

Abstract

The pre-clinical characterization of the aryl piperazinyl urea inhibitor of fatty acid amide hydrolase (FAAH) JNJ-42165279 is described. JNJ-42165279 covalently inactivates the FAAH enzyme, but is highly selective with regard to other enzymes, ion channels, transporters, and receptors. JNJ-42165279 exhibited excellent ADME and pharmacodynamic properties as evidenced by its ability to block FAAH in the brain and periphery of rats and thereby cause an elevation of the concentrations of anandamide (AEA), oleoyl ethanolamide (OEA), and palmitoyl ethanolamide (PEA). The compound was also efficacious in the spinal nerve ligation (SNL) model of neuropathic pain. The combination of good physical, ADME, and PD properties of JNJ-42165279 supported it entering the clinical portfolio.
Keywords: FAAH, covalent, ethanolamides, enzyme, anandamide
The fatty acid amide hydrolases (FAAH and FAAH-2)1,2 interrupt the actions, through degradation, of a variety of endogenous lipid signaling molecules.3 FAAH rapidly degrades several fatty acid ethanolamides, including FAAH’s primary substrate, AEA (N-arachidonyl ethanolamide or anandamide),4 PEA (N-palmitoyl ethanolamide),5,6 and OEA (N-oleoyl ethanolamide).7 In contrast, FAAH-2 catabolizes ethanolamides less efficiently, but will hydrolyze long-chain primary amides. The likely source of AEA’s analgesic pharmacology is its ability to agonize the cannabinoid receptor CB1.8−10 However, AEA is synthesized on demand and then rapidly broken down locally, which mitigates the side-effects observed as a result of systemic CB1 agonism (e.g., Δ9 THC pharmacology).
As AEA is synthesized in a localized manner, one might hypothesize that inhibiting FAAH could lead to elevated concentrations of AEA in relevant tissues. Indeed, prior reports have described an increase in AEA levels in the plasma and brains11−14 of rats and in the plasma of humans upon inhibition of FAAH.
Small molecule interruption of FAAH activity has been examined in numerous laboratories (Figure 1) including those of Boger,15−20 Piomelli,21−23 Sanofi,24 Pfizer,25,26 Takeda,27 and us.28 Each of the aforementioned laboratories focused on molecules that form covalent bonds with Ser241 within the FAAH active site, though recently Boger reported the preparation of compounds that could form an additional covalent bond with Cys269.29 OL-135 forms a reversible tetrahedral hemiketal intermediate derived from the attack of Ser241 onto the ketone. FAAH inhibitors prepared in the laboratories of Piomelli and Sanofi carbamylate the active site Ser241 of the FAAH enzyme with the ejection of an alcoholic or phenolic fragment.30 Urea derivatives as prepared by Pfizer, Takeda, and us operate via a similar mechanism but have aromatic amines as leaving groups rather than alcohols or phenols.31 Importantly, OL-135,32 URB597,33,34 JNJ-1661010,35 JNJ-40355003,36−38 and PF-0445784539 have all been found to exhibit analgesic pharmacology in various animal models without the motor impairment associated with direct CB1 agonism.
Figure 1.

Some known FAAH inhibitors.
In addition to the many covalent inactivators of FAAH, several competitive inhibitor classes have been reported by Abbott,40 Amgen,41 Renovis,42 and Johnson and Johnson.43
Several compounds have been profiled to some degree in the clinic. Pfizer’s PF-0445784544 was evaluated in phase 2 as an analgesic in subjects with osteoarthritis pain, but was apparently ineffective despite evidence of peripheral target engagement. Ongoing studies with PF-04457845 include evaluating the potential for treating cannabinoid dependence,45 PTSD,46 fear conditioning,47 and Tourette’s syndrome.48 Vernal is currently recruiting for a neuropathic pain trial49 involving V158866, and Sanofi-Aventis’ SSR411298 for major depressive disorder (phase II)50 and later for persistent cancer pain. The latter trial was discontinued for strategic reasons.51
In the course of preparing aryl piperazinyl ureas, one compound, JNJ-42165279, stood out with regard to selectivity and ADME properties. The optimized preparation (Scheme 1) of JNJ-42165279 involved a reductive amination of 2,2-difluorobenzo[d][1,3]dioxole-5-carbaldehyde with piperazine under continuous flow hydrogenation conditions. The resultant product (1) was then added to a freshly prepared solution of phenyl (4-chloropyridin-3-yl)carbamate (2) to yield JNJ-42165279 in good overall yield.
Scheme 1. Synthesis of JNJ-42165279.

