram.
Scheme 1. Coupling of the Opioid and NK1 Pharmacophores in Solution.

Reaction conditions: (a) DIC 1.5 equiv, HOBt 1.5 equiv in CH2Cl2, rt, on; (b) flash chromatography purification; (c) TFA/TES/H2O, 95:2.5:2.5 v/v, 3 h; (d) DIC 1.5 equiv, HOAt 1.5 equiv in CH2Cl2, rt, on.
The ligands 7–14 were evaluated in in vitro binding and tissue functional assays (Table 1). [Dmt]1-DALDA 3 is a highly potent MOR agonist that is able to cross the BBB and induce a potent analgesic effect in vivo.21 When this sequence was conjugated to the NK1 fragments 4–6, the resulting hybrids 7–9 showed high affinity for the NK1 receptor, but disappointingly, their affinity and activity at μ- and δ-opioid receptors was drastically reduced. For this reason these compounds were not considered for further testing, and potential antagonism at the NK1 receptor was not determined.
Table 1. Opioid and NK1 Receptor Affinity (nM) and Activity (nM) of the Synthesized Compounds.
| cmpd | pA2a (NK1R) | hNK1Rb (Ki) | GPIc (IC50) | MVDc (IC50) | MORc (Ki) | DORc (Ki) |
|---|---|---|---|---|---|---|
| 1 | 7.80 | 0.50 | 8.51 | 43.3 | 0.42 | 10.4 |
| 2 | 6.44 | 13.0 | 1.86 | 2.16 | 0.08 | 0.28 |
| 3 | 1.41 | 23.1 | 0.58 | 877 | ||
| 7 | nd | 0.99 | 137 | 384 | 9.91 | 533 |
| 8 | nd | 1.1 | 70.2 | 814 | 54.0 | >1000 |
| 9 | nd | 1.2 | 658 | 349 | 39.8 | 339 |
| 10 | nd | nd | 5.90 | 7.70 | 0.187 | 290 |
| 11 | 8.33 | 2.2 | 538 | 1660 | 20.2 | >1000 |
| 12 | 8.11 | 1.4 | 20.3 | 14.1 | 1.18 | 10.6 |
| 13 | 7.54 | 1.4 | 13.0 | 12.6 | 1.82 | 11.2 |
| 14 | 6.72 | 1.9 | 30.1 | 10.3 | 3.07 | 7.07 |
The pA2 values were calculated using Schild’s equation.
Binding affinities for hNK1 receptor in CHO cells determined by displacement of [3H]SP.
The GPI functional assay is representative of MOR activation, whereas the MVD is a DOR receptor-representative assay. Binding affinities for MOR and DOR opioid receptors were determined by displacement of [3H]DAMGO ([d-Ala2,NMePhe4,Gly-ol5]enkephalin) and [3H]DSLET ([d-Ser2,Leu5]enkephalin-Thr6) from rat brain membrane binding sites, respectively. Values represent means of 3–4 experiments. nd: not determined.
When a DML is designed, advantages linked to pharmacophore overlap (e.g., lower molecular weight) can be neutralized by mutual steric hindrance and, consequently, this may result in reduced drug–target interaction. This phenomenon was observed for the chimeric compound 1: reduced binding affinity at both μ and δ opioid receptors, compared to the parent opioid sequence, was observed.22,26 In the present study, the opioid and NK1 pharmacophores do not overlap, but are “fused” or in close proximity of each other. Nevertheless, for hybrids 7–9 it is apparent that the bulky NK1 antagonist component severely hinders opioid binding. In order to increase the distance between the opioid and the NK1 pharmacophores, an additional methylene group was inserted via use of a β-homo amino acid. The [Dmt]1-DALDA sequence 3 modified with a β3homo-Lys residue (i.e., peptide 10, Dmt-d-Arg-Phe-β3homoLys-NH2) proved to have a 3-fold better affinity for both the μ- and δ-opioid receptors than the parent peptide 3 (Table 1). Agonist activity was slightly decreased at MOR but significantly increased at DOR, thereby providing an ideal balanced μ-/δ-agonist profile. Unfortunately, when conjugated to NK1 fragment 6, the high opioid agonist activity of hybrid 11 was, again, severely reduced.