Conditions: (a) 4 equiv of piperazine, 18 h; (b) two cycles through an H-Cube 20% Pd(OH)2/C, 1 atm. H2 (10% excess), 70 °C, 6 mL/min, 83%; (c) 0.95 equiv of PhOCOCl, 1.20 equiv of pyridine, toluene, 2–5 °C, 7 h; (d) 1.0 equiv of 1, 1.5 equiv of K2CO3, water, 15 h, recrystallized from hot i-PrOAc, 50% over two steps.
The ability of JNJ-42165279 to inhibit recombinant human and rat FAAH was then quantified 1 h postincubation with the enzyme and the apparent IC50s found to be hFAAH = 70 ± 8 nM and rFAAH = 313 ± 28 nM.52 The expectation was that JNJ-42165279 would be a covalent inhibitor of FAAH and that the apparent IC50 would be dependent on the length of time it was incubated with the enzyme, and indeed, this was the case. Interestingly, JNJ-42165279 was not a completely irreversible inhibitor of FAAH as dialysis (Figure 1S) of rFAAH pretreated with JNJ-42165279 overnight at 20 °C yielded a partial return of enzymatic activity.
JNJ-42165279 exhibited high selectivity against a panel of 50 receptors, enzymes, transporters, and ion-channels at 10 μM, at which concentration it did not produce >50% inhibition of binding to any of the targets. Fortunately, JNJ-42165279 also did not inhibit CYPS (1A2, 2C8, 2C9, 2C19, 2D6, 3A4) or hERG when tested at a 10 μM compound concentration.
JNJ-42165279 possessed good physical properties,53 but exhibited some hydrolytic instability at pHs 2–10 at both 22 and 2 °C on a time scale of 1–4 weeks. The products of hydrolysis were 1 and 3-amino-4-chloropyridine.54 Additionally, JNJ-42165279 underwent slight degradation under fluorescent light (but not UV A) in the solid form. As hydrolytic instability would make development of JNJ-42165279 challenging, identifying formulations in which it was stable for the full duration of toxicological and clinical studies (weeks) was of paramount importance. Fortunately, a simple suspension of the free-base of JNJ-42165279 in 0.5% Methocel was developed, and the results from a preformulation assessment supported a >30-day shelf life of the formulated product when stored refrigerated and protected from light.
Preliminary characterization of the metabolic profile of JNJ-42165279 (10 μM) was conducted in vitro using liver microsomes (1 mg/mL) in the presence of NADPH, UDPGA, and GSH and in hepatocytes (1 million cells/mL). Five species (mouse, rat, dog, monkey, and human) were used for microsomal studies, and four species (rat, dog, monkey, and human) were used for hepatocyte studies. A catalogue of the detected metabolites is summarized in the Supporting Information (Table 1S), and a proposed biotransformation scheme for JNJ-42165279 is depicted in Figure 2. Multiple metabolites were observed in all species. Unknown metabolites M1, M2, M3, and M6 involve the loss of the chloro substituent at the pyridine ring. Mono-oxidation of JNJ-42165279 resulted in four metabolites, three localized to the substituted pyridine ring (M8, M10, and M11) and one localized to the piperazine linker (M14). M14 likely represents an N-oxide based on its longer retention time compared to parent. Sequential oxidations of these metabolites formed the dioxidation metabolites (M8 and M13). The dioxidation metabolite M9 was detected in human hepatocytes only.
Figure 2.

Proposed biotransformation scheme for JNJ-42165279 in mouse, rat, dog, monkey, and human. For metabolites not found in all species, the species of metabolite detection is denoted in parentheses. Ms, mouse; R, rat; D, dog; Mk, monkey; H, human. The locations at which biotransformations occurred was narrowed through the use of a MS–MS analysis of secondary ions. The boxed area is where the biotransformation took place.
In vivo, metabolites involving the loss of the chloro substituent from the pyridine ring were only found in rat (M2 and M5) and monkey (M2). Similar to in vitro findings, all four mono-oxidation metabolites (M8, M10, M11, and M14) were identified in rat, dog, and monkey plasma samples. Based on UV chromatograms at 254 nm, M10, M8, and M11 appear to be the major circulating mono-oxidation metabolites in rat (Figure 3S), dog (Figure 4S), and monkey (Figure 5S), respectively. Sequential oxidation metabolites were detected in dog (M13) and monkey (M12 and M13) but not in rat plasma samples. Monkey and dog plasma samples also had detectable levels of the glucuronide M7 derived from the mono-oxidative metabolite M8, M10, or M11.
A GSH adduct of JNJ-42165279 detected in rat liver microsomes and in rat plasma samples involved oxidative dechlorination and GS addition. This adduct was unique to the rat and not detected in vitro or in vivo in other species (Table S1).
JNJ-42165279 exhibited relatively rapid clearance in the course of rat pharmacokinetic experiments, manifesting as a low AUC and Cmax;55 however, sufficiently high exposures were obtainable to support preclinical animal models. In a subsequent higher dose (20 mg/kg) oral PK experiment, compound concentrations were determined both in the plasma and brain of rats (Figure 2S). JNJ-42165279 reached a maximum plasma concentration of 4.2 μM after 1 h, falling to about 130 nM at 8 h and decreasing to below the LLQ by 16 h. Observed concentrations in the brain were somewhat elevated relative to plasma at the Cmax (6.3 μM at 1 h), but those differences diminished by the 8 h time-point (167 nM).
In addition to measuring compound concentrations in the above experiment, the time at which the levels of FAAs in rat brains were maximal was determined. AEA levels increased >4-fold over the basal concentrations, reaching their maximal elevation 2 h postdose. As seen previously in dose–response FAA elevation studies, PEA and OEA concentrations increased to a much greater extent than AEA,56 with elevations reaching almost 10-fold and 12-fold over their respective basal levels. OEA and PEA reached their maximal fold elevation at 4 h postdosing. By the 24 h time-point, the concentrations of all three FAAs had returned to baseline levels.
The analgesic properties of JNJ-42165279 in the rat spinal nerve ligation (SNL or Chung) model of neuropathic pain were examined. In this model, long lasting tactile allodynia is induced in rats by performing tight ligations of the L5 and L6 lumbar spinal nerves. Tactile allodynia is measured by testing the sensitivity to von Frey filaments introduced to the hind paw. Animals developed robust tactile allodynia that was dose-dependently reversed by JNJ-42165279. In this stringent model, where reversion to preinjury paw withdrawal threshold constitutes the maximal possible effect (MPE), the greatest degree of reversal of allodynia at a dose of 60 mg/kg was seen 30 min after compound dosing and was about 59% of MPE (Figure 3, top). In a time-course study, peak efficacy was seen 30 min after dosing but significant efficacy was maintained for at least 2 h (Figure 3, bottom). The ED90 was 22 mg/kg, which corresponds to a plasma concentration of 2.5 μM at 30 min, the time at which efficacy measurements were taken. In comparison, the reference analgesic gabapentin produced a similar degree of efficacy (∼65% of MPE) in this model, but the ED90 for gabapentin was calculated to be 97 mg/kg.
Figure 3.
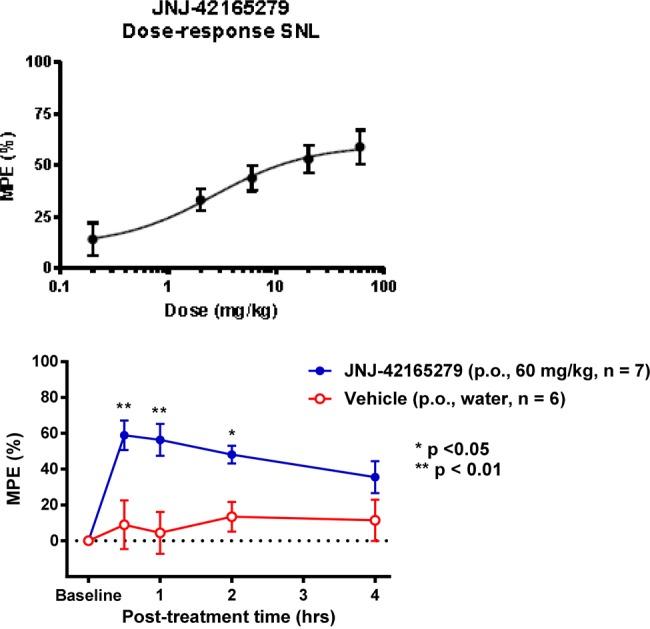
Efficacy of JNJ-42165279 in the rat SNL model of neuropathic pain. The data are expressed as the percentage of the maximum possible effect (%MPE). Top graph: JNJ-42165279 dose-dependently decreased the tactile allodynia associated with this model. The ED90 in this model was determined to be 22 mg/kg, which corresponds to a plasma concentration of 2.5 M at the 30 min time point. Bottom graph: the time course of the antiallodynic efficacy of JNJ-42165279 (60 mg/kg p.o.) in the SNL model.