We therefore switched to the opioid sequence Dmt-d-Arg-Aba-βAla-NH2, which was known to be tolerant to C-terminal conjugations.19,20,26 The functional activities at the μ and δ opioid receptors for these hybrid opioid-NK1 ligands 12–14 are significantly reduced, compared to the opioid parent compound Dmt-d-Arg-Aba-βAla-NH2 (GPI IC50 0.80 nM and MVD IC50 0.24 nM, data not shown).20 However, the MVD (DOR) activity of compounds 12–14 was improved compared to the previous lead hybrid compound 1, confirming that the replacement of Gly with the βAla residue is still beneficial to maintain the δ opioid activity. Whereas this replacement seemed to be detrimental for NK1 receptor antagonism in the previous compounds20 [pA2 from 7.80 (1) to 6.44 (2)], it does not have the same negative effect in this new series of compounds. The NK1 receptor affinity is maintained for compounds 12–14, and associated with potent antagonism for 12 and 13. Even though MOR agonism was slightly decreased in compounds 12 and 13, as compared to hybrid 1, improved DOR activity with concomitantly a potent antagonism at NK1 justified further in vivo evaluation of these ligands.
Compound 12 and 13 were selected for further in vivo evaluation.
The compounds were administered intrathecally (i.t.) at different doses, and the effect was measured by the tail-flick test (acute pain model). The analgesic effect of compounds 12 and 13 and of a representative NK1 pharmacophore (5) was compared to the effect of morphine in naïve rats (Figure 3) 30 min after the administration. The hybrid compounds 12 and 13 give potent antinociceptive responses (at 8 and 10 nmol doses, respectively) comparable to the effect of morphine used at dose 35 nmol; hence being 3.5 to 4.5-fold more effective than morphine in alleviating acute pain. In contrast, the analgesic effect of the parent NK1 antagonist 5 was very weak compared to its hybrid 13. The antinociceptive effect of compound 12 was recorded in function of time and gave significant antinociception up to 2 h postadministration (data not shown).
Figure 3.
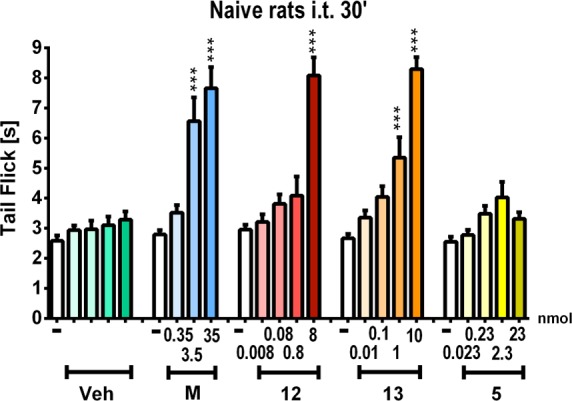
Effect of intrathecal (i.t.) administration of morphine (M), 12, 13, and the parent NK1 antagonist 5 as measured in the tail flick-test 30 min after administration in naïve rats; ***p < 0.001 vs vehicle (Veh)-treated naïve rats.
Additionally, the hybrid compounds 12 and 13, together with the parent NK1 antagonist 5, were tested in the chronic constriction injury (CCI) model of neuropathic pain after i.t. administration, in order to verify their ability to suppress allodynia or hyperalgesia (Figure 4). Seven days after CCI to the sciatic nerve in rats, compounds 12, 13, and 5, as well as morphine, were administered at increasing doses, and their effect on reducing allodynia (von Frey test) or hyperalgesia (cold plate test) was tested 30 min after administration. The antiallodynic effect of the hybrid compounds 12 and 13 was comparable to the one of morphine at a 3.5- to 4.5-fold lower dose, while compounds 12 and 13 were significantly stronger than morphine in the cold plate test, which shows the attenuation of hyperalgesia (Figure 4). From these data, the chimeric compound 12 seems to be the most potent hybrid, efficiently attenuating allodynia and hyperalgesia even at a low dose of 0.8 nmol. The efficiency of the opioid-NK1R hybrids in the applied neuropathic pain models clearly results from the added NK1R component, as the parent NK1 antagonist 5 was very potent in attenuating neuropathic pain symptoms, whereas this NK1R pharmacophore did not give any added value with regard to acute pain thresholds (cf. Figure 3).