The combination of good physical, ADME, and PD properties of JNJ-42165279 led to it being advanced to the clinic where it has completed several studies and continues to be evaluated for a variety of neurological conditions.57−61 Additional data supporting the entry of JNJ-42165279 into the clinic will be reported elsewhere.
Acknowledgments
We acknowledge the rest of the Pain and Related Disorders and Support teams for their valuable advice and technical expertise.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00353.
Detailed synthetic procedures, annotated 1H NMR, MS data for the described compounds, and metabolite I.D. chromatograms for JNJ-42165279 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Cravatt B. F.; Giang D. K.; Mayfield S. P.; Boger D. L.; Lerner R. A.; Gilula N. B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 1996, 384, 83–87. 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- Wei B. Q.; Mikkelsen T. S.; McKinney M. K.; Lander E. S.; Cravatt B. F. A second fatty acid amide hydrolase with variable distribution among placental mammals. J. Biol. Chem. 2006, 281, 36569–36578. 10.1074/jbc.M606646200. [DOI] [PubMed] [Google Scholar]
- Ezzili C.; Otrubova K.; Boger D. L. Fatty acid amide signaling molecules. Bioorg. Med. Chem. Lett. 2010, 20, 5959–5968. 10.1016/j.bmcl.2010.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devane W. A.; Hanus L.; Breuer A.; Pertwee R. G.; Stevenson L. A.; Griffin G.; Gibson D.; Mandelbaum A.; Etinger A.; Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Lambert D. M.; Vandevoorde S.; Jonsson K. O.; Fowler C. J. The palmitoylethanolamide family: a new class of anti-inflammatory agents?. Curr. Med. Chem. 2002, 9, 663–674. 10.2174/0929867023370707. [DOI] [PubMed] [Google Scholar]
- Verme J. L.; Fu J.; Astarita G.; La Rana G.; Russo R.; Calignano A.; Piomelli D. The nuclear receptor peroxisome proliferator-activated receptor-α mediates the anti- inflammatory actions of palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15–19. 10.1124/mol.104.006353. [DOI] [PubMed] [Google Scholar]
- Thabuis C.; Destaillats F.; Tissot-Favre D.; Martin J.-C. Oleoyl-ethanolamide (OEA): a bioactive lipid derived from oleic acid and phosphatidylethanolamine. Lipid Technol. 2007, 19, 225–227. 10.1002/lite.200700074. [DOI] [Google Scholar]
- Lichtman A. H.; Hawkins E. G.; Griffin G.; Cravatt B. F. Pharmacological activity of fatty acid amides is regulated, but not mediated, by fatty acid amide hydrolase in vivo. J. Pharmacol. Exp. Ther. 2002, 302, 73–79. 10.1124/jpet.302.1.73. [DOI] [PubMed] [Google Scholar]
- Steffens M.; Zentner J.; Honegger J.; Feuerstein T. Binding affinity and agonist activity of putative endogenous cannabinoids at the human neocortical CB1 receptor. Biochem. Pharmacol. 2005, 69, 169–178. 10.1016/j.bcp.2004.08.033. [DOI] [PubMed] [Google Scholar]
- Cravatt B. F.; Demarest K.; Patricelli M. P.; Bracey M. H.; Giang D. K.; Martin B. R.; Lichtman A. H. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 9371–9376. 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K.; Johnson D. S.; Mileni M.; Beidler D.; Long J. Z.; McKinney M. K.; Weerapana E.; Sadagopan N.; Liimatta M.; Smith S. E.; Lazerwith S.; Stiff C.; Kamtekar S.; Bhattacharya K.; Zhang Y.; Swaney S.; Becelaere K. V.; Stevens R. C.; Cravatt B. F. Discovery and Characterization of a Highly Selective FAAH Inhibitor that Reduces Inflammatory Pain. Chem. Biol. 2009, 16, 411–420. 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D. S.; Stiff C.; Lazerwith S.; Kesten S.; Fay L.; Morris M.; Beidler D.; Liimatta M.; Smith S.; Sadagopan N.; Bhattachar S.; Kesten S.; Dudley D.; Nomanbhoy T. K.; Cravatt B. F.; Ahn K. Discovery of PF-04457845: A Highly Potent, Orally Bioavailable, and Selective Urea FAAH Inhibitor. ACS Med. Chem. Lett. 2011, 2, 91–96. 