Figure 4.
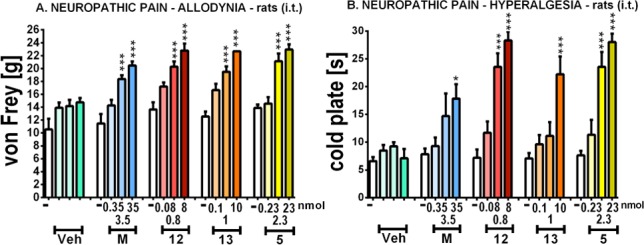
Effect of i.t. administration of morphine (M), hybrids 12 and 13, and the parent NK1 antagonist 5 on (A) allodynia, measured by von Frey test, and (B) hyperalgesia, measured by cold plate test, 30 min after administration in CCI-exposed rats; *p < 0.05, ***p < 0.001 vs Veh-treated CCI-exposed rats.
The hybrid compounds 12 and 13, parent NK1 antagonist 5, and morphine were injected i.t. for 6 consecutive days (once daily) starting from day 7 after CCI, in order to study the effect of chronic drug administration on allodynia and hyperalgesia. As can be noticed in Figure 5, all compounds lost their initial effectiveness, both in the von Frey and cold plate test, on the sixth day of repeated administrations, and the effect of the investigated compounds was similar to the long-term profile of morphine administration. Particularly in the cold plate, the hybrids 12 and 13 were more potent than morphine in attenuating hyperalgesia on the fourth and the fifth day of the experiment (Figure 5B).
Figure 5.
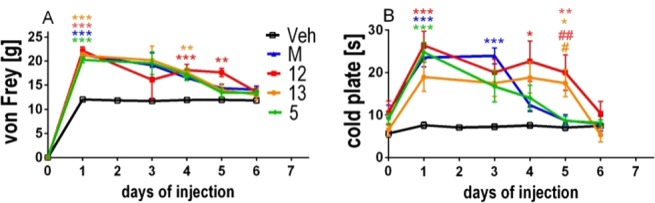
Development of tolerance to antiallodynic effect, measured by the von Frey test (A) and antihyperalgesic effect measured by the cold plate test (B) after repeated once daily (6 days) i.t. injections of morphine (M, 3.5 nmol), hybrid compounds 12 (0.8 nmol) and 13 (1 nmol), and the NK1 parent compound 5 (2.3 nmol); *p < 0.05, **p < 0.01, ***p < 0.001 vs Veh-treated CCI-exposed rats; #p < 0.05, ##p < 0.01 vs morphine-treated CCI-exposed rats.
The hybrid compounds 12, 13 and morphine were examined in a cross tolerance study (Figure 6). Starting from day 7 after CCI compounds 12 and 13 or morphine were daily administered to rats. As tolerance is developed within 6 days of repeated injections for all compounds (Figure 5), on day 7 of the repeated administration (day 14 after CCI) the animals that were tolerant to morphine were injected with a single dose of 12 or 13 (Figure 6A,D), and the animals tolerant to 12 or 13 were administered with a single dose of morphine (Figure 6B,E or C,F, respectively). When morphine is administered to animals tolerant to the hybrid compounds 12 and 13, the antinociceptive effect, especially for hyperalgesia (Figure 6E,F), increases. This indicates that no cross-tolerance occurs between the hybrid compounds and morphine. Similarly, when compound 12 is injected to an animal tolerant of morphine, it is also able to reduce allodynia (Figure 6A). This lack in cross-tolerance was even more pronounced in the cold plate test, a model of hyperalgesia (Figure 6D). In contrast, a single dose of compound 13 was not effective in rats that were rendered tolerant to morphine (Figure 6A,D).