10.1021/ml100190t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K.; Smith S. E.; Liimatta M. B.; Beidler D.; Sadagopan N.; Dudley D. T.; Young T.; Wren P.; Zhang Y.; Swaney S.; Van Becelaere K.; Blankman J. L.; Nomura D. K.; Bhattachar S. N.; Stiff C.; Nomanbhoy T. K.; Weerapana E.; Johnson D. S.; Cravatt B. F. Mechanistic and pharmacological characterization of PF-04457845: a highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. J. Pharmacol. Exp. Ther. 2011, 338, 114–124. 10.1124/jpet.111.180257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G. L.; Winter H.; Arends R.; Jay G. W.; Le V.; Young T.; Huggins J. P. Assessment of the pharmacology and tolerability of PF-04457845, an irreversible inhibitor of fatty acid amide hydrolase-1, in healthy subjects. Br. J. Clin. Pharmacol. 2012, 73, 706–716. 10.1111/j.1365-2125.2011.04137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger D. L.; Sato H.; Lerner A. E.; Hedrick M. P.; Fecik R. A.; Miyauchi H.; Wilkie G. D.; Austin B. J.; Patricelli M. P.; Cravatt B. F. Exceptionally potent inhibitors of fatty acid amide hydrolase: the enzyme responsible for degradation of endogenous oleamide and anandamide. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 5044–5049. 10.1073/pnas.97.10.5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger D. L.; Miyauchi H.; Du W.; Hardouin C.; Fecik R. A.; Cheng H.; Hwang I.; Hedrick M. P.; Leung D.; Acevedo O.; Guimaraes C. R.; Jorgensen W. L.; Cravatt B. F. Discovery of a Potent, Selective, and Efficacious Class of Reversible α-Ketoheterocycle Inhibitors of Fatty Acid Amide Hydrolase Effective as Analgesics. J. Med. Chem. 2005, 48, 1849–1856. 10.1021/jm049614v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero F. A.; Du W.; Hwang I.; Rayl T. J.; Kimball F. S.; Leung D.; Hoover H. S.; Apodaca R. L.; Breitenbucher J. G.; Cravatt B. F.; Boger D. L. Potent and Selective α-Ketoheterocycle-Based Inhibitors of the Anandamide and Oleamide Catabolizing Enzyme, Fatty Acid Amide Hydrolase. J. Med. Chem. 2007, 50, 1058–1068. 10.1021/jm0611509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan K. K.; Otrubova K.; Boger D. L. α-Ketoheterocycle inhibitors of fatty acid amide hydrolase: Exploration of conformational constraints in the acyl side chain. Bioorg. Med. Chem. 2014, 22, 2763–2770. 10.1016/j.bmc.2014.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileni M.; Garfunkle J.; DeMartino J. K.; Cravatt B. F.; Boger D. L.; Stevens R. C. Binding and Inactivation Mechanism of a Humanized Fatty Acid Amide Hydrolase by α-Ketoheterocycle Inhibitors Revealed from Cocrystal Structures. J. Am. Chem. Soc. 2009, 131, 10497–10506. 10.1021/ja902694n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimaraes C. R.; Boger D. L.; Jorgensen W. L. Elucidation of Fatty Acid Amide Hydrolase Inhibition by Potent α-Ketoheterocycle Derivatives from Monte Carlo Simulations. J. Am. Chem. Soc. 2005, 127, 17377–17384. 10.1021/ja055438j. [DOI] [PubMed] [Google Scholar]
- Fegley D.; Gaetani S.; Duranti A.; Tontini A.; Mor M.; Tarzia G.; Piomelli D. Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3′-carbamoyl-biphenyl-3-yl ester (URB597): Effects on anandamide and oleoylethanolamide deactivation. J. Pharmacol. Exp. Ther. 2005, 313, 352–358. 10.1124/jpet.104.078980. [DOI] [PubMed] [Google Scholar]
- Mileni M.; Kamtekar S.; Wood D. C.; Benson T. E.; Cravatt B. F.; Stevens R. C. Crystal structure of fatty acid amide hydrolase bound to the carbamate inhibitor URB597: discovery of a deacylating water molecule and insight into enzyme inactivation. J. Mol. Biol. 2010, 400, 743–754. 10.1016/j.jmb.2010.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krustev E.; Reid A.; McDougall J. J. Tapping into the endocannabinoid system to ameliorate acute inflammatory flares and associated pain in mouse knee joints. Arthritis Res. Ther. 2014, 16, 437. 10.1186/s13075-014-0437-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abouabdellah A.; Burnier P.; Hoornaert C.; Jeunesse J.; Puech F.. Piperidinyl- and piperazinyl-alkylcarbamates, their preparation and use as fatty acid amido hydrolase (FAAH) inhibitors for treating FAAH-related pathologies. PCT Int. Appl. 2004099176, 2004.