Figure 6.
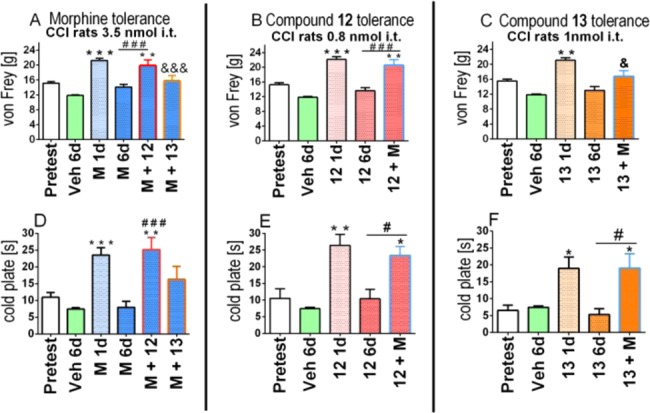
Cross-tolerance of hybrid compounds 12 and 13 with morphine (M) measured in neuropathic pain. The influence of repeated 6 days i.t. administration of morphine (3.5 nmol), 12 (0.8 nmol), and 13 (1 nmol) on the effect of a single dose of 12 (0.8 nmol) or 1 nmol of 13 (in morphine repeatedly treated CCI-exposed rats) or 3.5 nmol of morphine (in 12 or 13 repeatedly treated CCI-exposed rats) was measured on day 7. *p < 0.05, **p < 0.01, ***p < 0.001 indicates a significant difference compared with pretest. #p < 0.05, ###p < 0.001 indicates a significant difference compared with the effect of a drug on 6th day of repeated once daily administration. &p < 0.05, &&&p < 0.001 indicates a significant difference compared with the effect of a drug on the first day of repeated administration.
In conclusion, hybrid opioid agonist-neurokinin 1 antagonist ligands were prepared by fusion of peptide opioid pharmacophores and nonpeptide neurokinin 1 receptor antagonists. Hybridization of the pharmacophores gratifyingly maintained the desired dual profile in vitro. Furthermore, this fusion resulted in compounds that manage to alleviate acute pain, an effect mostly induced by the opioid subunit of the chimeric compounds, and neuropathic pain in vivo. From the data it is clear that NK1R antagonism contributes to the observed effect in the neuropathic pain model. In comparison to our earlier reported ligands (Figure 1), these ligands manage to suppress to some extent of tolerance to antihyperalgesic effects (cf. Figure 5), and compounds 12 and 13 diminish cross tolerance with morphine. Therefore, a mixed opioid agonism-neurokinin 1 antagonist profile with highly potent NK1R antagonism represents a viable approach toward analgesics that remain orthogonal to the existing clinically applied painkillers.
Glossary
ABBREVIATIONS
- Aba
4-amino-2-benzazepinone scaffold
- BBB
blood–brain barrier
- CCI
chronic constriction injury
- CNS
central nervous system
- DIC
N,N′-dicyclohexylcarbodiimide
- DIPEA
diisoproypethylamine
- DML
designed multiple ligand
- Dmt
2′,6′-dimethyl tyrosine
- DOR
δ opioid receptor
- GPI
guinea pig ileum
- HOAt
1-hydroxyazabenzotriazole
- HOBt
1-hydroxybenzotriazole
- MOR
μ opioid receptor
- MVD
mouse vas deferens
- NK1
neurokinin-1
- RP-HPLC
reverse phase high pressure liquid chromatography
- SP
substance P
- SPPS
solid phase peptide synthesis
- TBTU
O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate
- TES
triethylsilane
- TFA
trifluoroacetic acid
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00359.