- Ahn K.; Johnson D. S.; Fitzgerald L. R.; Liimatta M.; Arendse A.; Stevenson T.; Lund E. T.; Nugent R. A.; Nomanbhoy T. K.; Alexander J. P.; Cravatt B. F. Novel Mechanistic Class of Fatty Acid Amide Hydrolase Inhibitors with Remarkable Selectivity. Biochemistry 2007, 46, 13019–13030. 10.1021/bi701378g. [DOI] [PubMed] [Google Scholar]
- Johnson D. S.; Ahn K.; Kesten S.; Lazerwith S. E.; Song Y.; Morris M.; Fay L.; Gregory T.; Stiff C.; Dunbar J. B.; Liimatta M.; Beidler D.; Smith S.; Nomanbhoy T. K.; Cravatt B. F. Benzothiophene piperazine and piperidine urea inhibitors of fatty acid amide hydrolase (FAAH). Bioorg. Med. Chem. Lett. 2009, 19, 2865–2869. 10.1016/j.bmcl.2009.03.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T.; Kori M.; Miyazaki J.; Kiyota Y.. Preparation of piperidinecarboxamides and piperazinecarboxamides as fatty acid amide hydrolase (FAAH) inhibitors. PCT Int. Appl. 2006054652, 2006.
- Keith J. M.; Apodaca R.; Xiao W.; Seierstad M.; Pattabiraman K.; Wu J.; Webb M.; Karbarz M. J.; Brown S.; Wilson S.; Scott B.; Tham C.-S.; Luo L.; Palmer J.; Wennerholm M.; Chaplan S.; Breitenbucher J. G. Thiadiazolopiperazinyl ureas as inhibitors of fatty acid amide hydrolase. Bioorg. Med. Chem. Lett. 2008, 18, 4838–4843. 10.1016/j.bmcl.2008.07.081. [DOI] [PubMed] [Google Scholar]
- Otrubova K.; Cravatt B. F.; Boger D. L. Design, Synthesis, and Characterization of α-Ketoheterocycles That Additionally Target the Cytosolic Port Cys269 of Fatty Acid Amide Hydrolase. J. Med. Chem. 2014, 57, 1079–1089. 10.1021/jm401820q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander J. P.; Cravatt B. F. Mechanism of Carbamate Inactivation of FAAH: Implications for the Design of Covalent Inhibitors and In Vivo Functional Probes for Enzymes. Chem. Biol. 2005, 12, 1179–1187. 10.1016/j.chembiol.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileni M.; Johnson D. S.; Wang Z.; Everdeen D. S.; Liimatta M.; Pabst B.; Bhattacharya K.; Nugent R. A.; Kamtekar S.; Cravatt B. F.; Ahn K.; Stevens R. C. Structure-guided inhibitor design for human FAAH by interspecies active site conversion. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 12820–12824. 10.1073/pnas.0806121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman A. H.; Leung D.; Shelton C. C.; Saghatelian A.; Hardouin C.; Boger D. L.; Cravatt; Benjamin F. Reversible Inhibitors of Fatty Acid Amide Hydrolase That Promote Analgesia: Evidence for an Unprecedented Combination of Potency and Selectivity. J. Pharmacol. Exp. Ther. 2004, 311, 441–448. 10.1124/jpet.104.069401. [DOI] [PubMed] [Google Scholar]
- Piomelli D.; Tarzia G.; Duranti A.; Tontini A.; Mor M.; Compton T. R.; Dasse O.; Monaghan E. P.; Parrott J. A.; Putman D. Pharmacological profile of the selective FAAH inhibitor KDS-4103 (URB597). CNS Drug Rev. 2006, 12, 21–38. 10.1111/j.1527-3458.2006.00021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fegley D.; Gaetani S.; Duranti A.; Tontini A.; Mor M.; Tarzia G.; Piomelli D. Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3′-carbamoyl-biphenyl-3-yl ester (URB597): effects on anandamide and oleoylethanolamide deactivation. J. Pharmacol. Exp. Ther. 2005, 313, 352–358. 10.1124/jpet.104.078980. [DOI] [PubMed] [Google Scholar]
- Kono M.; Matsumoto T.; Imaeda T.; Kawamura T.; Fujimoto S.; Kosugi Y.; Odani T.; Shimizu Y.; Matsui H.; Shimojo M.; Kori M. Design, synthesis, and biological evaluation of a series of piperazine ureas as fatty acid amide hydrolase inhibitors. Bioorg. Med. Chem. 2014, 22, 1468–1478. 10.1016/j.bmc.2013.12.023. [DOI] [PubMed] [Google Scholar]