Experimental procedures as well as compound characterization data (PDF)
Author Contributions
C.B., L.F., and A.N. were in charge of the chemical synthesis of all GPCR ligands. B.P., J.S., J.M., and W.M. were in charge of the evaluation of in vivo potency of all ligands. N.N.C. performed the in vitro functional opioid assays. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The work of C.B., S.B., D.T., and P.W.S. was supported by a collaboration convention between the Ministère du Développement Economique, de l’Innovation et de l’Exportation du Québec, and the Research Foundation–Flanders (FWO Vlaanderen). The work of B.P., J.S., J.M., and W.M. was supported by MAESTRO grant 2012/06/A/NZ4/00028 and statutory funds; J.S. is a Ph.D. student and has a scholarship from the KNOW funds sponsored by Ministry of Science and Higher Education, Poland. The work of P.W.S. was also supported by grants from the NIH (DA004443) and the Canadian Institutes of Health Research (MOP-89716).
The authors declare no competing financial interest.
Supplementary Material
References
- Pasternak G. W. Opioids and their receptors: Are we there yet?. Neuropharmacology 2014, 76, 198–203. 10.1016/j.neuropharm.2013.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Largent-Milnes T.; Yamamoto T.; Nair P.; Moulton J.; Hruby V.; Lai J.; Porreca F.; Vanderah T. Spinal or systemic TY005, a peptidic opioid agonist/neurokinin 1 antagonist, attenuates pain with reduced tolerance. Br. J. Pharmacol. 2010, 161, 986–1001. 10.1111/j.1476-5381.2010.00824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T.; Nair P.; Jacobsen N. E.; Kulkarni V.; Davis P.; Ma S.-w.; Navratilova E.; Yamamura H. I.; Vanderah T. W.; Porreca F.; Lai J.; Hruby V. J. Biological and conformational evaluation of bifunctional compounds for opioid receptor agonists and neurokinin 1 receptor antagonists possessing two penicillamines. J. Med. Chem. 2010, 53, 5491–5501. 10.1021/jm100157m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mika J.; Osikowicz M.; Makuch W.; Przewlocka B. Minocycline and pentoxifylline attenuate allodynia and hyperalgesia and potentiate the effects of morphine in rat and mouse models of neuropathic pain. Eur. J. Pharmacol. 2007, 560, 142–149. 10.1016/j.ejphar.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Przewłocki R.; Przewłocka B. Opioids in chronic pain. Eur. J. Pharmacol. 2001, 429, 79–91. 10.1016/S0014-2999(01)01308-5. [DOI] [PubMed] [Google Scholar]
- Mika J.; Zychowska M.; Popiolek-Barczyk K.; Rojewska E.; Przewlocka B. Importance of glial activation in neuropathic pain. Eur. J. Pharmacol. 2013, 716, 106–119. 10.1016/j.ejphar.2013.01.072. [DOI] [PubMed] [Google Scholar]
- King T.; Gardell L. R.; Wang R.; Vardanyan A.; Ossipov M. H.; Malan T. P.; Vanderah T. W.; Hunt S. P.; Hruby V. J.; Lai J.; Porreca F. Role of NK-1 neurotransmission in opioid-induced hyperalgesia. Pain 2005, 116, 276–288. 10.1016/j.pain.2005.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasabov S. G.; Rogers S. D.; Ghilardi J. R.; Peters C. M.; Mantyh P. W.; Simone D. A. Spinal neurons that possess the substance P receptor are required for the development of central sensitization. J. Neurosci. 2002, 22, 9086–9098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y. J.; Arttamangkul S.; Evans C. J.; Williams J. T.; von Zastrow M. Neurokinin 1 receptors regulate morphine-induced endocytosis and desensitization of μ-opioid receptors in CNS neurons. J. Neurosci. 2009, 29, 222–233. 10.1523/JNEUROSCI.4315-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foran S. E.; Carr D. B.; Lipkowski A. W.; Maszczynska I.; Marchand J. E.; Misicka A.; Beinborn M.; Kopin A. S.; Kream R. M. Inhibition of morphine tolerance development by a substance P-opioid peptide chimera. J. Pharmacol. Exp. Ther. 2000, 295, 1142–1148. [PubMed] [Google Scholar]
- Tumati S.; Largent-Milnes T. M.; Keresztes A. I.; Yamamoto T.; Vanderah T. W.; Roeske W. R.; Hruby V. J.; Varga E. V. Tachykinin NK1 receptor antagonist co-administration attenuates opioid withdrawal-mediated spinal microglia and astrocyte activation. Eur. J. Pharmacol. 2012, 684, 64–70. 10.1016/j.ejphar.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleczkowska P.; Lipkowski A. W.; Tourwé D.; Ballet S. Hybrid opioid/non-opioid ligands in pain research. Curr. Pharm. Des. 2013, 19, 7435–7450. 10.2174/138161281942140105165646. [DOI] [PubMed] [Google Scholar]
- Fujii H.Twin and triplet drugs in opioid research. In Chemistry of Opioids; Springer: New York, 2011; pp 239–275. [DOI] [PubMed] [Google Scholar]
- Costantino L.; Barlocco D. Designed multiple ligands: basic research vs clinical outcomes. Curr. Med. Chem. 2012, 19, 3353–3387. 10.2174/092986712801215883. [DOI] [PubMed] [Google Scholar]
- Foran S. E.; Carr D. B.; Lipkowski A. W.; Maszczynska I.; Marchand J. E.; Misicka A.; Beinborn M.; Kopin A. S.; Kream R. M. A substance P-opioid chimeric peptide as a unique nontolerance-forming analgesic. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 7621–7626. 10.1073/pnas.130181897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardanyan R.; Kumirov V. K.; Nichol G. S.; Davis P.; Liktor-Busa E.; Rankin D.; Varga E.; Vanderah T.; Porreca F.; Lai J.; Hruby V. J. Synthesis and biological evaluation of new opioid agonist and neurokinin-1 antagonist bivalent ligands. Bioorg. Med. Chem. 2011, 19, 6135–6142. 10.1016/j.bmc.2011.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T.; Nair P.; Largent-Milnes T. M.; Jacobsen N. E.; Davis P.; Ma S.-W.; Yamamura H. I.; Vanderah T. W.; Porreca F.; Lai J.; Hruby V. J. Discovery of a potent and efficacious peptide derivative for δ/μ opioid agonist/neurokinin 1 antagonist activity with a 2′, 6′-dimethyl-L-tyrosine: in vitro, in vivo, and NMR-based structural studies. J. Med. Chem. 2011, 54, 2029–2038. 10.1021/jm101023r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T.; Nair P.; Vagner J.; Largent-Milnes T.; Davis P.; Ma S.-w.; Navratilova E.; Moye S.; Tumati S.; Lai J. A structure–activity relationship study and combinatorial synthetic approach of C-terminal modified bifunctional peptides that are δ/μ opioid receptor agonists and neurokinin 1 receptor antagonists. J. Med. Chem. 2008, 51, 1369–1376. 10.1021/jm070332f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballet S.; Feytens D.; Buysse K.; Chung N. N.; Lemieux C.; Tumati S.; Keresztes A.; Van Duppen J.; Lai J.; Varga E.; Porreca F.; Schiller P. W.; Vanden Broeck J.; Tourwé D. Design of novel neurokinin 1 receptor antagonists based on conformationally constrained aromatic amino acids and discovery of a potent chimeric opioid agonist-neurokinin 1 receptor antagonist. J. Med. Chem. 2011, 54, 2467–2476. 