- Keith J. M.; Apodaca R.; Tichenor M.; Xiao W.; Jones W.; Pierce J.; Seierstad M.; Palmer J.; Webb M.; Karbarz M.; Scott B.; Wilson S.; Luo L.; Wennerholm M.; Chang L.; Brown S.; Rizzolio M.; Rynberg R.; Chaplan S.; Breitenbucher J. G. Aryl Piperazinyl Ureas as Inhibitors of Fatty Acid Amide Hydrolase (FAAH) in Rat, Dog, and Primate. ACS Med. Chem. Lett. 2012, 3, 823–827. 10.1021/ml300186g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tichenor M.; Keith J. M.; Jones W. M.; Pierce J. M.; Merit J.; Hawryluk N.; Seierstad M.; Palmer J. A.; Webb M.; Karbarz M. I.; Wilson S. J.; Wennerholm M. L.; Woestenborghs F.; Beerens D.; Lou L.; Brown S. M.; De Boeck M.; Chaplan S. R.; Breitenbucher J. G. Heteroaryl urea inhibitors of fatty acid amide hydrolase: Structure-mutagenicity relationships for arylamine metabolites. Bioorg. Med. Chem. Lett. 2012, 22, 7357–7362. 10.1016/j.bmcl.2012.10.076. [DOI] [PubMed] [Google Scholar]
- Hill M. N.; Kumar S. A.; Filipski S. B.; Iverson M.; Stuhr K. L.; Keith J. M.; Cravatt B. F.; Hillard C. J.; Chattarji S.; McEwen B. S. Disruption of fatty acid amide hydrolase activity prevents the effects of chronic stress on anxiety and amygdalar microstructure. Mol. Psychiatry 2013, 18, 1125–1135. 10.1038/mp.2012.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G. L.; Winter H.; Arends R.; Jay G. W.; Le V.; Young T.; Huggins J. P. Assessment of the pharmacology and tolerability of PF-04457845, an irreversible inhibitor of fatty acid amide hydrolase-1, in healthy subjects. Br. J. Clin. Pharmacol. 2012, 73, 706–716. 10.1111/j.1365-2125.2011.04137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Sarris K.; Kage K.; Zhang D.; Brown S. P.; Kolasa T.; Surowy C.; El Kouhen O. F.; Muchmore S. W.; Brioni J. D.; Stewart A. O. Synthesis and evaluation of benzothiazole-based analogues as novel, potent, and selective fatty acid amide hydrolase inhibitors. J. Med. Chem. 2009, 52, 170–180. 10.1021/jm801042a. [DOI] [PubMed] [Google Scholar]
- Gustin D. J.; Ma Z.; Min X.; Li Y.; Hedberg C.; Guimaraes C.; Porter A. C.; Lindstrom M.; Lester-Zeiner D.; Xu G.; Carlson T. J.; Xiao S.; Meleza C.; Connors R.; Wang Z.; Kayser F. Identification of potent, noncovalent fatty acid amide hydrolase (FAAH) inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2492–2496. 10.1016/j.bmcl.2011.02.052. [DOI] [PubMed] [Google Scholar]
- Kelly M. G.; Kincaid J.; Gowlugari S.; Kaub C.. Preparation of substituted naphthyridines as FAAH modulators. PCT Int. Appl. 2009, WO 2009011904.
- Keith J. M.; Hawryluk N.; Apodaca R. L.; Chambers A.; Pierce J. M.; Seierstad M.; Palmer J. A.; Webb M.; Karbarz M. J.; Scott B. P.; Wilson S. J.; Luo L.; Wennerholm M. L.; Chang L.; Rizzolio M.; Chaplan S. R.; Breitenbucher J. G. 1-Aryl-2-((6-aryl)pyrimidin-4-yl)amino)ethanols as competitive inhibitors of fatty acid amide hydrolase. Bioorg. Med. Chem. Lett. 2014, 24, 1280–1284. 10.1016/j.bmcl.2014.01.064. [DOI] [PubMed] [Google Scholar]
- Huggins J. P.; Smart T. S.; Langman S.; Taylor L.; Young T. An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain 2012, 153, 1837–1846. 10.1016/j.pain.2012.04.020. [DOI] [PubMed] [Google Scholar]
- Safety and Efficacy of a FAAH-Inhibitor to Treat Cannabis Withdrawal. https://www.clinicaltrials.gov/ct2/show/NCT01618656 (accessed May 2, 2015).