10.1021/jm1016285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemyn K.; Kleczkowska P.; Lesniak A.; Dyniewicz J.; Van der Poorten O.; Van den Eynde I.; Keresztes A.; Varga E.; Lai J.; Porreca F.; Chung N. N.; Lemieux C.; Mika J.; Rojewska E.; Makuch W.; Van Duppen J.; Przewlocka B.; Vanden Broeck J.; Lipkowski A. W.; Schiller P. W.; Tourwé D.; Ballet S. Synthesis and biological evaluation of compact, conformationally constrained bifunctional opioid agonist – Neurokinin-1 antagonist peptidomimetics. Eur. J. Med. Chem. 2015, 92, 64–77. 10.1016/j.ejmech.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller P. W.; Nguyen T. M.-D.; Berezowska I.; Dupuis S.; Weltrowska G.; Chung N. N.; Lemieux C. Synthesis and in vitro opioid activity profiles of DALDA analogues. Eur. J. Med. Chem. 2000, 35, 895–901. 10.1016/S0223-5234(00)01171-5. [DOI] [PubMed] [Google Scholar]
- Ballet S.; Feytens D.; Wachter R. D.; Vlaeminck M. D.; Marczak E. D.; Salvadori S.; de Graaf C.; Rognan D.; Negri L.; Lattanzi R.; Lazarus L. H.; Tourwé D.; Balboni G. Conformationally constrained opioid ligands: The Dmt-Aba and Dmt-Aia versus Dmt-Tic scaffold. Bioorg. Med. Chem. Lett. 2009, 19, 433–437. 10.1016/j.bmcl.2008.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballet S.; Frycia A.; Piron J.; Chung N.; Schiller P. W.; Kosson P.; Lipkowski A.; Tourwe D. Synthesis and biological evaluation of constrained analogues of the opioid peptide H-Tyr-d-Ala-Phe-Gly-NH2 using the 4-amino-2-benzazepin-3-one scaffold. J. Pept. Res. 2005, 66, 222–230. 10.1111/j.1399-3011.2005.00291.x. [DOI] [PubMed] [Google Scholar]
- Ballet S.; Misicka A.; Kosson P.; Lemieux C.; Chung N. N.; Schiller P. W.; Lipkowski A. W.; Tourwé D. Blood– brain barrier penetration by two dermorphin tetrapeptide analogues: role of lipophilicity vs structural flexibility. J. Med. Chem. 2008, 51, 2571–2574. 10.1021/jm701404s. [DOI] [PubMed] [Google Scholar]
- Ballet S.; Marczak E. D.; Feytens D.; Salvadori S.; Sasaki Y.; Abell A. D.; Lazarus L. H.; Balboni G.; Tourwé D. Novel multiple opioid ligands based on 4-aminobenzazepinone (Aba), azepinoindole (Aia) and tetrahydroisoquinoline (Tic) scaffolds. Bioorg. Med. Chem. Lett. 2010, 20, 1610–1613. 10.1016/j.bmcl.2010.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemyn K.; Kleczkowska P.; Novoa A.; Vandormael B.; Van den Eynde I.; Kosson P.; Asim M. F.; Schiller P. W.; Spetea M.; Lipkowski A. W.; Tourwé D.; Ballet S. In vivo antinociception of potent mu opioid agonist tetrapeptide analogues and comparison with a compact opioid agonist-neurokinin 1 receptor antagonist chimera. Mol. Brain 2012, 5, 4. 10.1186/1756-6606-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens F.; Sommen F.; De Boeck B. C. A. G.; Leenaerts J. E.; Van Roosbroeck Y. E. M.; Meert T. F.. Novel formulations of opioid-based treatments of pain comprising substituted 1,4-di-piperidin-4-yl-piperazine derivatives. WO 2004/110415 A2, 2004.
- Kolczewski S.; Roever S.; Schnider P.. Piperidine derivatives as neurokinin 1 antagonists. WO 02/062784 A1, 2002.
- Miyake T.; Yamanaka T.; Asai H.; Terakawa Y.. Piperidine compound and process for preparing the same. WO 2006/004195 A1, 2006.
- Shirai J.; Sugiyama H.; Morimoto S.; Maezaki H.; Yamamoto Y.; Okanishi S.; Kamo I.; Matsumoto S.; Ishigami K.; Inatomi N.; Imanishi A.; Kawamoto M.; Tarui N.; Hashimoto T.; Ikeura Y. Novel 3-phenylpiperidine-4-carboxamides as highly potent and orally long-acting neurokinin-1 receptor antagonists with reduced CYP3A induction. Bioorg. Med. Chem. 2012, 20, 962–977. 10.1016/j.bmc.2011.11.048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.