- A Study To Assess Effects Of PF-04457845 On Blood-Oxygen-Level Dependent Functional Magnetic Resonance Imaging In Subjects With Post Traumatic Stress Disorder. https://www.clinicaltrials.gov/ct2/show/NCT02216097 (accessed May 2, 2015).
- Cannabinoid Augmentation of Fear Response in Humans. https://www.clinicaltrials.gov/ct2/show/NCT01665573 (accessed May 2, 2015).
- FAAH Inhibitor Trial for Adults With Tourette Syndromehttps://www.clinicaltrials.gov/ct2/show/NCT02134080 (accessed May 2, 2015).
- A Safety, Tolerability and Efficacy Study of V158866 in Central Neuropathic Pain Following Spinal Cord Injury. https://www.clinicaltrials.gov/ct2/show/NCT01748695 (accessed May 2, 2015).
- An Eight-week Study of SSR411298 as Treatment for Major Depressive Disorder in Elderly Patients (FIDELIO). https://www.clinicaltrials.gov/ct2/show/NCT00822744 (accessed May 2, 2015).
- A Clinical Trial to Assess the Clinical Benefit of SSR411298 as Adjunctive Treatment for Persistent Cancer Pain (ACT11705). https://www.clinicaltrials.gov/ct2/show/NCT01439919 (accessed May 2, 2015).
- Wilson S. J.; Lovenberg T. W.; Barbier A. J. A high-throughput-compatible assay for determining the activity of fatty acid amide hydrolase. Anal. Biochem. 2003, 318, 270–275. 10.1016/S0003-2697(03)00217-3. [DOI] [PubMed] [Google Scholar]
- pKa 1 = 6.47 ± 0.15, pKa 2 = 3.47 ± 0.15; Log P = 3.35 ± 0.15; Log D7.4 = 3.25; two crystalline free-base forms identified, form 1 was a crystalline anhydrate, mp = 126 °C, hygroscopicity (40–90% RH) = +0.44%; form 2 was a crystalline anhydrate, mp = 102 °C, hygroscopicity (40–90% RH) = +3.89%; solubility at pH7.34 = 0.067 mg/mL; at pH1.15 > 100 mg/mL (bis-HCl salt).
- Both (1) and 3-amino-4-chloropyridine were found to be nongene toxic in an Ames II assay.
- I.V. (2 mg/kg as the HCl salt in water, N = 6): CL (mL/min/kg) = 60 ± 9, VSS (L/kg) = 2.5 ± 0.5, T1/2 (h) = 1.1 ± 0.5, AUCinf (h·μg/mL) = 0.6 ± 0.1. P.O. (10 mg/kg as an HPMC suspension, N = 6): Cmax (μg/mL) = 0.5 ± 0.06, AUCinf (h·μg/mL) = 0.92 ± 0.13, Tmax (h) = 0.50 ± 0, %F = 32 ± 4.
- Ueda N.; Kurahashi Y.; Yamamoto K.; Yamamoto S.; Tokunaga T. N-Arachidonoylethanolamine (anandamide), an endogenous cannabinoid receptor ligand, and related lipid molecules in the nervous tissues. J. Lipid Mediators Cell Signalling 1996, 14, 57–61. 10.1016/0929-7855(96)00509-3. [DOI] [PubMed] [Google Scholar]
- A Study to Investigate the Effects of Itraconazole on the Pharmacokinetics of JNJ-42165279 in Healthy Male Participants. https://www.clinicaltrials.gov/ct2/show/NCT02065739 (accessed May 2, 2015).
- Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of JNJ-42165279 in Healthy Young and Elderly Participants. https://www.clinicaltrials.gov/ct2/show/NCT01964651 (accessed May 2, 2015).
- A Study to Investigate the Regional Brain Kinetics of the Positron Emission Tomography Ligand 11C-MK-3168 and the Blocking of the Retention of the Ligand in the Human Brain by JNJ-42165279. https://www.clinicaltrials.gov/ct2/show/NCT02169973 (accessed May 2, 2015).
- A Single Ascending Dose Study to Investigate the Safety, Tolerability, and Pharmacokinetics of JNJ-42165279 in Healthy Male Participants. https://www.clinicaltrials.gov/ct2/show/NCT01650597 (accessed May 2, 2015).
- The Effects of JNJ-42165279 on the Neural Basis of Anxiety Disorders in Healthy Male Volunteers. https://www.clinicaltrials.gov/ct2/show/NCT01826786 (accessed May 2, 2015).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.